

Abstract
Notch signaling plays an emerging role in the regulation of immune cell development and function during inflammatory response. Activation of the ras homolog gene family, member A (RhoA)/Rho-associated protein kinase (ROCK) pathway promotes leukocyte accumulation in tissue injury. However, it remains unknown whether Notch signaling regulates RhoA/ROCK-mediated immune responses in liver ischemia and reperfusion injury (IRI). This study investigated intracellular signaling pathways regulated by Notch receptors in the IR-stressed liver and in vitro. In a mouse model of IR-induced liver inflammatory injury, we found that mice with myeloid specific Notch1 knockout (Notch1M-KO) showed aggravated hepatocellular damage, with increased serum ALT levels, hepatocellular apoptosis, macrophage/neutrophil trafficking, and pro-inflammatory mediators compared to the Notch1-proficient (Notch1FL/FL) controls. Unlike in the Notch1FL/FL controls, myeloid Notch1 ablation diminished hairy and enhancer of split-1 (Hes1) and augmented c-Jun N-terminal kinase (JNK)/stress-activated protein kinase-associated protein 1 (JSAP1), JNK, ROCK1, and PTEN activation in ischemic livers. Disruption of JSAP1 in Notch1M-KO livers improved hepatocellular function and reduced JNK, ROCK1, PTEN, and TLR4 activation. Moreover, ROCK1 knockdown inhibited PTEN and promoted Akt, leading to depressed TLR4. In parallel in vitro studies, transfection of lentivirus-expressing Notch1 intracellular domain (NICD) promoted Hes1 and inhibited JSAP1 in LPS-stimulated bone marrow-derived macrophages (BMMs). Hes1 deletion enhanced JSAP1/JNK activation whereas CRISPR/Cas9-mediated JSAP1 knockout diminished ROCK1/PTEN and TLR4 signaling.
Conclusions
Myeloid Notch1 deficiency activates the RhoA/ROCK pathway and exacerbates hepatocellular injury by inhibiting transcriptional repressor Hes1 and inducing scaffold protein JSAP1 in IR-triggered liver inflammation. Our findings underscore the crucial role of the Notch-Hes1 axis as a novel regulator of innate immunity-mediated inflammation and imply the therapeutic potential for the management of organ IRI in transplant recipients.
Keywords: Innate Immunity, Hes1, JSAP1, PTEN, Liver Injury
Ischemia and reperfusion injury is an innate immunity-dominated local inflammation response. It remains the major cause of organ dysfunction and failure in liver transplantation (1). Innate immune cells and signaling pathways recognize exogenous danger signals such as pathogen-derived molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) that are released from stressed, injured or dying cells (2-4). Macrophages are key components of the innate immune system and contribute to liver inflammatory response (5). We have demonstrated that hepatic IR activates liver macrophages (Kupffer cells) and triggers toll-like receptor 4 (TLR4) or NLRP3-driven inflammation (6-8). Indeed, macrophage activation increases the release of oxygen free radicals and different types of cytokines such as tumor necrosis factor (TNF-α), which triggers the apoptotic pathway leading to the death of hepatocytes (9, 10).
Recent evidence suggests that RhoA/Rho kinase (ROCK) may act as a “molecular switch” in the activation and synthesis of LPS-mediated monocyte proinflammatory response (11). Specific inhibition of the ROCK pathway prevents NF-κB activation and inflammatory response in different inflammatory diseases (12, 13). Activation of RhoA/ROCK signaling increases hepatic stellate cell (HSC) susceptibility in steatotic livers to IR injury (14). Moreover, activation of RhoA downstream effector ROCK might initiate PTEN activity to promote leukocyte migration during inflammation (15). We have shown that PTEN/PI3K signaling plays an important role in the regulation of TLR4-mediated innate immune response in hepatic IRI (6). Thus, RhoA/ROCK activation may be critical for triggering IR-induced liver inflammation.
Notch signaling is highly conserved and critically involved in cell growth, differentiation, and survival (16). Four distinct Notch receptors (Notch 1-4) and five Notch ligands (Jagged1, Jagged2, Delta-like 1 (DLL1), DLL3, and DLL4) have been identified in mammalian cells (17, 18). The interaction between Notch receptors and their ligands leads to two proteolytic cleavage steps by a disintegrin and metalloprotease (ADAM) family proteases and by the intracellular γ-secretase complex that releases the Notch intracellular domain (NICD). The NICD then translocates to the nucleus and binds to the recombinant recognition sequence binding protein at the Jκ site (RBP-J, also named CSL or CBF1), a potent DNA-binding transcription factor that is associated with a large number of chromatin regulators, corepressors, and coactivators (19). This interaction results in the activation of Notch target genes (20). In the immune system, Notch signaling controls the homeostasis of several innate cell populations and regulates immune cell development and function (21). Activation of Notch1 and its ligand Jagged-1 increases cell growth and differentiation during liver regeneration (22). Disruption of the transcription factor RBP-J increases cell apoptosis/necrosis and inflammatory response, leading to aggravated liver injury (23). Although these studies have shown that hepatocellular protection is correlated with Notch signaling during liver inflammation, the molecular mechanisms and crosstalk between transcription targets and signaling pathways of Notch-mediated regulation in liver inflammation remain largely unknown.
Experimental Procedures
Animals
The floxed Notch1 (Notch1FL/FL, catalog number 007181) mice (The Jackson Laboratory, Bar Harbor, ME) and the mice expressing Cre recombinase under the control of the Lysozyme 2 (Lyz2) promoter (LysM-Cre; catalog number 004781, The Jackson Laboratory) were used to generate myeloid-specific Notch1 knockout (Notch1M-KO) mice (Supplementary Figure 3). Two steps were used to generate Notch1M-KO mice. First, a homozygous loxP-flanked Notch1 mouse was mated with a homozygous Lyz2-Cre mouse to generate the F1 mice that were heterozygous for a loxP-flanked Notch1 allele and heterozygous for the Lyz2-Cre. Next, these F1 mice were backcrossed to the homozygous loxP-flanked Notch1 mice, resulting in generation of Notch1M-KO (25% of the offspring), which were homozygous for the loxP-flanked Notch1 allele and heterozygous for the Lyz2-Cre allele (Supplementary Figure 4). This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals published by the NIH. The study protocols were approved by the IACUC of The University of California at Los Angeles, University of Southern California, and Nanjing Medical University in China. See Supplementary Materials.
Mouse liver IRI model and treatment
We used an established mouse model of warm hepatic ischemia followed by reperfusion, as described (24). Some animals were injected via tail vein with JSAP1 siRNAs, ROCK1 siRNA or non-specific (control) siRNA, (2 mg/kg) (Santa Cruz Biotechnology, CA) mixed with mannose-conjugated polymers at a ratio according to the manufacturer's instructions 4h prior to ischemia as described (8). See Supplementary Materials.
Hepatocellular function assay
Serum alanine aminotransferase (sALT) levels, an indicator of hepatocellular injury, were measured by IDEXX Laboratories (Westbrook, ME).
Histology, immunohistochemistry, immunofluorescence staining
Liver sections were stained with hematoxylin and eosin (H&E). The severity of IRI was graded using Suzuki's criteria (25). Liver macrophages and neutrophils were detected using primary rat anti-mouse CD11b+ and Ly6G mAbs for immunofluorescence or immunohistochemistry staining. See Supplementary Materials.
TUNEL assay
The Klenow-FragEL DNA Fragmentation Detection Kit (EMD Chemicals, Gibbstown, NJ) was used to detect the DNA fragmentation characteristic of oncotic necrosis/apoptosis in formalin-fixed paraffin-embedded liver sections (7). See Supplementary Materials.
Caspase-3 activity assay
Caspase-3 activity was performed and determined by an assay kit (Calbiochem, La Jolla, CA) as described (26). See Supplementary Materials.
