

Abstract
Advances in genomics technology have provided the means to probe myriad chromatin interactions at unprecedented spatial and temporal resolution. This has led to a profound understanding of nucleosome organization within the genome, revealing that nucleosomes are highly dynamic. Nucleosome dynamics are governed by a complex interplay of histone composition, histone post-translational modifications, nucleosome occupancy and positioning within chromatin, which are influenced by numerous regulatory factors, including general regulatory factors, chromatin remodellers, chaperones and polymerases. It is now known that these dynamics regulate diverse cellular processes ranging from gene transcription to DNA replication and repair.
The basic unit of chromatin is a nucleosome composed of two copies of each of the four core histones: H2A, H2B, H3 and H4, together with ~147 bp of DNA that wraps around the histone core1–3. Nucleosomes are highly dynamic. First, they are positionally malleable and can slide along DNA4,5. Second, they can also fully or partially disassemble6. Finally, they are often subject to post-translational modifications (PTMs)7,8, and their component histones can be replaced by their sequence variants9. These dynamics are intimately involved in genome regulation.
Transcriptionally active gene promoters are characterized by the presence of a nucleosome-free region (NFR) (or a nucleosome-depleted region (NDR)) in their core region; this local nucleosome depletion ensures that DNA is accessible to proteins, including various chromatin regulators, as well as transcription and replication machineries10,11. A highly regulated and well-positioned nucleosome, known as the +1 nucleosome, typically, resides at a canonical distance downstream (in the direction of transcription) of each major transcriptional start site (TSS) and forms the downstream border of the NFR. A well-positioned −1 nucleosome forms the upstream border (FIG. 1a). NFRs or NDRs are also found in the regions of active enhancers. Moving down-stream from the +1 nucleosome (into the gene body), well-positioned arrays of nucleosomes are initially observed, and they are typically positioned at defined intervals (~165 bp dyad-to-dyad in Saccharomyces cerevisiae, for example)12,13. However, this well-defined nucleosome positioning dissipates further into gene bodies (FIG. 1a). The position and histone composition of the +1 nucleosome are important for transcription, as these factors affect both RNA polymerase II passage through the gene body and binding of transcription factors14–17. The regular spacing of nucleosomal arrays is also highly dynamic, and the spacing observed at any given position and at any given time point is a result of cooperative and often redundant activity of numerous factors18. The maintenance of regularly spaced nucleosomes in an array has been suggested to regulate transcription by either promoting or inhibiting transcription initiation17,19–21.
Figure 1. Nucleosome organization as a combination of nucleosome occupancy and positioning.
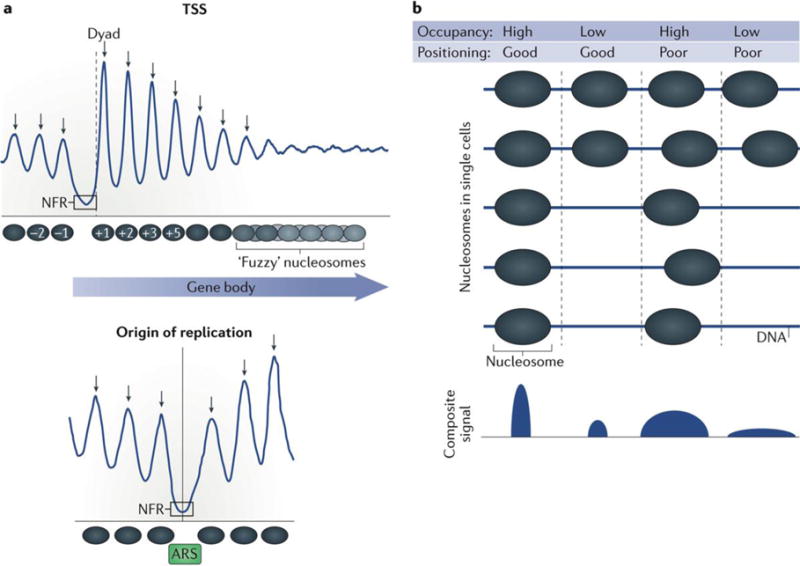
a | Nucleosomes can be found at conserved distances from genomic loci such as the transcription start site (TSS) and origins of replication. The blue lines represent the typical occupancy and positioning of nucleosomes relative to common genomic positions. The peaks and valleys correspond to locations of high and low nucleosome occupancy, respectively. The width and narrowness of the peaks correspond to the relative positioning of those nucleosomes. The arrows correspond to predicted dyads. Just downstream of the nucleosome-free region (NFR) (in the direction of transcription) a well-positioned nucleosome, termed ‘+1’ is present11. The +1 nucleosome serves as the downstream border of the NFR. A well-positioned −1 nucleosome forms the upstream border of the NFR. Nucleosomes form well-positioned arrays downstream of the +1 nucleosome into the bodies of genes. However, this positioning dissipates further into gene bodies, becoming ‘fuzzy’. Origins of replication are also characterized by well-positioned nucleosomes that flank NFRs, which encompass an autonomous replicating sequence (ARS) consensus motif29. b | Nucleosome occupancy is defined as the probability of a nucleosomes being present over a specific genomic region within a population of cells and is often measured in sequencing-based experiments by the number of aligned sequencing reads mapped to this region. Nucleosome positioning is defined as the probability of a nucleosome reference point (for example, a dyad) being at a specific genomic coordinate relative to surrounding coordinates. Good nucleosome positioning can be biologically interpreted as a nucleosome dyad occurring at the same genomic coordinate every time it is present. Poor positioning or ‘fuzziness’ can be interpreted as a nucleosome dyad occupying a range of positions within the same general footprint of an entire nucleosome. Nucleosome occupancy and positioning are independent metrics of nucleosome organization.
Although nucleosomes are largely symmetrical with regard to the internal organization of histones, transcription (and replication) proceeds directionally through a nucleosome, and so polymerases interact with one side of a nucleosome differently than with the other side15. Reflecting this intrinsic directionality, histone marks and variants are asymmetrically placed within nucleosomes, allowing for precise regulation of chromatin-binding proteins6. For example, the histone- variant H2A.Z is preferentially located on the promoter-distal side of many +1 nucleosomes and may function to destabilize the promoter-proximal side of the nucleosome, allowing for easier passage of polymerase through the nucleosome6,16,22.
The presence or absence of a nucleosome can affect chromatin properties and functions. For example, it may reflect different regulation of promoters. Most promoters contain an NFR at all times. In yeast, for example, this is the case under rich growth conditions, under stress or stationary growth conditions, and when their genomes are being compacted for sporulation21. Nonetheless, a subset of genes (for example, genes that are stress-regulated) do have a nucleosome in their promoter region (they contain an NDR not an NFR), and this nucleosome is depleted only during gene activation (by a cue, such as stress signals). And so NFR and NDR terms, even though often used interchangeably, reflect underlying chromatin regulatory mechanisms that are inherent to distinct classes of promoters. NFRs are constitutive, whereas NDRs are regulated.