Quantitative RT-PCR analysis
Quantitative real-time PCR was performed as described (27). Primer sequences used for the amplification are shown in Supplementary Table 1. See Supplementary Materials.
Western blot analysis
Protein was extracted from liver tissue or cell cultures as described (27). The monoclonal rabbit anti-mouse Notch1, NICD, Hes1, p-JNK, JNK, ROCK1, PTEN, p-Akt, Akt, TLR4, cleaved caspase-3, p-IκBα, and β-actin Abs (Cell Signaling Technology, MA) and mouse monoclonal antibody JSAP1 (Santa Cruz Biotechnology) were used. See Supplementary Materials.
Lentiviral vector construction
The pSin-NICD vector, which expresses the NICD that contains EF2 promoter and puromycin gene, was constructed. psPAX2 and pCMV-VSV-G are lentiviral packaging plasmids. The 293T Cells were co-transfected with pSin-NICD, psPAX2, and pCMV-VSV-G using lipofectamine LTX Plus reagent to package lentiviruses according to the manufacturer's instructions. See Supplementary Materials.
The lentiviral CRISPR Hes1 knockout (KO) or JSAP1 KO vector was constructed by the first cloning of Hes1 or JSAP1 single guide RNA (sgRNA) sequences into the site of BsmBI of LentiCRISPRv2 vector as described (28). Lentiviral vectors were produced as described above. The Lenti-CRISPRv2-Hes1 KO (LV-Hes1 KO) or Lenti-CRISPRv2-JSAP1 KO (LV-JSAP1 KO), psPAX2, and pCMV-VSV-G were used for packaging viruses. Lenti-CRISPRv2 without gRNA virus was used as a control. See Supplementary Materials.
Isolation of hepatocyte and liver macrophages
Primary hepatocytes and liver macrophages (Kupffer cells) from Notch1FL/FL and Notch1M-KO mice were isolated, as described (27, 29). The purity of macrophages in ischemic livers was 80% as assessed by immunofluorescence staining for CD11b+. See Supplementary Materials.
BMM isolation and in vitro transfection
Murine bone-derived macrophages (BMMs) were generated, as described (26). Cells (1×106/well) were cultured for 7 days and then transduced with lentivirus-expressing NICD, CRISPR/Cas9-Hes1 KO, CRISPR/Cas9-JSAP1 KO or control vector. See Supplementary Materials.
ELISA assay
Murine serum and BMM culture supernatants were harvested for cytokine analysis. ELISA kits were used to measure TNF-α, IL-1β, MCP-1, and IL-6 levels. See Supplementary Materials.
Reactive oxygen species (ROS) assay
ROS production in BMMs was measured using the Carboxy-H2DFFDA kit as described (8). ROS produced by BMMs were analyzed and quantified by fluorescence microscopy according to the manufacturer's instructions. See Supplementary Materials.
Statistical analysis
Data are expressed as mean±SD and analyzed by Permutation t-test and Pearson correlation. Per comparison two-sided p values less than 0.05 were considered statistically significant. Multiple group comparisons were made using one-way ANOVA followed by Bonferroni's post hoc test. When groups showed unequal variances, we applied Welch's ANOVA to make multiple group comparisons. All analyses were used by SAS/STAT software, version 9.4.
Results
Myeloid-specific Notch1 deficiency aggravates IR-induced hepatocellular damage
The myeloid-specific Notch1-deficient (Notch1M-KO) and Notch1-proficient (Notch1FL/FL) mice were subjected to 90min of warm ischemia followed by 6h or 24h of reperfusion. We isolated both hepatocytes and liver macrophages (Kupffer cells) from these ischemic livers. Indeed, Notch1M-KO did not change hepatocyte Notch1 expression. However, the Notch1 expression was lacking in liver macrophages from the Notch1M-KO mice but not from the Notch1FL/FL mice (Figure 1A). Hepatocellular functions were evaluated by measuring the serum ALT (sALT) levels (IU/L) (Figure 1B). Disruption of myeloid Notch1 increased sALT levels at 6h and 24h post liver reperfusion in the Notch1M-KO mice compared to the Notch1FL/FL controls (6h, 10824±1473 vs. 5911±1109, respectively; p<0.01; 24h, 3733±469 vs. 1555±530, respectively; p<0.01). These data correlated with Suzuki's histological grading of liver IRI (Figure 1C). Unlike the Notch1FL/FL controls, which showed mild to moderate edema, sinusoidal congestion, and mild necrosis (6h, score=2.2±0.25; 24h, score=1.6±0.21), the Notch1M-KO mouse livers displayed severe edema, sinusoidal congestion, and extensive hepatocellular necrosis (6h, score=3.62±1.23, p<0.01; 24h, score=2.45±0.38, p<0.05). Consistent with the histopathological and hepatocellular function data, the MPO levels, which reflect liver neutrophil activity (U/g), were significantly elevated in the Notch1M-KO group but not in the Notch1FL/FL group (Figure 1D, 6h, 3.93±0.72 vs. 1.92±0.67, respectively; p<0.05; 24h, 3.03±0.42 vs. 1.65±0.22, respectively; p<0.01).
Figure 1. Myeloid-specific Notch1 deficiency aggravates IR-induced hepatocellular damage.
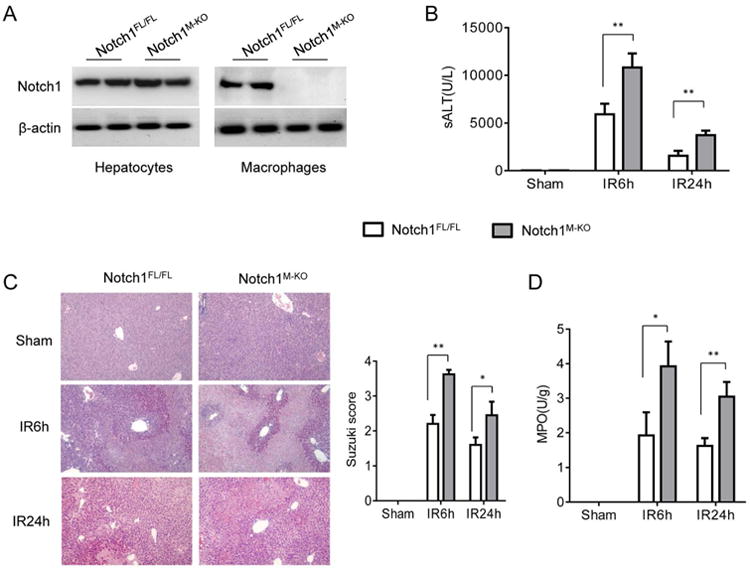
Mice were subjected to 90min of partial liver warm ischemia, followed by 6h or 24h of reperfusion. (A) The Notch1 expression was detected in hepatocytes and liver macrophages by Western blot assay. Representative of three experiments. (B) Liver function in serum samples was evaluated by sALT levels (IU/L). Results expressed as mean±SD (n=4-6 samples/group). **p<0.01. (C) Representative histological staining (H&E) of ischemic liver tissue. Results representative of 4-6 mice/group; original magnification ×100. Liver damage, evaluated by Suzuki's histological score. **p<0.01. (D) Liver neutrophil accumulation, analyzed by MPO activity (U/g). Mean±SD (n=4-6 samples/group). *p<0.05.
Myeloid-specific Notch1 deficiency increases macrophage/neutrophil infiltration and proinflammatory mediators in liver IRI
Having shown that myeloid Notch1 deficiency enhances hepatocellular damage, we then analyzed macrophage and neutrophil accumulation in IR-stressed livers at 6h of reperfusion by immunofluorescence or immunohistochemistry staining. The Notch1M-KO ischemic livers showed increased CD11b+ macrophages infiltration compared to the Notch1FL/FL controls (Figure 2A, 205±60 vs. 75±19, p<0.05). To evaluate the phenotype of macrophages, liver macrophages (Kupffer cells) were isolated from Notch1M-KO and Notch1FL/FL livers at 6h of reperfusion after 90min of ischemia. Notch1-deficient macrophages from Notch1M-KO mice increased S100A9 while reducing arginase-1 (Arg-1) and CD206 expression compared to the Notch1-proficient macrophages from Notch1FL/FL mice (Supplementary Figure 1A). Consistent with this data, the mRNA levels coding for TNF-α, IL-1β, and MCP1 were significantly increased in the Notch1M-KO but not in the Notch1FL/FL livers (Figure 2B). Moreover, the Notch1M-KO ischemic livers exhibited increased neutrophil accumulation compared to the Notch1FL/FL controls (Figure 2C, 136±47 vs. 30.0±8.16, p<0.01).
Figure 2. Myeloid-specific Notch1 deficiency increases macrophage/neutrophil infiltration and proinflammatory mediators in liver IRI.
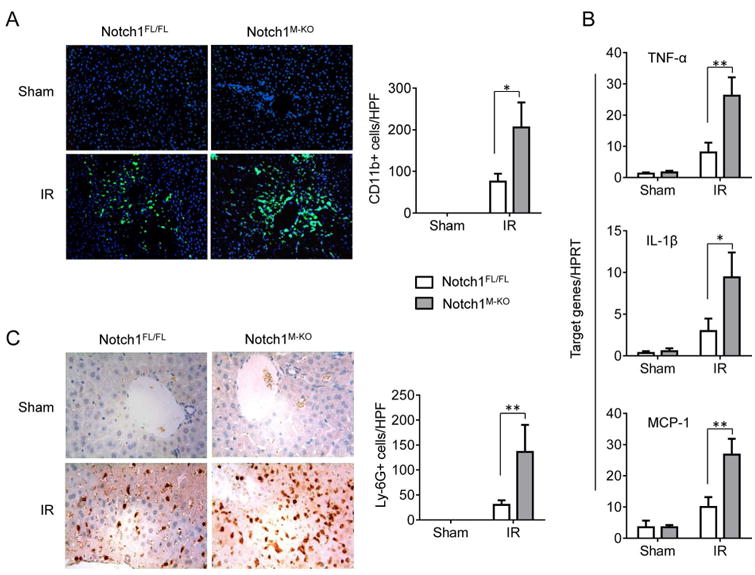
By 6h of reperfusion after 90min of ischemia, liver macrophages and neutrophils were detected by immunofluorescence and immunohistochemistry staining using mAbs against mouse CD11b+ and Ly6G in Notch1FL/FL (
 ) and Notch1M-KO (
) and Notch1M-KO (
 ) mice. (A) Immunofluorescence staining of CD11b+ macrophages in ischemic livers. Quantification of CD11b+ macrophages per high power field. Results scored semi-quantitatively by averaging number of positively-stained cells (mean±SD)/field at 200×magnification. Representative of 4-6 mice/group. *p<0.05. (B) Quantitative RT-PCR-assisted detection of TNF-α, IL-1β, and MCP-1 in mouse livers. Each column represents the mean±SD (n=3-4 samples/group). *p<0.05, **p<0.01. (C) Immunohistochemistry staining of Ly6G+ neutrophils in ischemic livers. Quantification of Ly6G+ neutrophils per high power field (original magnification ×200). Representative of 4-6 mice/group. **p<0.01.
) mice. (A) Immunofluorescence staining of CD11b+ macrophages in ischemic livers. Quantification of CD11b+ macrophages per high power field. Results scored semi-quantitatively by averaging number of positively-stained cells (mean±SD)/field at 200×magnification. Representative of 4-6 mice/group. *p<0.05. (B) Quantitative RT-PCR-assisted detection of TNF-α, IL-1β, and MCP-1 in mouse livers. Each column represents the mean±SD (n=3-4 samples/group). *p<0.05, **p<0.01. (C) Immunohistochemistry staining of Ly6G+ neutrophils in ischemic livers. Quantification of Ly6G+ neutrophils per high power field (original magnification ×200). Representative of 4-6 mice/group. **p<0.01.
Myeloid-specific Notch1 deficiency depresses Hes1 but induces RhoA/ROCK activation in IR-stressed liver
Next, we analyzed whether Notch1 may influence its target gene Hes1 and RhoA/ROCK pathway in IR-induced liver injury. By 6h of reperfusion after 90min of ischemia, Notch1M-KO diminished Hes1 and increased RhoA mRNA expression in the ischemic livers compared to the Notch1FL/FL controls (Figure 3A). The Western blotting analysis revealed reduced NICD and Hes1, while augmented JSAP1, p-JNK, and ROCK1 protein levels in the Notch1M-KO but not in the Notch1FL/FL livers (Figure 3B). Moreover, Notch1M-KO increased IL-1β, MCP-1, and IL-6 production compared to the Notch1FL/FL controls after hepatic IR (Figure 3C).
Figure 3. Myeloid-specific Notch1 deficiency depresses Hes1 but induces RhoA/ROCK activation in IR-stressed liver.
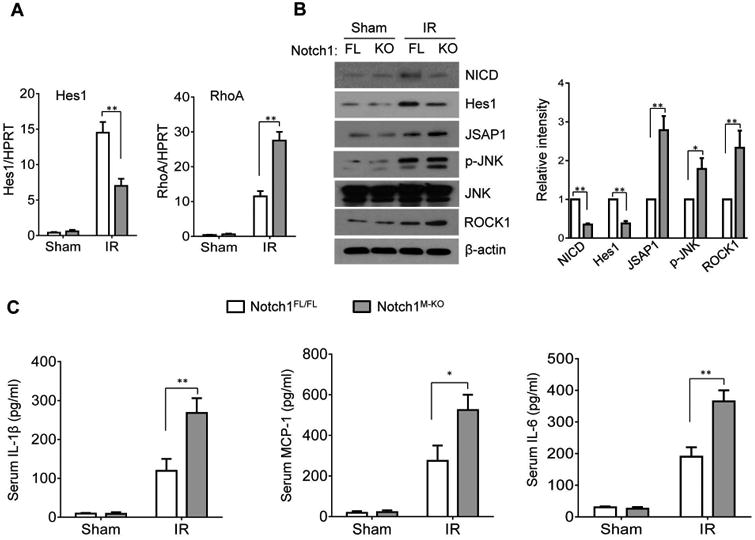
(A) Quantitative RT-PCR-assisted detection of mRNA coding for Hes1 and RhoA in mouse livers at 6h of reperfusion followed by 90min of ischemia. Each column represents the mean±SD (n=3-4 samples/group). **p<0.01. (B) Western-assisted analysis and relative density ratio of NICD, Hes1, JSAP1, p-JNK, and ROCK1. Representative of three experiments. *p<0.05, **p<0.01. (C) ELISA analysis of IL-1β, MCP-1, and IL-6 levels in animal serum. Mean±SD (n=3-4 samples/group), *p<0.05, **p<0.01.
Myeloid-specific Notch1 deficiency increases hepatocellular apoptosis in IR-stressed liver
To determine whether disruption of myeloid Notch1 may affect hepatic IR-induced apoptosis, we analyzed the hepatocellular apoptosis/necrosis in ischemic livers by TUNEL staining. By 6h of reperfusion after 90min of ischemia, livers in Notch1M-KO revealed an increased frequency of apoptotic TUNEL+ cells compared to the Notch1FL/FL livers after IR (Figure 4A, 52.5±17.7 vs. 25±7, p<0.05). This data was confirmed by increased caspase-3 activity in the Notch1M-KO mice, compared to the Notch1FL/FL mice (Figure 4B: 8.25±1.8 vs. 4.1±1.41, p<0.01). Western blot analysis showed that Notch1M-KO upregulated PTEN and TLR4 expression but downregulated Akt phosphorylation compared to the Notch1FL/FL controls (Figure 4C). Moreover, increased TNF-α production was observed in the Notch1M-KO mice but not in the Notch1FL/FL mice (Figigure 4D, 128.8±19.6 vs. 67.4±12.1, p<0.01).