Nucleosomes can be dynamically regulated at a subnucleosomal level through PTMs, assembly of histones into canonical octamers or alternative (non-canonical) structures, as well as incorporation of histone variants. Organization of these nucleosomes within chromatin is in turn carefully regulated through the underlying and surrounding DNA sequence, as well as numerous proteins, including chromatin remodellers, histone chaperones, PTMs and histone variant readers. These numerous and semi-redundant mechanisms operate to achieve nucleosomal positioning and spacing that is optimal for specific chromatin functions18,20,23–26. Through chromatin remodellers and histone chaperones, nucleosomes may move towards or away from nearby TSSs, as well as dissociate from DNA, either in whole or in part, as part of transcriptional regulation14,20,27. Likewise, genome-wide rearrangement of nucleosomes must be properly regulated during DNA replication28,29. Overall, there is now a large body of evidence that nucleosome movement, dissociation and alteration of nucleosomal structure are crucial features by which chromatin regulates gene expression and DNA replication. Disruption of these processes has been linked to multiple disease states30–32.
In this Review, we discuss our current understanding of genome-scale nucleosome dynamics as reflected in nucleosome occupancy (driven by their regulated turnover), their positioning and composition. Aspects of nucleosome composition involving histone modifications and histone variants are not discussed in detail here, and the reader is directed to excellent reviews on these topics33–35. We discuss how these dynamics are achieved and measured, and how they relate to cellular processes including DNA replication, transcription and DNA repair.
Overview of nucleosome dynamics
Nucleosome dynamics can be viewed as the interplay between nucleosome occupancy and positioning. Both of these properties have been well defined11,36–39, but they warrant further investigation to determine their respective roles in nucleosome dynamics and DNA site accessibility. Nucleosome occupancy is essentially the average number of nucleosomes measured within a specified genomic region in a cellular population and so is related to the probability of a nucleosome being present at the analysed site. Occupancy is generally a relative metric, as uncertainty in how measurements of nucleosomes relate to the true number of nucleosomes at a particular genomic region in a cellular population makes it difficult to precisely determine absolute levels of nucleosomes at particular locations.
Nucleosome positioning is related to the probability of a reference point on a nucleosome (typically its dyad) existing at a specific genomic coordinate relative to being present at surrounding coordinates, in a given population of cells. The concept of positioning is generally not associated with sequence-specific DNA-binding proteins because such proteins typically bind only to their cognate motif and do not generally bind to adjacent out-of-frame sequences. Thus, nucleosome organization can be described by a combination of occupancy and positioning (FIG. 1b), although more-detailed properties such as subnucleosomal structures, histone variants and modifications also contribute to nucleosome organization. Numerous genomic assays have been developed to quantify these interactions (TABLE 1).
Table 1.
Genomic assays used to measure nucleosome dynamics
| Genomic assay | Description | Features | Drawbacks | Refs |
|---|---|---|---|---|
| ChIP followed by deep sequencing: ChIP–seq |
|
|
|
135, 169 |
| ChIP followed by exonuclease digestion and deep sequencing: ChIP–exo |
|
|
|
135 |
| MNase digestion followed by deep sequencing: MNase–seq |
|
|
|
170, 171 |
| MNase digestion followed by ChIP and deep sequencing: MNase–ChIP–seq |
|
|
172 | |
| Time–ChIP |
|
|
|
68 |
| NOME–seq |
|
|
|
173 |
| MPE sequencing MPE–ChIP–seq |
|
|
|
174 |
| ATAC–seq |
|
|
|
175, 176 |
| Chemical mapping |
|
|
|
177 |
| NChAP and MINCE–seq |
|
|
|
166, 167 |
| Micrococcal nuclease digestion with Hi-C:Micro-C |
|
|
|
178 |
ATAC, assay for transposable-accessible chromatin; ChIP, chromatin immunoprecipitation; dNTP, deoxynucleoside 5′-triphosphate; EdU, 5-ethynyl-2 deoxyuridine; MINCE, mapping in vivo nascent chromatin with EdU; MNase, micrococcal nuclease; MPE, methidiumpropyl-EDTA; NChAP, nascent chromatin avidin pull-down; NOME-seq, nucleosome occupancy and methylome sequencing.
Nucleosome occupancy
The presence or absence of a nucleosome at particular locations can greatly affect the accessibility of the underlying DNA to proteins and, as a result, nucleosome occupancy is a key parameter that influences chromatin functions. Here, we describe how the presence (that is, occupancy) or absence of a nucleosome at a particular position can be measured. We also discuss the regulation of nucleosome occupancy — in particular, how nucleosome turnover is mediated by chromatin remodellers and how these processes affect chromatin organization.
Mapping nucleosome occupancy
Nucleosome locations are mapped genome-wide through a variety of methods, each of which has advantages and disadvantages (TABLE 1). The key steps include chromosome fragmentation to separate adjacent nucleosomes, optionally enriching for specific nucleosomal variants and then sequencing nucleosomal DNA and comparing its locations to existing genomic features (such as TSSs).
One of the most common methods of nucleosome mapping, chromatin immunoprecipitation followed by deep sequencing (ChIP–seq), involves the use of formaldehyde to trap histone–DNA interactions in vivo, followed by sonication to fragment chromatin, and then use of immobilized antibodies directed against histones or their PTMs to purify the histone–DNA complexes40,41. The DNA is then sequenced, generating 10–300 million sequencing reads depending on several factors that include genome size and the cost and throughput limits of available deep sequencers. Budding yeast haploid genomes contain about 60,000 nucleosomes12, whereas human diploid genomes have close to 30 million42. Thus, each nucleosome location within a population of cells is measured, in principle, about 10–100 times. However, although each read may correspond to an independent nucleosome measurement in a population of cells, amplification of isolated nucleosomal DNA that precedes the sequencing step often produces multiple copies of the same chromatin region that do not represent independent measurements and therefore are not useful. The depth of independent measurements, known as library complexity, is important in obtaining high-quality maps of nucleosome organization. ChIP–seq is useful for measuring nucleosome occupancy levels but poorly resolves neighbouring nucleosomes, and therefore it is more useful for assessing general chromatin landscapes. In the case of assays that analyse the occupancy of histone modifications using modification- specific antibodies, it is important to know that low levels of detection of a histone modification may be due to the absence of a nucleosome rather than solely the absence of the modification. Occupancy levels are therefore normalized by dividing regional signals obtained from modification-specific antibodies by the corresponding regional signals obtained using an antibody that recognizes the core sequence of this histone.
Another common technique used to assess the presence or absence of a nucleosome involves the use of micrococcal nuclease (MNase) to cleave accessible linker DNA located between nucleosomes43,44. Compared with ChIP–seq, MNase digestion followed by sequencing (MNase–seq) has substantially higher resolution (several base pairs versus hundreds of base pairs) owing to preferred digestion of naked DNA by MNase, which results in the remaining DNA ends being enriched for the entry and exit sites of nucleosomes45. Although the technical simplicity of MNase–seq makes it attractive, this technique suffers from an inability to distinguish nucleosomes from other large non-nucleosomal complexes that protect DNA from digestion. This may be remedied by including a histone ChIP step after MNase digestion. As discussed further below, MNase can be both a boon and a bane of chromatin studies, depending how the resulting data are interpreted.