Figure 4. Myeloid-specific Notch1 deficiency increases hepatocellular apoptosis in IR-stressed liver.
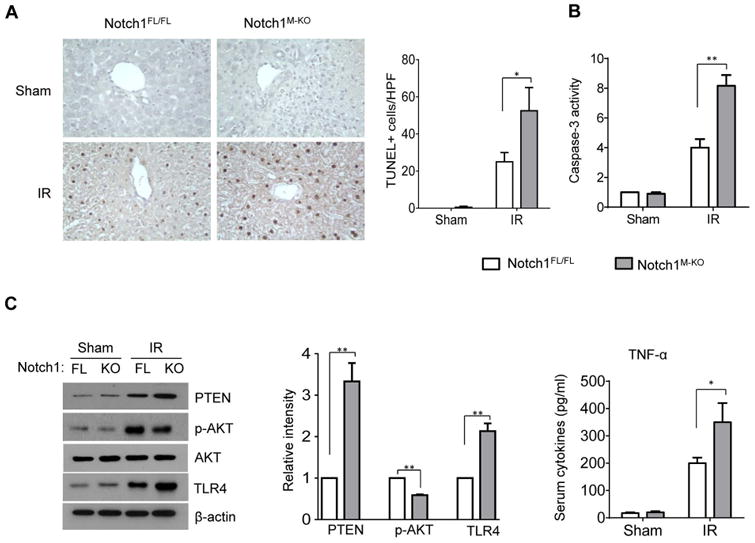
(A) Liver apoptosis by TUNEL staining in mouse liver at 6h of reperfusion followed by 90min of ischemia. Results scored semi-quantitatively by averaging the number of apoptotic cells (mean±SD) per field at 200× magnification. Representative of 4-6 mice/group, *p<0.05. (B) Caspase-3 activity. Mean±SD; n=4-6 samples/group. **p<0.01. (C) Western-assisted analysis and relative density ratio of PTEN, p-Akt, and TLR4 Representative of three experiments. **p<0.01. (C) ELISA analysis of TNF-α levels in animal serum. Mean±SD (n=3-4 samples/group), **p<0.01.
Activation of RhoA/ROCK pathway triggers innate immune response and IR-induced inflammatory injury in myeloid Notch1-deficient liver
To evaluate whether the activation of RhoA/ROCK pathway in the Notch1M-KO livers may trigger liver innate immune response and IR-induced inflammatory injury, we disrupted ROCK1 in the Notch1M-KO livers with an in vivo mannose-mediated ROCK1 siRNA delivery system that specifically deliver to macrophages by expressing a mannose-specific membrane receptor as previously described (8, 30). The mannose receptor is a C-type lectin primarily present on the surface of macrophages. Indeed, knockdown of ROCK1 with the mannose-mediated siRNA treatment in the Notch1M-KO mice reduced IR-induced liver damage as evidenced by the decreased Suzuki's histological score (Figure 5A, score=1.475±0.55 vs. 3.275±0.88, p<0.05) and sALT levels (Figure 5B, 4875.5±1444.9 vs. 9310.3±1982, p<0.01) compared to the non-specific (NS) siRNA-treated controls. Moreover, ROCK1 siRNA treatment in the Notch1M-KO ischemic livers decreased CD11b+ macrophages (Figure 5C, 52.3±22.6 vs. 180.7±37.1, p<0.01) and neutrophil (Figure 5D, 40.67±10.2 vs. 120±30.3, p<0.05) accumulation compared to the NS siRNA-treated controls. Moreover, liver macrophages from ROCK1 siRNA treated-Notch1M-KO mice diminished S100A9 and increased Arg-1 and CD206 expression compared to the NS siRNA-treated controls at 6h of reperfusion after 90min of ischemia (Supplementary Figure 1B). Consistent with these data, ROCK1 knockdown reduced PTEN and TLR4, and increased Akt expression in the Notch1M-KO mice (Figure 5E), which was accompanied by reduced liver TNF-α, IL-1β and MCP-1 mRNA levels compared to the controls (Figure 5F).
Figure 5. Activation of RhoA/ROCK pathway triggers innate immune response and IR-induced inflammatory injury in myeloid Notch1-deficient liver.
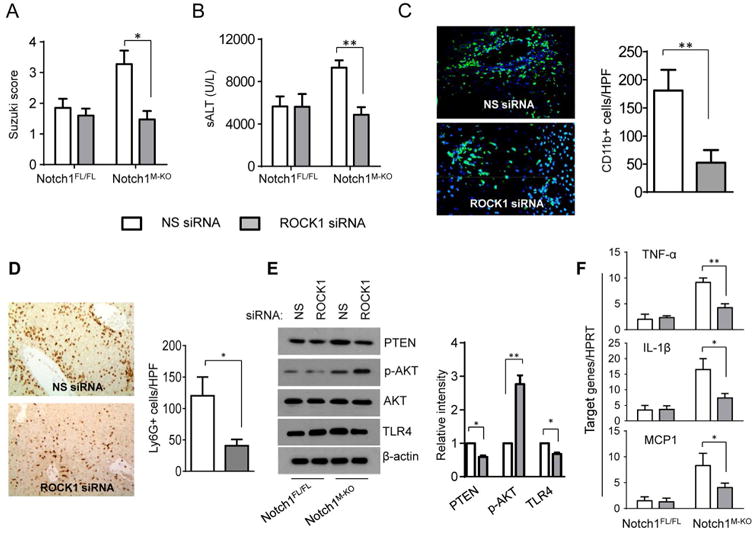
The Notch1M-KO and Notch1FL/FL mice were injected via tail vein with nonspecific (NS) control siRNAs (
) or ROCK1 siRNA (
) (2 mg/kg) mixed with mannose-conjugated polymers at 4h prior to ischemia. (A) The severity of liver IRI was evaluated by the Suzuki's histological grading at 6h of reperfusion followed by 90min of ischemia. *p<0.05. (B) Hepatocellular function was evaluated by sALT levels (IU/L). Results expressed as mean±SD (n=4-6 samples/group). **p<0.01. (C) Immunofluorescence staining of CD11b+ macrophages in ischemic livers. Quantification of CD11b+ macrophages per high power field. Results scored semi-quantitatively by averaging number of positively-stained cells (mean±SD)/field at 200×magnification. Representative of 4-6 mice/group. **p<0.01. (D) Immunohistochemistry staining of Ly6G+ neutrophils in ischemic livers. Quantification of Ly6G+ neutrophils per high power field (original magnification ×200). Representative of 4-6 mice/group. *p<0.05. (E) Western blots analysis and relative density ratio of PTEN, p-Akt, and TLR4. Representative of three experiments. *p<0.05, **p<0.01. (F) Quantitative RT-PCR-assisted detection of mRNA coding for TNF-α, IL-1β, and MCP-1. Each column represents the mean±SD (n=3-4 samples/group). *p<0.05, **p<0.01.
Myeloid-specific Notch1 deficiency activates RhoA/ROCK pathway via a JSAP1-dependent manner in IR-stressed liver
Because myeloid-specific Notch1 deficiency promoted JSAP1, which is a JNK-binding protein and functions as a scaffold factor in the JNK signaling pathway (31), we examined whether JSAP1 is required for activation of RhoA/ROCK pathway in IR-stressed livers. We disrupted JSAP1 in Notch1M-KO livers using a mannose-mediated JSAP1 siRNA in vivo. Livers in the Notch1M-KO mice with non-specific (NS) siRNA treatment revealed significant edema, severe sinusoidal congestion/cytoplasmic vacuolization, and extensive (30-50%) necrosis (Figure 6A, score=3.67±1.5). In contrast, the livers in the mice treated with mannose-mediated JSAP1 siRNA showed mild to moderate edema without necrosis (Figure 6A, score=2.11±0.27, p<0.01). Consistent with these data, the sALT levels were significantly decreased in the JSAP1 siRNA-knockdown mice compared to the NS siRNA-treated controls (Figure 6B, 5375±843 vs. 11060±1473, p<0.01). Moreover, JSAP1 siRNA treatment in the Notch1M-KO livers reduced serum TNF-α release (Figure 6C, 210±74 vs. 450±91, p<0.01) and p-JNK, ROCK1, PTEN, TLR4 and cleaved caspase-3 protein expression (Figure 6D), which led to decreased RhoA, IL-1β, and MCP1 mRNA expression compared to the NS siRNA-treated group (Figure 6E).
Figure 6. Myeloid-specific Notch1 deficiency activates RhoA/ROCK pathway via a JSAP1-dependent manner in IR-stressed livers.
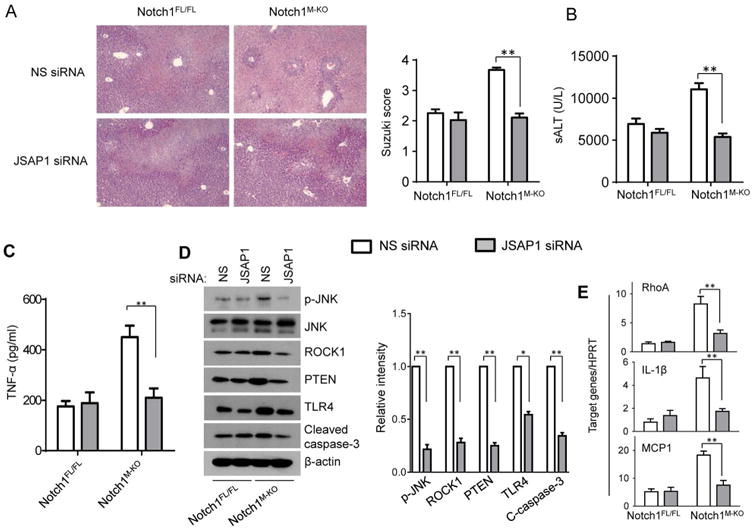
The Notch1M-KO and Notch1FL/FL mice were injected via tail vein with nonspecific (NS) control siRNAs (
) or JSAP1 siRNA (
) (2 mg/kg) mixed with mannose-conjugated polymers at 4h prior to ischemia. (A) Representative histological staining (H&E) of ischemic liver tissue at 6h of reperfusion followed by 90min of ischemia. Results representative of 4-6 mice/group; original magnification ×100. The severity of liver IRI was evaluated by the Suzuki's histological grading. **p<0.01. (B) Hepatocellular function was evaluated by sALT levels (IU/L). Results expressed as mean±SD (n=4-6 samples/group). **p<0.01. (C) ELISA analysis of TNF-α levels in animal serum. Mean±SD (n=3-4 samples/group), **p<0.01. (D) Western blots analysis and relative density ratio of p-JNK, ROCK1, PTEN, TLR4, and cleaved caspase-3. Representative of three experiments. *p<0.05, **p<0.01. (E) Quantitative RT-PCR-assisted detection of mRNA coding for RhoA, IL-1β, and MCP-1. Each column represents the mean±SD (n=3-4 samples/group). **p<0.01.
Myeloid Notch1-Hes1 axis is crucial in the regulation of JSAP1-dependent RhoA/ROCK activation in macrophages
To elucidate the mechanisms of Notch signaling in regulating RhoA/ROCK-mediated immune response, we cultured BMMs from Notch1M-KO mice and then transfected them with the lentivirus expressing NICD (LV-pSIN-NICD) or the control vector (LV-control) followed by LPS stimulation. Clearly, LV-pSin-NICD transfection in the Notch1M-KO cells markedly increased Hes1 and reduced JSAP1 expression compared to the LV-control-transfected cells (Figure 7A). In contrast to the LV-controls, transfection of lentivirus CRISPR/Cas9-mediated Hes1 knockout (LV-Hes1 KO) enhanced JSAP1, ROCK1, and PTEN (Figure 7B), resulting in augmented TNF-α release (Figure 7C) and IL-1β, and MCP-1 expression (Figure 7D) in LPS-stimulated Notch1FL/FL macrophages. To determine the crosstalk between JSAP1 and RhoA/ROCK activation in Notch signaling-mediated immune regulation, we disrupted JSAP1 by using a CRISPR/Cas9 JSAP1 knockout vector (LV-JSAP1 KO) in Notch1M-KO macrophages. Interestingly, JSAP1 deficiency in LV-JSAP1 KO-treated cells led to decreased ROCK1, PTEN, TLR4, and p-IκBα (Figure 7E), which were accompanied by reduced ROS production (Figure 7F, 51.5±9.82 vs. 265.0±40.4, p<0.01) and mRNA levels coding for TNF-α, IL-1β, and MCP-1 (Figure 7G) in LPS-stimulated macrophages, as compared with the control groups.
Figure 7. Myeloid Notch1-Hes1 axis is crucial in the regulation of JSAP1-dependent RhoA/ROCK activation in macrophages.

(A) BMMs from Notch1M-KO mice were transfected with the lentivirus expressing NICD (LV-pSIN-NICD) or the control vector (LV-control) followed by LPS (100 ng/ml) stimulation. Western-assisted analysis and relative density ratio of Hes1, and JSAP1. Representative of three experiments. **p<0.01. (B) BMMs from Notch1FL/FL mice were transfected with the lentiviral-mediated CRISPR/Cas9-mediated Hes1 knockout (LV-Hes1 KO) or the LentiCRISPRv2 vector without gRNA sequence control (LV-control) followed by LPS (100 ng/ml) stimulation. Western-assisted analysis and relative density ratio of Hes1, JSAP1, ROCK1, and PTEN. Representative of three experiments. **p<0.01. (C) ELISA-assisted production of TNF-α in cell culture supernatants. Mean±SD (n=3-4 samples/group). **p< 0.01. (D) Quantitative RT-PCR-assisted detection of mRNA coding for IL-1β and MCP-1. Each column represents mean±SD (n=3-4 samples/group). *p<0.05. (E) BMMs from Notch1M-KO mice were transfected with the lentiviral-mediated CRISPR/Cas9-mediated JSAP1 knockout (LV-JSAP1 KO) or the LentiCRISPRv2 vector without gRNA sequence control (LV-control) followed by LPS (100 ng/ml) stimulation. Western-assisted analysis and relative density ratio of JSAP1, ROCK1, PTEN, TLR4, and p-IκBα. Representative of three experiments. **p<0.01. (F) ROS production was detected by Carboxy-H2DFFDA in LPS-stimulated BMMs from Notch1M-KO mice. Positive green fluorescent-labeled cells were counted blindly in 10 HPF/section (×200). Quantification of ROS-producing BMMs (green) per high power field (×200). **p<0.01. (G) Quantitative RT-PCR-assisted detection of mRNA coding for TNF-α, IL-1β and MCP-1. Each column represents mean±SD (n=3-4 samples/group). *p<0.05, **p<0.01. (
) Cells only; (
 ) LV-pSin-NICD; (
) LV-pSin-NICD; (
 ) LPS + LV-Hes1 KO; (
) LPS + LV-Hes1 KO; (
 ) LPS + LV-control; (
) LPS + LV-control; (
 ) LPS + LV-JSAP1 KO.
) LPS + LV-JSAP1 KO.