Regulation of nucleosome occupancy by nucleosome turnover
DNA on nucleosomes becomes unwrapped during the course of DNA transcription, replication, recombination and repair14,46,47. Although a full nucleosome is typically restored to its original position afterwards, it may no longer possess the same chemically modified states or even be composed of its original histones48. The ability of factors to compete with histone assembly may be further regulated through intracellular signalling14,17,49. Importantly, even in the absence of transcription and replication events, there exists a basal level of association–dissociation dynamics between histones and DNA, locally changing nucleosome occupancy. This is important because these local dynamics enable proteins that lack an intrinsic ability to evict nucleosomes to access the underlying DNA.
Molecular mechanisms of nucleosome dynamics involve varying contributions of a myriad of factors. Whereas nucleosomes will assemble on nearly any DNA sequence, nucleosomes within AT-rich sequences tend to be less stable50,51, and therefore DNA sequence may contribute passively to nucleosome dynamics. Indeed, simply mixing pure histones and pure genomic DNA together partially reconstitutes in vivo chromatin organization, whereby nucleosomes are partially depleted from AT-rich promoter regions47,52,53. Nucleosome occupancy is also influenced by the activity of histone-specific chaperones that guide assembly and disassembly of the histone core of the nucleosome. The main regulators of nucleosome occupancy are chromatin remodellers that use energy from ATP hydrolysis to physically remove or deposit histones, even against thermodynamic gradients. How these factors work together is currently an active area of research.
Histone turnover may be best illustrated by the S. cerevisiae multisubunit Swr1 remodelling complex (SWR-C)26, which mostly targets well-positioned nucleosomes that flank promoter NFRs (FIG. 2a). These nucleosomes selectively contain the H2A.Z variant of the H2A histone54, and SWR-C mediates the exchange of H2A–H2B to H2A.Z–H2B. Specificity is achieved through SWR-C subunit interactions with the NFR, and this helps to place the rest of the complex over the adjacent nucleosome. There, ATP hydrolysis is used to dislodge the resident H2A–H2B histone dimer. Part of the SWR-C captures chaperone-bound H2A.Z–H2B and delivers it to the now vacated H2A– H2B site. Much of this is further regulated through histone acetylation of H2A.Z and H3 and reversed by another remodeller INO80 ( REF. 55), although this point has been controversial56,57. The replacement of the H2A histone with H2A.Z histone variants results in increased turnover of the full nucleosome octamer, possibly owing to increased exposure of the remaining core histones during replacement26,56. INO80 also has roles beyond H2A.Z–H2B dimer exchange at the +1 and the −1 nucleosomes. INO80 is involved in DNA replication elongation, implicating it in general nucleosome mobilization that promotes DNA accessibility for replication factors58. INO80 and its partners are also involved in DNA damage repair59, which is likely to resolve the inhibitory effect of nucleosomes on the repair proteins60.
Figure 2. Nucleosome occupancy as a function of histone turnover.
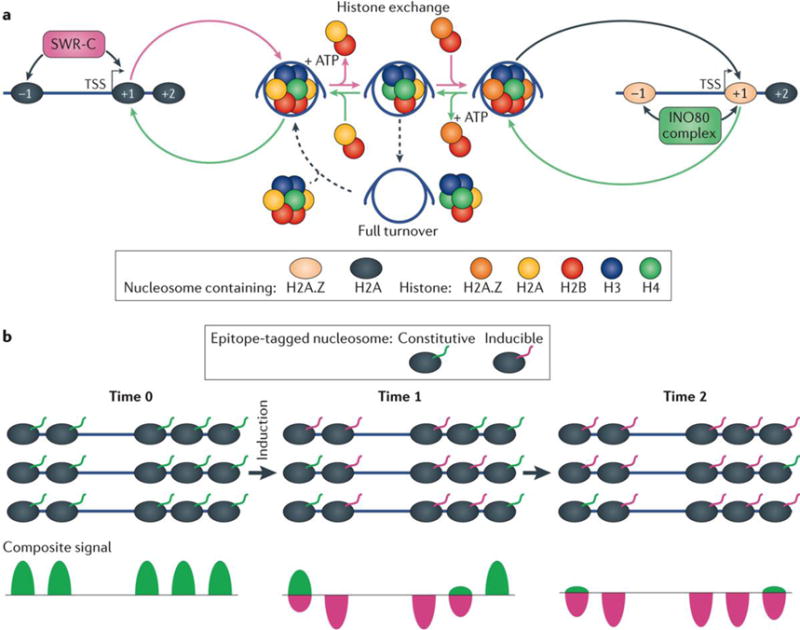
a | The Swr1 remodelling complex (SWR-C) targets the well-positioned +1 and −1 nucleosomes flanking nucleosome-free regions (NFRs) of active promoters, which typically contain the H2A.Z variant of the H2A histone26,54. SWR-C mediates the exchange of H2A–H2B to H2A.Z–H2B. Subunits of the SWR-C bind within the NFR and assist with positioning the rest of the complex over adjacent nucleosomes. Once positioned, ATP hydrolysis is used to dislodge the resident H2A–H2B histone dimer. Subunits of the SWR-C capture chaperone-bound H2A.Z–H2B and deliver it to the now vacated H2A–H2B site. The reversal of this process may be mediated by INO80 ( REF. 55). INO80-mediated H2A.Z exchange results in increased turnover of the full nucleosome octamer, possibly owing to increased exposure of H3–H4 during H2A.Z–H2B eviction26,56. b | In an inducible histone turnover system, newly synthesized histones are typically distinguished from existing histones by a distinct epitope tag (magenta)61. The zero time point is typically a time point of tagged-histone induction. The rate of incorporation of the epitope tagged-histones into chromatin is then monitored over time.
The turnover rate at which nucleosomes or histones are replaced at any given locus is measured by assaying new histone incorporation over time. New histones are typically distinguished from existing histones by inducing expression of a uniquely tagged histone, defined as the zero time point, and following its temporal incorporation into chromatin (FIG. 2b). For example61, in a S. cerevisiae strain harbouring constitutive expression of a MYC-tagged H3, which represents existing histones, newly deposited histones can be monitored by a FLAG-epitope tag placed on a galactose-inducible H3. At defined time points following galactose addition, cells are harvested and separate MNase–ChIP–seq analysis is performed using anti-MYC and anti-FLAG antibodies. At each consensus nucleosome locus, the number of MYC-tagged nucleosomes is expected to decrease with time, whereas the number of FLAG-tagged nucleosomes increases. Importantly, each locus has its characteristic rate of exchange, with the 5′ ends of genes being most dynamic. Exchange is even more rapid when transcription is occurring in these regions. Similar turnover studies have been conducted in flies, mice and humans, including expanded studies to monitor turnover of histone variants62–65, as well as the turnover rates of PTMs66. Together, these studies have revealed that genomic regions with the highest rates of nucleosome (and histone) turnover are biochemically active. In addition to promoters, these regions include enhancers and origins of replication.
Alternative methods of measuring nucleosome turnover, such as use of SNAP chemistry, involves the covalent addition of an epitope (GFP, FLAG, MYC, and so on) to a protein of interest using the SNAP-tag67. The epitope is pulsed into live cells, where it is covalently bound to a SNAP-tagged histone and then immunoprecipitated in a time series. The decreasing detected levels of epitope labelled histones as normal nucleosome turnover occurs can be interpreted as the relative turnover rates of histones. For example, this method has been applied to measuring H3.3 histone turnover at enhancers and promoters of differentiating mouse embryonic stem cells68, revealing that enhancers (and super enhancers in particular) are sites of high nucleosome turnover. This turnover is associated with increased accessibility of DNA to transcription factors and active transcription of genes that are important for pluripotency (in embryonic stem cells) or differentiated states (in differentiated progenitors).
This finding that rapid nucleosome turnover is associated with high rates of transcription seems to be incongruent with other findings, indicating that nucleosomes are retained at the site of transcription by histone chaperones and other factors15,69. This discrepancy may be reconciled by the possibility that histone exchange might not always be tied to full nucleosome turnover. Therefore, looking at individual histones might not provide a complete picture of whole nucleosome dynamics. Another confounding variable in assessing whether histones and nucleosomes are retained may lie in the experimental design. In particular, histone exchange rates are often measured by histone overexpression. The resulting increased local histone concentration might allow levels of exchange that could not be supported under normal basal levels of histone expression. Therefore, although histones have the potential to exchange, and do so when free histones are available, they may not do so under normal conditions in which there are very few free histones to exchange.
Nucleosome positioning
The position of nucleosomes is regulated by many complex processes, which can be distilled into predominately two apects: cis-acting (DNA sequence) and trans-acting (protein)70,71. One trans-acting process, gene transcription, is initially disruptive to chromatin structure but is then followed by rapid and proper reassembly and positioning of nucleosomes. The role of transcription in nucleosome positioning is currently much debated18,38,72.
Sequence determinants of nucleosome positioning
The role of DNA sequence in determining nucleosome organization had been a source of some controversy over the years37,73. DNA sequences can intrinsically favour or disfavour nucleosome formation, and have some intrinsic ability to influence the position of nucleosomes. Extensive analysis of DNA sequence has revealed particular aspects that support wrapping the histone octamer50 (FIG. 3a). Specifically, nucleosomal DNA is more enriched with AA, TT, TA and AT dinucleotides at a 10 bp periodicity (or approximately one helical twist of DNA) than non-nucleosomal DNA36,54,74; this is most evident at −1 and +1 nucleosomes at TSSs18,50. DNA sequences possessing these characteristics make preferred bends that accommodate their wrap on the histone octamer surface, thereby producing a stronger rotational setting (FIG. 3b). However, a strong rotational setting need not be tied to a strong translational setting (narrow positioning in the genome), in that a DNA sequence can have a single predominant orientation on a histone octamer but in roughly 10 bp intervals it can adopt multiple alternative positions along the histone octamer surface54. In in vitro studies, DNA sequences containing relatively high GC content tend to wrap nucleosomes with greater affinity37. However, the CpG-rich islands in mammalian genomes, although enriched with histones, do not appear to form canonical nucleosomes75,76.
Figure 3. Determinants of nucleosome positioning.
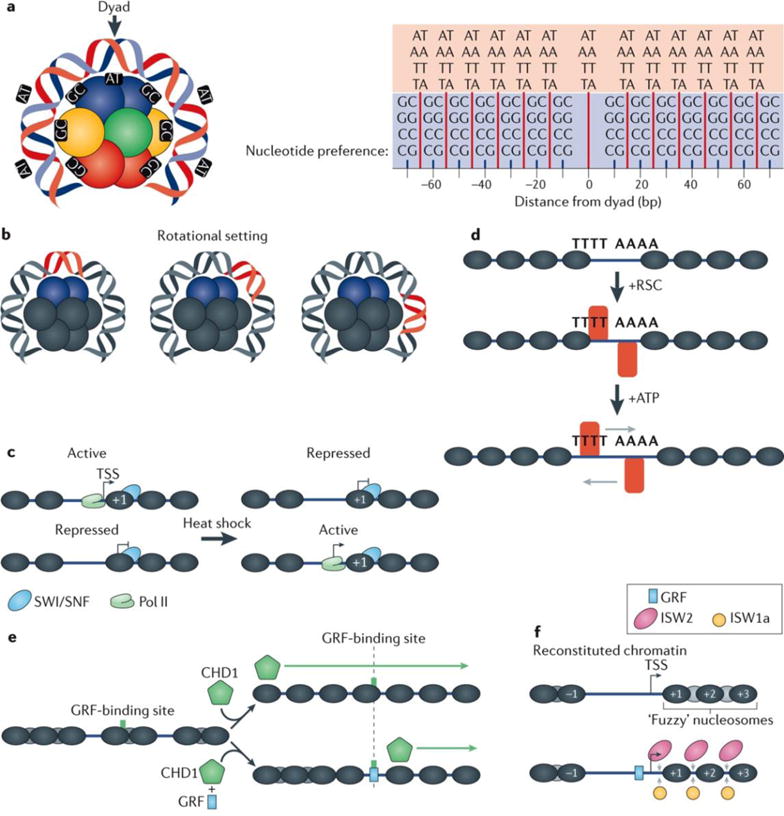
a | Nucleosomal DNA, particularly at +1 and −1 positions with respect to transcription start sites (TSSs), is modestly enriched with AA, TT, AT, and TA dinucleotides at a 10 bp periodicity and GG, CC, GC, and CG dinucleotides offset by 5 bp from those dinucleotides and also possessing a 10 bp periodicity36,38,50,54,74. These periodicities result in the formation of preferred bends in DNA, thereby defining how DNA wraps around the histone octamer. Point 0 represents the dyad with preference for AA, TT, AT and TA dinucleotides, and preference in 10 bp periodicity at 15, 25, 35, 45, 55 and 65 bp from the dyad are represented by the red lines. The GG, CC, GC and CG dinucleotide preferences are represented by the short blue lines at points 10, 20, 30, 40, 50, 60 and 70 bp from the dyad. b | The position of DNA relative to the histone octamer defines its rotational setting. DNA sequences possessing the nucleotide periodicities shown in part a possess a strong rotational setting, which means that they typically wrap around the octamer in a preferred orientation. However, for a single rotational setting, multiple translational settings can exist, typically in 10 bp intervals, reflecting the nucleotide preference periodicities in the DNA. c | The switch/sucrose non-fermenting (SWI/SNF) chromatin remodelling complex helps to regulate various cellular processes, including stress responses, such as heat shock. It may do so by altering the accessibility of promoter DNA to regulatory proteins and the transcription machinery (contributing to both activation and repression of transcription)14,17. d | The highly abundant and essential remodelling the structure of chromatin (RSC) complex acts on poly(dA:dT) tracts, which are common in Saccharomyces cerevisiae promoters. RSC regulates the size of the nucleosome-free region in these promoters by repositioning and/or evicting nucleosomes located near poly(dA:dT) tracts in an ATP-dependent manner18,90,91. e | The chromodomain helicase DNA binding 1 (CHD1) remodeller produces regularly spaced nucleosome arrays in vitro using ATP95. The targeting of nucleosome remodelling activity of CHD1 is suspected to be linked to transcription98,99, which explains its apparent lack of DNA-binding specificity in genome-wide in vitro nucleosome assembly assays18. f | In S. cerevisiae the imitation SWI (ISWI) family of chromatin remodellers are enriched at the 5′ ends of gene bodies25. In vitro experiments have revealed that ISW1a and ISW2 are crucial for establishing proper in vivo nucleosome spacing18. Spacing is further improved with the addition of general transcription factors (GRFs), which supports the predicted role of GRFs as anchor points, or boundary elements, to guide the directionality for ISW1a112. See also part e. Pol II, RNA polymerase II.