Discussion
This study is the first to document the key role of myeloid Notch signaling in regulating RhoA/ROCK-mediated innate immune responses in sterile inflammatory liver injury. First, myeloid Notch1 deficiency promotes liver inflammation through the depression of its target gene Hes1. Second, inhibition of Hes1 induces the scaffold protein JSAP1, which is required for the activation of the RhoA/ROCK pathway. Third, activated RhoA downstream effector ROCK1 is crucial for triggering the TLR4-driven inflammatory response. Our results highlight the importance of the myeloid Notch-Hes1 axis as a key regulator of the RhoA/ROCK function in IR-triggered liver inflammation.
Notch signaling has varied roles in regulating inflammatory response and tissue homeostasis. Under inflammatory conditions, it is conceivable that Notch signaling in myeloid cells could be promoted by various stimuli, such as exogenous pathogens and/or endogenous mediators. Notch signaling can be activated through the TLR signaling cascade, which is involved in proinflammatory response (32). However, Notch1 signaling exerts an immunoregulatory effect by inducing regulatory T cell (Treg) production both in vitro and in vivo (33, 34). Overexpression of Notch intracellular domain (NICD) reduced TLR4-mediated proinflammatory cytokine production in vitro (35). Notch and TLR pathways cooperate to activate canonical Notch target genes, including transcriptional repressor Hes1, which can regulate proinflammatory cytokines via an inhibitory feedback loop (36). The suppressive function of Hes1 in inflammatory response is associated with transcription regulation (37). Thus, Notch signaling serves as a dual functional regulator of inflammatory response in various animal models. In our current study, we analyzed the myeloid specific Notch1 function in mediating its immunomodulation on RhoA/ROCK activation during liver IRI. We found that myeloid Notch1 deficiency promoted JNK binding protein JSAP1, RhoA/ROCK and PTEN activation by inhibiting Hes1 expression, which led to increased IR-triggered liver inflammation. Our findings demonstrate the ability of myeloid Notch signaling in modulating innate immunity and inflammation cascades in IR-stressed livers.
Attenuation of RhoA/ROCK activation by inhibiting Rho kinase has indicated that the RhoA/ROCK pathway is an important mediator during T cell-mediated inflammatory response (38). Inhibition of Rho kinase prevents NF-κB activation and proinflammatory cytokine production in intestinal inflammation (12). However, it is unknown how myeloid Notch signaling influences the RhoA/ROCK-mediated innate immune response in IR-induced liver injury. Based on the findings from the present study, the transcriptional repressor Hes1 is a key determinant of Notch signaling-mediated immune regulation. We found that induction of Hes1 by NICD overexpression selectively inhibited the expression of RhoA downstream effector ROCK1, resulting in reduced PTEN and increased Akt, which in turn regulated TLR4 signaling via a negative feedback mechanism (6). Notably, CRISPR/Cas9-mediated Hes1 knockout activated JSAP1, whereas knockdown of JSAP1 reduced JNK phosphorylation, inflammatory cytokine expression, and caspase-3 activation. In addition, although Notch signaling can also be activated in liver macrophages after liver IR, disruption of myeloid Notch1 or macrophage Hes1 promoted RhoA/ROCK pathway both in vitro and in vivo. These results suggest the important regulatory role of the myeloid Notch1-Hes1 axis on RhoA/ROCK function during liver IRI.
One striking finding was that RhoA/ROCK activation was inhibited by disrupting JSAP1. JSAP1 did not interrupt myeloid Notch-Hes1 signaling (data not shown) but instead triggered RhoA/ROCK activation. Because RhoA/ROCK-mediated PTEN activity was required for inflammatory cell accumulation (15) and myeloid PTEN promoted tissue inflammation (39), suppression of the RhoA/ROCK pathway may provide a new mechanism by which Notch1-Hes1 signaling modulates TLR responses by specifically suppressing JSAP1 activation in IR-stressed livers. Furthermore, our data demonstrated the ability of JSAP1 to mediate JNK activation and increase TLR-induced production of MCP1. Indeed, inflammatory cell migration is critical for tissue inflammation. During liver IRI, the infiltration of monocytes/macrophages plays an important role in the initiation of local inflammatory injury (40). MCP-1 is a potent chemoattractant for monocytes/macrophages and has been shown to be involved in macrophage recruitment in acute liver injury (41). We found that myeloid Notch1 deficiency increased macrophage infiltration whereas ROCK1 disruption blunted macrophage infiltration and MCP-1 expression in ischemic livers. This suggests that inhibition of macrophage accumulation was due to suppression of MCP-1. In addition, ROCK1 knockdown significantly reduced the gene expression of TNF-α and IL-1β in myeloid Notch1-deficent livers. These data indicate that activation of the JSAP1-mediated RhoA/ROCK pathway may be crucial for macrophage accumulation in IR-triggered liver inflammation and inhibition of this cell-specific signaling pathway may provide a possible targeting strategy.
Although inhibition of the RhoA/ROCK pathway ameliorates liver injury, the role of RhoA/ROCK in mediating innate immune response is less clear. Previous studies have reported that Rho kinase is involved in the activation of the NF-κB pathway (42, 43). Our results showed that JSAP1 was involved in ROCK-dependent TLR4 activation during inflammatory response and that utilization of the CRISPR/Cas9-mediated JSAP1 knockout can inhibit RhoA/ROCK-mediated TLR4 signaling, which was accompanied by decreased NF-κB-induced inflammatory cytokines and chemokines. The involvement of JSAP1 and RhoA/ROCK in TLR4 activation indicated the existence of mechanistic links between JSAP1-mediated RhoA/ROCK pathway and innate immunity during liver IRI. Further evidence revealed that disruption of ROCK1 suppressed PTEN and TLR4 activation, resulting in reduced liver inflammatory injury. The inhibitory effect of ROCK1 on TLR4 activation is associated with increased Akt activity. Although there could be many causes for TLR4 activation, it is clear that the JSAP1-mediated RhoA/ROCK pathway is an important mechanism for activating TLR4 in liver IRI. This was further supported by the in vitro study, which showed that reduced TLR4 expression occurred after JSAP1 knockout in Notch1-deficient macrophages. Thus, our findings reveal an essential role for JSAP1 in activating the RhoA/ROCK pathway and TLR4 signaling during liver inflammation.
It is worth noting that the myeloid Notch-Hes1 axis, identified here as likely novel players in cytoprotection, could be involved in apoptotic pathways during liver IRI. Indeed, both programmed cell death (apoptosis) and necrosis are known to occur following liver IR. Previous studies suggest that the Rho/ROCK pathway is activated during the execution phase of apoptosis to stimulate apoptotic membrane blebbing (44). ROCK1 is a direct target of caspase activity, whereby caspase 3 cleavage of ROCK1 occurs in early apoptosis (45). ROCK1 cleavage also increases caspase-3 activity, which coincides with the activation of PTEN and the subsequent inhibition of Akt activity (45). Consistent with this report, we examined apoptotic liver cell death by TUNEL staining. The percentage of apoptotic TUNEL+ cells was markedly increased in the Notch1M-KO livers but not in the Notch1FL/FL livers after IR. Further evidence was supported by caspase-3 activity assay, which showed that Notch1M-KO significantly increased caspase-3 activity in ischemic livers compared to the Notch1FL/FL controls. Our findings demonstrate an unexpected role for the myeloid Notch-Hes1 axis in negatively modulating IR-induced hepatocellular apoptosis/necrosis. The regulation of apoptosis/necrosis by myeloid Notch signaling in ischemic livers may depend on several factors. First, macrophage Notch1 deficiency increases TNF-α release, which leads to increased hepatocellular apoptosis via a JNK-dependent pathway. Because JSAP1 is a scaffold protein that interacts with specific components of the JNK signaling pathway (31), JSAP1 appears to be involved in the TNF-α-mediated apoptosis in liver IRI. Indeed, knockdown of JSAP1 inhibited JNK activation and reduced pro-apoptotic caspase-3 expression, which was accompanied by decreased TNF-α release in Notch1M-KO livers. This result suggests that JSAP1 may represent a key target in strategies to limit cell apoptosis. Second, ROS production and oxidant stress are the most invoked disease mechanisms in liver IRI. ROS formation by Kupffer cells initiates cellular injury and activates a cascade of mediators leading to increased apoptosis/necrosis and acute inflammatory response (46). Rho GTPases are key components of activated NADPH oxidase (NOX) complexes and the subsequent generation of ROS (47). Increased ROS production activates the RhoA/ROCK pathway (48). Thus, a crosstalk between ROS and Rho/ROCK signaling plays a pivotal role in activating apoptosis. Our results showed that increased NICD expression promoted Hes1 and inhibited JSAP1, whereas disruption of Hes1 augmented JSAP1 and ROCK1 with increased TNF-α release from macrophages. This data was strengthened by our observations of in vivo mannose-mediated JSAP1 knockdown or in vitro CRISPR/Cas9-mediated JSAP1 knockout, which showed a reduction of ROCK1, cleaved caspase-3, and macrophage ROS production. Taken together, these findings suggest that the anti-apoptotic effect of myeloid Notch-Hes1 signaling is likely through regulation of JSAP1-mediated ROCK signaling in liver IRI.