The effect of DNA sequence on nucleosome organization has been quantified using purified nucleosomes assembled onto DNA through the well-established method of salt-gradient dialysis, whereby histones and DNA in a high-salt solution assemble into nucleosomes as the salt is dialysed away47,52. In this setting, nucleosomes are placed in their thermodynamically favoured locations. Notably, this thermodynamically favoured nucleosome distribution does not fully resemble the positioning observed in vivo. This is because, in vivo, contributions of DNA sequence are mostly superseded by the action of trans-acting factors, including chromatin remodellers and general regulatory factors (GRFs)36,38,52,77.
Role of chromatin remodelers
The role of chromatin remodellers in positioning and restructuring nucleosomes has been studied extensively in vivo and in vitro4,18,20,25,26,76,78,79. Many chromatin remodellers have the ability to respond to cell stresses and can be triggered on demand to dynamically restructure the chromatin landscape14,80,81. In vivo, these chromatin remodellers appear to be enriched at specific nucleosome positions relative to genomic regions such as TSSs and origins of replication, and they possess both redundant and distinct functionality25,58,79.
The switch/sucrose non-fermenting (SWI/SNF) complex is highly conserved across eukaryotes and is involved in regulating basic metabolic processes, such as sugar metabolism82. In S. cerevisiae, SWI/SNF is also involved in regulating stress responses (for example, to heat shock) by remodelling chromatin to either repress or activate certain classes of genes to promote cell survival14,83,84 (FIG. 3c). Under non-stress conditions, SWI/SNF is generally enriched at nucleosomes that flank the NFR25. In mammals, the SWI/SNF complex also has a role in the heat shock response85 and has a cell-type-specific role in tumour suppression86,87.
Structurally related to the SWI/SNF complex, the RSC (remodelling the structure of chromatin) complex is a highly abundant chromatin remodeller and is required for cell viability88. RSC displaces nucleosomes at promoter regions in an ATP-dependent manner, contributing to the establishment of NFRs at transcriptionally active promoters78,89. This is consistent with RSC being enriched in the vicinity of promoters25,76. S. cerevisiae promoters also tend to be enriched with poly(dA:dT) tracts, which have been implicated in RSC activation as a potential binding location for the remodeller90,91 (FIG. 3d). Indeed, the activity of RSC alone is able to induce NFR formation at promoters both in vitro and in vivo, in a manner that is positionally linked to poly(dA:dT) tracts18,92. As RSC is able regulate NFR width, it is likely to be involved in gene repression as well as activation.
The chromodomain-helicase-DNA-binding (CHD) family of chromatin remodellers is implicated in DNA replication and repair pathways, and it is related to the SNF2 family of chromatin remodellers93,94. CHD1 remodels nucleosomes in gene bodies similarly to other chromatin remodellers79. The effects of purified CHD1 on nucleosome positioning have also been studied in vitro with what would seem to be conflicting results. In a nonspecific spacing assay, CHD1 addition induces regularly spaced arrays95 (FIG. 3e). By contrast, in an assay that measures spacing relative to fixed DNA sequences, CHD1 seems to have no effect on nucleosome positioning18. These potentially contradictory findings may be reconciled with the idea that CHD1 produces properly spaced arrays in vitro regardless of the underlying DNA sequences and may require additional guidance to position them relative to specific genomic reference points. Experiments have shown that by either adding a barrier element such as a DNA-binding protein or fusing a sequence-specific DNA-binding domain to CHD1, the nucleosome remodelling activity of CHD1 could be directed to specific locations and be driven only in a specific direction96,97. So, in vivo, the activity of CHD1 is most likely to be guided by other factors (such as transcriptional regulators). The potential ability of CHD1 to position nucleosomes relative to each other on the same DNA molecule, and its guidance by DNA-binding proteins, makes it ideally suited for restoring nucleosomal arrays in the wake of transcription98,99.
Remodeller complexes containing the imitation SWI (ISWI) class of chromatin remodellers are crucial for transcription, DNA replication and, in mammals, the DNA damage response100–104. ISWI remodellers reposition nucleosomes somewhat similarly to SWI/SNF by ratcheting DNA along the nuclesome105, although they are functionally distinct from SWI/SNF106. They additionally contain a HAND-SANT-SLIDE domain that may serve as a molecular ruler for proper nucleosome spacing107. ISWI-containing chromatin remodellers are conserved across budding yeast, flies and mammals80,108–110. In S. cerevisiae, ISW1 exists in two variant forms, ISW1a and ISW1b111. ISW1a spaces nucleosomes into arrays24 but is unable to remodel nucleosomes past bound GRFs112, indicating that GRFs act as barriers for nucleosome remodelling. Further evidence for GRFs acting as barrier elements for nucleosome positioning comes from in vitro experiments wherein in the presence of GRFs and ISW1a or ISW2 remodellers could recapitulate precise in vivo +1 nucleosome positioning18 (FIG. 3f).
Subnucleosomal composition
Nucleosome dynamicity is also in part governed by nucleosome composition. As mentioned above, one way by which nucleosome composition can be affected is through incorporation of sequence variants of histones (see REFS 33–35 for reviews). However, the changes in overall core histone composition that lead to the formation of non-canonical nucleosomes have also been described.
The structure of a canonical nucleosome consisting of two copies of the four core histones plus 147 bp of DNA has long been established and is thought to occupy the vast majority of eukaryotic genomes14,113. Much of this has come from the observation that such canonical nucleosomes are the major histone- containing entities isolated from cells and can also be faithfully reproduced and visualized in vitro. However, early studies describing half-nucleosomes, consisting of one copy of each of the four core histones and half of the amount of DNA have suggested that at least a small fraction of nucleosomes present in cells does not exhibit this canonical composition114–117. More recently, in vitro and in vivo evidence has emerged for the existence of a wide range of subnucleosomal structures (FIG. 4a), some of which may populate biologically important regions of the genome such as gene bodies and centromeres116. The presence and functions of these structures are still controversial, perhaps mostly owing to how methods and data that define these structures are interpreted. Subnucleosomal structures can minimally be thought of as having some portion of their DNA detectably not in contact with the underlying histones, which may contain or lack the full complement of histones.
Figure 4. Examination of nucleosome substructures and methods to detect them.