Another important implication of our results is that myeloid Notch1 deficiency may result in cholangiocyte injury, as evidenced by increased serum levels of alkaline phosphatase (ALP) and direct bilirubin (DBIL) after liver IRI (Supplementary Figure 2). It is known that Notch and Wnt signaling are required for hepatic progenitor cell (HPC) differentiation and proliferation (49, 50). HPCs are activated and able to differentiate into hepatic parenchymal cells, hepatocytes, and/or bile ductular epithelial cells after liver IRI. In the injured liver, different cell types such as hepatocytes, cholangiocytes, endothelial cells, macrophages and other inflammatory cells can potentially interact with HPCs. Macrophages are important cell components of the HPC niche in stimulating and initiating a liver regenerative response (49). Hence, we speculate that macrophage Notch signaling may be involved in HPC-mediated regeneration in response to IR-induced liver damage.
Figure 8 depicts putative molecular mechanisms by which myeloid Notch1 signaling may regulate RhoA/ROCK-mediated innate immune response in liver IRI. Notch1 can be activated in IR-stressed livers. Upon ligand binding, Notch1 is cleaved by γ-secretase, releasing the intracellular domain (NICD), which translocates into the nucleus and forms a complex with the CSL DNA-binding protein (known as RBPJ in mouse), and activates its target gene Hes1. Induction of Hes1 inhibits JNK binding protein JSAP1-mediated ROCK1 activation. Blockade of ROCK1 reduces PTEN and augments Akt activity, leading to suppressed TLR4 signaling in liver IRI. Moreover, the Notch-Hes1 axis inhibits JSAP1-dependent ROCK1 and caspase-3 activity, resulting in reduced hepatocellular apoptosis/necrosis in IR-triggered liver inflammation.
Figure 8. Schematic illustration of myeloid Notch1 signaling in the regulation of innate immune response in IR-triggered liver inflammation.
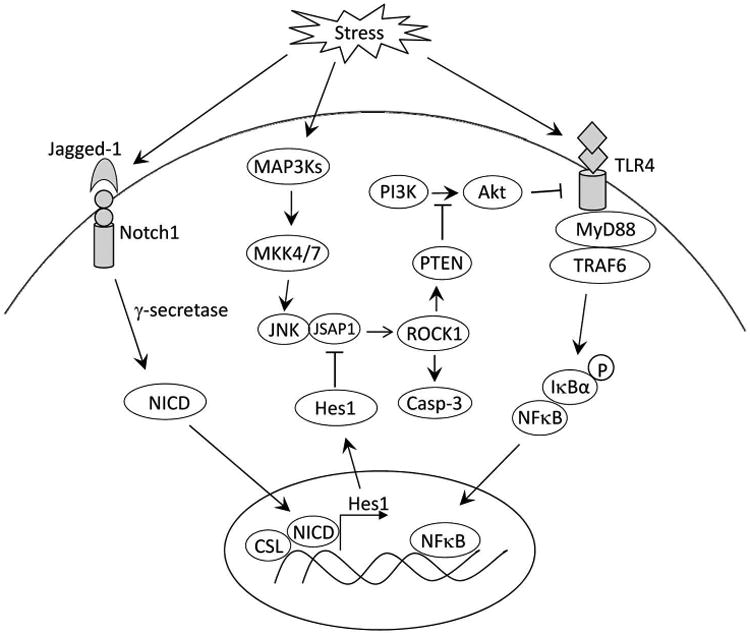
Notch1 can be activated in IR-stressed livers. Upon ligand binding, Notch1 is cleaved by γ-secretase leading to a release of the intracellular domain (NICD), which translocates into the nucleus and forms a complex with the CSL DNA-binding protein and activates its target gene Hes1. Induction of Hes1 inhibits JNK binding protein JSAP1-mediated ROCK1 activation. Blockade of ROCK1 reduces PTEN and augments Akt activity, leading to suppressed TLR4 signaling in liver IRI. In addition, the Notch-Hes1 axis inhibits JSAP1-dependent ROCK1 and caspase-3 activity, resulting in reduced hepatocellular apoptosis/necrosis in IR-triggered liver inflammation.
It should be stressed that the results of this study are from a whole animal myeloid knockout. It is possible that extrahepatic events may drive this response. Indeed, activation of the innate immune system in response to hepatic IR represents a key process determining the development of liver damage. Hepatic IR triggers inflammatory response and recruitment of infiltrating cells (macrophages, neutrophils, etc) of extrahepatic origins leading to differently influencing the organization of the hepatic immune cascade.
In conclusion, we demonstrate that myeloid Notch1 deficiency promotes the JSAP1-mediated RhoA/ROCK signaling pathway and exacerbates liver damage by depressing its target gene Hes1 in IR-stressed livers. By identifying molecular pathways by which myeloid Notch-Hes1 signaling regulates RhoA/ROCK-mediated innate immunity, our findings provide the rationale for novel therapeutic approaches in ameliorating sterile inflammatory liver injury.
Supplementary Material
Acknowledgments
NIH Grants R21AI112722, R21AI115133 (B.KE), RO1DK062357, RO1DK102110, RO1DK107533 (J.W.KW), California Institute for Regenerative Medicine (CIRM) RT3-07949 (Q-L.YING), National Natural Science Foundation of China 81100270, 1310108001, 81210108017, National Science Foundation of Jiangsu Province BK20131024, BE2016766, 863 Young Scientists Special Fund grant SS2015AA0209322 and the Foundation of Jiangsu Collaborative Innovation Center of Biomedical Functional Materials (L.LU), and The Dumont Research Foundation.