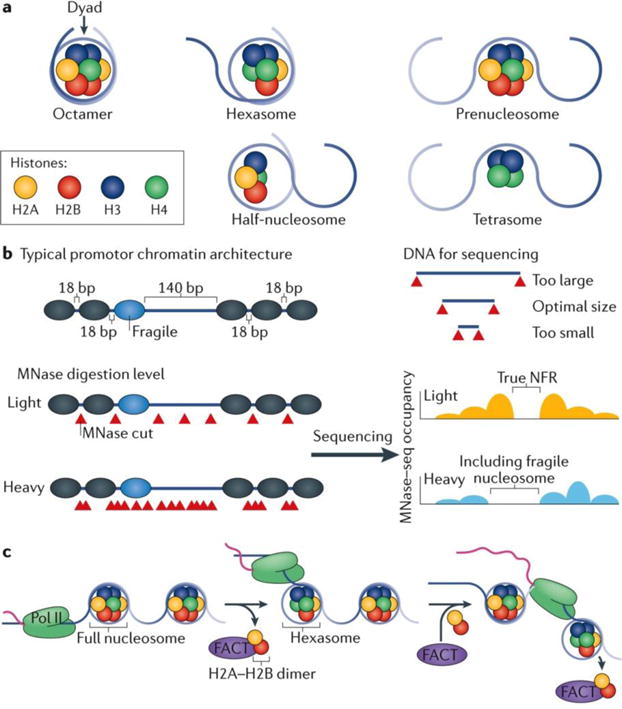
a | There is now evidence that besides the canonical histone octamer, a variety of subnucleosomal structures exist. The hexasome comprises a H3–H4 tetramer and a single H2A–H2B dimer, and is a functional intermediate, possibly resulting from the removal a H2A–H2B dimer by RNA polymerase II (Poll II) during transcription137 or through its intrinsic dynamics imparted by chromatin remodellers. A prenucleosome contains the full complement of the octamer but is wrapped by only 80 bp of DNA130. Half-nucleosomes (also known as hemisomes) comprise a single copy of each histone core particle. There is evidence that half-nucleosomes might accumulate at certain regions of DNA6. Tetrasomes are H3–H4 tetramers, which exist as possible intermediates during DNA replication (nucleosomes which are partially disassembled ahead of the replication fork and partially reassembled after the fork has passed)158,159. b | The extent of micrococcal nuclease (MNase) digestion affects the relative enrichment of nucleosomes at certain positions. Lighter digestion (with lower amounts of MNase) has the potential to enrich for open DNA not bound by nucleosomes, especially when chromatin immunoprecipitation is not used in conjunction. Heavier MNase digestion tends to better resolve true nucleosomes at the potential cost of losing the ability to detect fragile nucleosomes. Independent of size-selection criteria during experimental design, both current sequencing technology and PCR include an intrinsic DNA-size bias for library construction and sequencing cluster formation168. c | During transcription, Pol II must transcribe nucleosomal DNA. To facilitate this, nucleosomes are at least partially unravelled during transcription and an H2A–H2B dimer is removed forming a temporary hexasome. The FACT (facilitates chromatin transcription) complex is believed to interact with the free H2A–H2B dimers and assists with the nucleosome disassembly and reassembly137–139.
Non-canonical nucleosome structures
Considering the subnucleosome continuum from full nucleosomes to free DNA, perhaps the simplest first step towards generating subnucleosomes from full nucleosomes occurs when DNA near the nucleosome entry and exit sites intrinsically and transiently lifts off from the core histones (termed ‘breathing’) as a consequence of thermodynamics118. This breathing may be promoted by histone chaperones, such as the FACT (facilitates chromatin transcription) complex and nucleosome assembly protein (Nap1)119,120, by histone acetylation and/or by sequence-specific DNA-binding proteins, the cognate sites of which reside just within the entry and exit sites54,121–123. Chromatin remodellers may also lift DNA off from the octamer surface, far from the entry and exit sites4,105. Although these breathing events are often detected as an increase in nuclease accessibility to DNA (leading to increased digestion) — and are therefore assessed on the level of DNA — recent advances in single-molecule fluorescence resonance energy transfer (FRET) technology are now being applied to ascertain the underlying histone composition during nucleosome remodelling124.
Non-canonical nucleosomes have been reported in vivo6,125,126, with a most notable example of ‘fragile nucleosomes’, which have been described as genomic nucleosome locations that are particularly sensitive to MNase digestion76,90,127–129 (FIG. 4b). Fragile nucleosomes often reside at the −1 nucleosome position adjacent to NFRs upstream of TSSs, which often may be the terminal nucleosome of an upstream gene. The region is often mistakenly considered as part of the NFR because no nucleosome is detected there when high levels of MNase are used17,90 (FIG. 4b). This has led to the confusing notion that NFRs contain nucleosomes. They do not, as long as NFRs are defined using an appropriate level of MNase, and MNase assays should be carried out in conjunction with histone ChIP assays. What makes a fragile nucleosome particularly sensitive to MNase? Several explanations have been put forward, including roles for the associated DNA sequence, transcription factor binding and chromatin remodellers making the DNA on the nucleosome surface more accessible to MNase.
Recent reports130 have identified, in vitro, a ‘prenucleosome’ containing an assembled octamer that protects only 80 bp rather than 147 bp of DNA (FIG. 4a). Addition of the motor protein ATP-dependent chromatin assembly factor (ACF) and ATP readily converts such prenucleosomes to a canonical nucleosome. Prenucleosomes, although not yet identified in vivo, might be related to fragile nucleosomes as predicted by their shared MNase sensitivity and may exist as intermediates during the cellular processes of transcription and replication when nucleosome structure is perturbed to allow access of proteins to DNA.
Further evidence that subnucleosomes exist in vivo comes from the analysis of the histone composition of Drosophila melanogaster and S. cerevisiae centromeric nucleosomes, which contain only one copy of each of the four core histones125,131. These half-nucleosomes (also known as hemisomes; FIG. 4a) are thought to be intrinsically unstable, requiring centromere binding factor 1 (Cbf1) for stabilization in S. cerevisiae132, but their existence remains controversial133. Nevertheless, such half-nucleosomes can be assembled in vitro with purified proteins, whether using the centromeric- specific histone cenH3 together with canonical histones or canonical core histones alone125,134. Thus, half-nucleosomes can be potentially established even without the aid of other proteins.
Potential functions of subnucleosomes
Evidence for the genome-wide occurrence of half-nucleosomes in vivo has been obtained through high-resolution exonuclease digestion experiments6, termed ChIP–exo135 (TABLE 1). ChIP–exo mapping of histones in S. cerevisiae suggests that about 10% of +1 nucleosomes have an unbalanced histone H3–H4 composition, indicating that, at least in part, they exist as half-nucleosomes. This has been supported by orthogonal approaches such as chemical mapping (TABLE 1) and MNase digestion6,136. ChIP–exo examination of H2A–H2B suggests an even more widespread imbalance of these dimers on each side of +1 nucleosomes. A greater imbalance of H2A– H2B over the imbalance of H3–H4 suggests that hexasomes (nucleosomes lacking one copy of H2A–H2B) may be prevalent. Indeed, their existence at the 5′ ends of genes, where histone exchange is more dynamic, may correspond to nucleosome subassembly intermediates. Whether such subnucleosomal structures exist across gene bodies is less clear, as fuzziness in positioning precludes confident identification of distinct sides of the nucleosomes.
The function of subnucleosomes that may be lacking histones, apart from representing potential assembly intermediates, is unclear. They do not seem to be necessarily linked to the rate of transcription, and thus may be largely a manifestation of dynamic chromatin that exists constitutively within cells rather than chromatin dynamics associated with transcription. However, accessibility produced within such dynamic chromatin may create alternative sites for transcription complex assembly and hence may be responsible for some alternative TSSs. This would in essence alter the composition of the 5′ untranslated regions of mRNAs, and thus their post-transcriptional regulation. It may be relevant that in the case of stress-induced genes in S. cerevisiae, which often have complex modes of regulation, including at the mRNA level, nucleosome asymmetry — indicative of the presence of subnucleosomes — tends to be high6.
The evidence so far does not exclude a role for subnucleosomes during transcription, particularly as transcription events may be relatively infrequent compared with the availability of constitutively dynamic chromatin. The notion of the existence of hexasomes fits well with a current model of how RNA polymerase II navigates across nucleosomes during transcription, whereby it has been proposed that one H2A–H2B dimer may be displaced by the transiting RNA polymerase137. Any such transcription-coupled displacement and replacement of histones by RNA polymerase II is likely to be assisted by chaperone complexes such as FACT138,139 (FIG. 4c).
What is currently unclear is the extent to which the various subnucleosomal structures are related to each other. The difficulty lies in the different methodologies and conditions used to detect each of them, as the same subnucleosomal structure may manifest quite differently in different assays. This may be resolved in part by conducting relevant orthogonal assays on the same chromatin preparation, then determining whether the different metrics indicate the presence of a particular structure at a particular nucleosome location.
Functional implications
Nucleosomes exist in vivo at conserved positions relative to TSSs12,54 and origins of replication29,140. The position and composition of these nucleosomes are tightly controlled and have an active role in regulating transcription and replication77,141.
Dynamics of gene transcription
Transcription begins with recruitment of the pre-initiation complex (PIC) components, cofactors and the appropriate RNA polymerase at the promoter of genes142,143. PIC formation may require the +1 nucleosome to be positioned at a specific locus relative to the TSS144,145 (FIG. 1a). In turn, direct perturbations to PIC formation result in changes to nucleosome dynamics. Specifically, the H2A variant, H2A.Z, displays reduced rates of turnover when PIC assembly is blocked146. The regulation of H2A.Z dynamics by PIC assembly may be reciprocally related to H2A.Z regulating the passage of RNA polymerase II through the +1 nucleosome16. H2A.Z might destabilize these nucleosomes and thus allow RNA polymerase II to penetrate them more readily, thereby creating more histone turnover. That H2A.Z dynamics are a regulated component of transcription further underscores the importance of controlling nucleosome composition and positioning for transcriptional programmes. These dynamics may contribute to rapid induction of gene expression147.
Even seemingly small movements in the position of the +1 nucleosome may affect transcription. High-resolution mapping of nucleosomes and transcription factors at S. cerevisiae ribosomal protein promoters under heat-shock conditions reveals a decrease in transcription that is associated with eviction of specific DNA-binding factors and a positional shift of the +1 nucleosome in the upstream direction17,49 (FIG. 5). This suggests a model in which the upstream shift of the +1 nucleosome interferes with PIC formation through a direct steric occlusion of the PIC binding. This model is supported by evidence showing that PIC formation competes with nucleosomes at certain genes148. However, global changes in nucleosome positioning do not necessarily translate to changes in transcription, potentially owing to redundant regulatory mechanisms overcoming the repressive effect of a misplaced +1 nucleosome on PIC formation24,97.
Figure 5. Nucleosome dynamics of transcription at ribosomal protein genes.
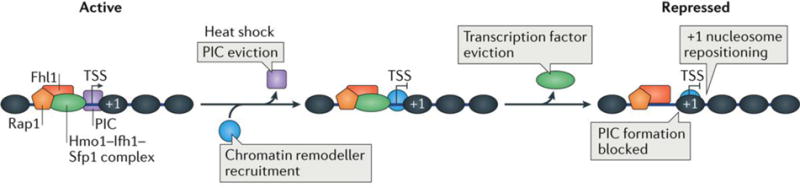
Ribosomal protein genes are among highly transcribed genes in Saccharomyces cerevisiae although they are silenced immediately when cells are stressed14. Repressor/activator protein 1 (Rap1) binds upstream of the transcription start site (TSS) at ribosomal protein genes and forms the anchoring point for binding transcription factors forkhead-like 1 (Fhl1), high mobility group 1 (Hmo1), interacts with forkhead 1 (Ifh1) and split finger protein (Sfp1). Together, these transcription factors enable the assembly of the pre-initiation complex (PIC) at the TSS. Upon heat-shock stress, components of the PIC are evicted and chromatin remodellers are recruited to the +1 nucleosome. Transcription factors are then removed from the promoter, and the +1 nucleosome is then shifted upstream into the promoter region17,49. This supports the model whereby the specific position of the +1 nucleosome can contribute to overall gene expression by either allowing or interfering with PIC formation.
Dynamics of DNA replication
DNA replication begins with the binding of the origins of replication complex (ORC) to an appropriate genomic locus followed by the recruitment of the MCM helicase (mini- chromosome maintenance helicase) complex and associated cofactors29,149,150. Similar to the +1 nucleosomes at the TSS, nucleosome positioning is conserved at origins of replication62,140 (FIG. 1a). In S. cerevisiae, there are well-positioned H2A.Z-containing nucleosomes flanking an NFR that contains an autonomous replicating sequence (ARS)54. Although there are numerous ARS consensus sequences (ACSs) present in the S. cerevisiae genome, many are not functionally active29,151. These non- functional origins of replication are partially overlaid by nucleosomes, implicating nucleosome positioning in the regulation of replication29 (FIG. 6a).
Figure 6. Nucleosome dynamics of DNA replication.
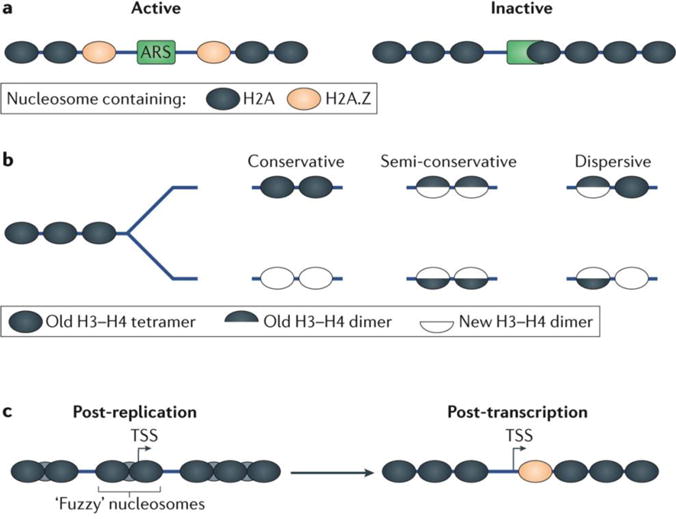
a | Active origins of replication in Saccharomyces cerevisiae are characterized by an ARS consensus sequence (ACS) located in a nucleosome-depleted region flanked by well-positioned nucleosomes containing the H2A.Z histone variant. Many non-functional ACSs exist in the S. cerevisiae genome and contain occluding nucleosomes. This implicates nucleosome positioning in regulating functional origins of replication29. b | The models for H3–H4 tetramer inheritance during replication include the conservative, semi-conservative and dispersive models. The conservative model is defined as the deposition of the entire tetramer on one daughter strand and another newly synthesized H3–H4 tetramer is deposited on another158. In the semi-conservative model, the tetramer is broken into its component H3–H4 dimers and each dimer is placed on a daughter strand with a new H3–H4 dimer141. The dispersive model is a combination of the conservative and semi-conservative model wherein the tetramer is either maintained or split. Evidence showing certain tetramers evenly split between daughter strands159 and other tetramers entirely segregated to a single strand160 suggest a guided dispersive model wherein tetramers are subject to different outcomes depending on their histone variant composition. c | Nucleosomes in gene bodies feature poor positioning immediately after replication, which is likely to be due to some level of random deposition immediately following the replication fork. At highly transcribed genes, this positioning is rapidly re-ordered, leading to even nucleosomal arrays with the +1 nucleosome containing H2A.Z variant, indicating that transcription supports nucleosome positioning166. TSS, transcription start site.