Abbreviations
- BMMs
bone marrow-derived macrophages
- CRISPR
clustered regularly interspaced short palindromic repeats
- Cas9
CRISPR associated protein 9
- Hes1
hairy and enhancer of split-1
- JNK
c-Jun N-terminal kinase
- JSAP1
JNK/stress-activated protein kinase-associated protein 1
- MKK4
MAPK kinase 4
- Notch1FL/FL
the floxed Notch1
- Notch1M-KO
myeloid specific Notch1 knockout
- NICD
Notch intracellular domain
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- RBP-J
recombinant recognition sequence binding protein at the Jκ site
- RhoA
ras homolog gene family, member A
- ROCK
Rho-associated protein kinase
- sALT
serum alanine aminotransferase
- TUNEL
terminal deoxyribonucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick end labeling
Footnotes
Author names in bold designate shared co-first authorship
References
- 1.Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec-Weglinski J, Zhai Y. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation. 2016;100:2601–2610. doi: 10.1097/TP.0000000000001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62:600–614. doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 6.Ke B, Shen XD, Ji H, Kamo N, Gao F, Freitas MC, Busuttil RW, et al. HO-1-STAT3 axis in mouse liver ischemia/reperfusion injury: regulation of TLR4 innate responses through PI3K/PTEN signaling. J Hepatol. 2012;56:359–366. doi: 10.1016/j.jhep.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ke B, Shen XD, Kamo N, Ji H, Yue S, Gao F, Busuttil RW, et al. beta-catenin regulates innate and adaptive immunity in mouse liver ischemia-reperfusion injury. Hepatology. 2013;57:1203–1214. doi: 10.1002/hep.26100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yue S, Zhu J, Zhang M, Li C, Zhou X, Zhou M, Ke M, et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology. 2016;64:1683–1698. doi: 10.1002/hep.28739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–936. doi: 10.1016/s0016-5085(03)01048-5. [DOI] [PubMed] [Google Scholar]
- 10.Jiang W, Bell CW, Pisetsky DS. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol. 2007;178:6495–6503. doi: 10.4049/jimmunol.178.10.6495. [DOI] [PubMed] [Google Scholar]
- 11.Chen LY, Zuraw BL, Liu FT, Huang S, Pan ZK. IL-1 receptor-associated kinase and low molecular weight GTPase RhoA signal molecules are required for bacterial lipopolysaccharide-induced cytokine gene transcription. J Immunol. 2002;169:3934–3939. doi: 10.4049/jimmunol.169.7.3934. [DOI] [PubMed] [Google Scholar]
- 12.Segain JP, Raingeard de la Bletiere D, Sauzeau V, Bourreille A, Hilaret G, Cario-Toumaniantz C, Pacaud P, et al. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: evidence in Crohn's disease and experimental colitis. Gastroenterology. 2003;124:1180–1187. doi: 10.1016/s0016-5085(03)00283-x. [DOI] [PubMed] [Google Scholar]
- 13.Mong PY, Wang Q. Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J Immunol. 2009;182:2385–2394. doi: 10.4049/jimmunol.0802811. [DOI] [PubMed] [Google Scholar]
- 14.Kuroda S, Tashiro H, Igarashi Y, Tanimoto Y, Nambu J, Oshita A, Kobayashi T, et al. Rho inhibitor prevents ischemia-reperfusion injury in rat steatotic liver. J Hepatol. 2012;56:146–152. doi: 10.1016/j.jhep.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 16.Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32:14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 18.Hansson EM, Lendahl U, Chapman G. Notch signaling in development and disease. Semin Cancer Biol. 2004;14:320–328. doi: 10.1016/j.semcancer.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 21.Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13:427–437. doi: 10.1038/nri3445. [DOI] [PubMed] [Google Scholar]
- 22.Kohler C, Bell AW, Bowen WC, Monga SP, Fleig W, Michalopoulos GK. Expression of Notch-1 and its ligand Jagged-1 in rat liver during liver regeneration. Hepatology. 2004;39:1056–1065. doi: 10.1002/hep.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu HC, Qin HY, He F, Wang L, Fu W, Liu D, Guo FC, et al. Canonical notch pathway protects hepatocytes from ischemia/reperfusion injury in mice by repressing reactive oxygen species production through JAK2/STAT3 signaling. Hepatology. 2011;54:979–988. doi: 10.1002/hep.24469. [DOI] [PubMed] [Google Scholar]
- 24.Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, Busuttil RW, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Ke B, Shen XD, Gao F, Ji H, Qiao B, Zhai Y, Farmer DG, et al. Adoptive transfer of ex vivo HO-1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Mol Ther. 2010;18:1019–1025. doi: 10.1038/mt.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ke B, Shen XD, Zhang Y, Ji H, Gao F, Yue S, Kamo N, et al. KEAP1-NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J Hepatol. 2013;59:1200–1207. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Froh M, Konno A, Thurman RG. Isolation of liver Kupffer cells. Curr Protoc Toxicol. 2003;Chapter 14 doi: 10.1002/0471140856.tx1404s14. Unit14 14. [DOI] [PubMed] [Google Scholar]
- 30.Yu SS, Lau CM, Barham WJ, Onishko HM, Nelson CE, Li H, Smith CA, et al. Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm. 2013;10:975–987. doi: 10.1021/mp300434e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, et al. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, Osborne BA. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol. 2008;38:174–183. doi: 10.1002/eji.200636999. [DOI] [PubMed] [Google Scholar]
- 33.Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Ray P, Ray A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. J Clin Invest. 2006;116:996–1004. doi: 10.1172/JCI26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Q, Li C, Wang K, Yue S, Jiang L, Ke M, Busuttil RW, et al. Phosphatase and tensin homolog-beta-catenin signaling modulates regulatory T cells and inflammatory responses in mouse liver ischemia/reperfusion injury. Liver Transpl. 2017;23:813–825. doi: 10.1002/lt.24735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Wang C, Liu Z, Liu X, Han C, Cao X, Li N. Notch signal suppresses Toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor kappaB activation. J Biol Chem. 2012;287:6208–6217. doi: 10.1074/jbc.M111.310375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, Tateya T, et al. Integrated regulation of Toll-like receptor responses by Notch and interferon-gamma pathways. Immunity. 2008;29:691–703. doi: 10.1016/j.immuni.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shang Y, Coppo M, He T, Ning F, Yu L, Kang L, Zhang B, et al. The transcriptional repressor Hes1 attenuates inflammation by regulating transcription elongation. Nat Immunol. 2016;17:930–937. doi: 10.1038/ni.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tharaux PL, Bukoski RC, Rocha PN, Crowley SD, Ruiz P, Nataraj C, Howell DN, et al. Rho kinase promotes alloimmune responses by regulating the proliferation and structure of T cells. J Immunol. 2003;171:96–105. doi: 10.4049/jimmunol.171.1.96. [DOI] [PubMed] [Google Scholar]
- 39.Schabbauer G, Matt U, Gunzl P, Warszawska J, Furtner T, Hainzl E, Elbau I, et al. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J Immunol. 2010;185:468–476. doi: 10.4049/jimmunol.0902221. [DOI] [PubMed] [Google Scholar]
- 40.Tomiyama K, Ikeda A, Ueki S, Nakao A, Stolz DB, Koike Y, Afrazi A, et al. Inhibition of Kupffer cell-mediated early proinflammatory response with carbon monoxide in transplant-induced hepatic ischemia/reperfusion injury in rats. Hepatology. 2008;48:1608–1620. doi: 10.1002/hep.22482. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012;3:56. doi: 10.3389/fphys.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 43.Montaner S, Perona R, Saniger L, Lacal JC. Multiple signalling pathways lead to the activation of the nuclear factor kappaB by the Rho family of GTPases. J Biol Chem. 1998;273:12779–12785. doi: 10.1074/jbc.273.21.12779. [DOI] [PubMed] [Google Scholar]
- 44.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 45.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A. 2006;103:14495–14500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando) 2012;26:103–114. doi: 10.1016/j.trre.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 48.Kajimoto H, Hashimoto K, Bonnet SN, Haromy A, Harry G, Moudgil R, Nakanishi T, et al. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: a newly recognized mechanism for sustaining ductal constriction. Circulation. 2007;115:1777–1788. doi: 10.1161/CIRCULATIONAHA.106.649566. [DOI] [PubMed] [Google Scholar]
- 49.Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, Ridgway RA, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spee B, Carpino G, Schotanus BA, Katoonizadeh A, Vander Borght S, Gaudio E, Roskams T. Characterisation of the liver progenitor cell niche in liver diseases: potential involvement of Wnt and Notch signalling. Gut. 2010;59:247–257. doi: 10.1136/gut.2009.188367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.