The dynamic processes of disassembly and reassembly of nucleosomes are mediated by numerous chaperone proteins. The histone chaperone anti-silencing function protein 1 (ASF1) binds the MCM2 helicase. It is believed to shuttle H3–H4 dimers and tetramers from disrupted nucleosomes to the chromatin assembly factor (CAF1) chaperone for re-assembly behind the replication fork27,152,153. The FACT complex has also been implicated in DNA replication, and mutations of this complex result in impaired H3–H4 deposition after replication34,154.
Immediately after the DNA replication fork, the recycled histones are reassembled into nucleosomes. However, this nucleosome restoration presents a unique problem for nucleosome inheritance by two DNA molecules. Instead of restoration of nucleosome positioning on a single duplex DNA (like in gene transcription), nucleosome assembly must occur on the two separate daughter duplex strands46,126,155. This presents additional difficulties such as how the inheritance of nucleosome variants and histone modifications occurs156. Similarly to the original proposed models of DNA replication, models for the H3–H4 tetramer inheritance on daughter strands can be summarized as conservative, semi-conservative and dispersive46,141,157 (FIG. 6b). Although the conservative model has been proposed as the dominant method of H3–H4 inheritance158, the specific mechanism of inheritance to daughter cells varies depending on the specific nucleosome modifications and histone variants159,160. Inheritance of histone variants may be subject to different mechanisms of deposition depending on their genomic locus. In the case of the H2A.Z histone variant, it has been shown to partition evenly (that is, randomly) at promoters during replication161. H2A.Z is also recruited to the centromeres during mitosis and thus serves a potential role in chromosome stability162. The mechanism of histone modification inheritance is further developed whereby downstream mechanisms recognize the diluted levels of the modifications in daughter cells and restore the pre-replication levels of the modification163,164.
Several recent papers have examined nucleosome dynamics of DNA replication using a novel run-on assay termed nascent chromatin avidin pull-down (NChAP)165–167 (TABLE 1). These methods have revealed that nucleosome positioning, although initially fuzzy after replication, is more rapidly restored at highly transcribed genes compared with regions with low transcriptional activity166 (FIG. 6c). This indicates that transcription is linked to proper positioning of nucleosomes, although, as in vitro studies18 show, this is probably only one of many mechanisms that contribute to establishing nucleosome organization in cells. Further advances in technology should allow us to de-convolute the complexity of chromatin dynamics in replication, including a better understanding of how the histone code is inherited by daughter cells.
Conclusions and perspectives
Originally viewed as a rather static mechanism of chromatin packaging, the nucleosome core complex is now well recognized as one of the key regulatory components of the genome. We also now see that instead of static protein complexes, nucleosomes are in fact exceptionally dynamic and that their positioning and composition are crucial for genome regulation. As such, the study of nucleosome dynamics is essentially the study of genome regulation. The complex interaction between nucleosome occupancy and positioning allows the cell to properly regulate accessibility of various proteins and their complexes to DNA and thus to regulate gene expression programmes. A variety of regulatory cofactors such as chromatin remodellers, chaperones and GRFs operates both independently and synergistically to maintain the precise organization and composition of nucleosome arrays at specific genomic loci. This dynamic environment probably exists so that the genome may respond and adapt quickly to both external stimuli as well as be able to quickly recover from chromatin-disruptive activities such as transcription and replication. The rapid improvements in technology have dramatically increased both our spatial and temporal resolution of nucleosome dynamics. In the future, the use of in vitro biochemistry applied to a genomic scale will detail mechanisms of chromatin assembly and regulation within the more native chromosomal context.
Acknowledgments
The authors thank the members of B. F. Pugh’s laboratory and the Center for Eukaryotic Gene Regulation for their helpful comments and feedback. This work was supported by the US National Institutes of Health (NIH) grant GM059055.
Glossary
- Nucleosome-free region (NFR)
A region of DNA that is constitutively nucleosome-free, such as promoter regions
- Nucleosome-depleted region (NDR)
A region of DNA that has regulated nucleosome occupancy
- Dyad
The midpoint of a canonical nucleosome, which creates mirrored pseudosymmetry
- SNAP-tag
An artificially engineered enzyme capable of covalently adding any compatible epitope on demand
- Super enhancers
Regions of the genome where clusters of enhancers are located
- CpG-rich islands
A dinucleotide combination of 5′-CG-3′. Prevalent and often methylated in mammalian promoter regions
- General regulatory factors (GRFs)
DNA-binding proteins known to regulate and assist directly and indirectly in the positioning of nucleosomes
- HAND-SANT-SLIDE domain
The protein domain of the imitation SWI (ISWI) family involved in DNA translocation around a nucleosome
- Fluorescence resonance energy transfer (FRET)
A biophysical assay able to determine close (that is, nanometre scale) proximity of molecules in vivo or in vitro
- Pre-initiation complex
The complex of general transcription factor proteins assembled at transcription start sites
- MCM helicase
(Mini-chromosome maintenance helicase). A DNA helicase protein complex responsible for unwinding of the DNA helix during replication
- Autonomous replicating sequence (ARS)
A DNA sequence that allows a plasmid to replicate in Saccharomyces cerevisiae. It is often bound by origin of replication complex proteins
- ARS consensus sequences (ACSs)
Consensus DNA motifs found in ARS
Footnotes
Competing interests statement
The authors declare competing interests: see Web version for details.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 3.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 4.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou CY, Johnson SL, Gamarra NI, Narlikar GJ. Mechanisms of ATP-dependent chromatin remodeling motors. Annu Rev Biophys. 2016;45:153–181. doi: 10.1146/annurev-biophys-051013-022819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhee HS, Bataille AR, Zhang L, Pugh BF. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell. 2014;159:1377–1388. doi: 10.1016/j.cell.2014.10.054. Non-canonical nucleosomal structures are identified in vivo at specific genomic regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8.Pokholok DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014;28:672–682. doi: 10.1101/gad.238873.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rando OJ, Ahmad K. Rules and regulation in the primary structure of chromatin. Curr Opin Cell Biol. 2007;19:250–256. doi: 10.1016/j.ceb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang C, Pugh BF. A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol. 2009;10:R109. doi: 10.1186/gb-2009-10-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui F, Cole HA, Clark DJ, Zhurkin VB. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res. 2012;40:10753–10764. doi: 10.1093/nar/gks870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shivaswamy S, et al. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6:e65. doi: 10.1371/journal.pbio.0060065. Nucleosomes are dynamically regulated in response to stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kulaeva OI, Hsieh FK, Chang HW, Luse DS, Studitsky VM. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim Biophys Acta. 2013;1829:76–83. doi: 10.1016/j.bbagrm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber CM, Ramachandran S, Henikoff S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell. 2014;53:819–830. doi: 10.1016/j.molcel.2014.02.014. The +1 nucleosome serves as a barrier to RNA polymerase II transcription. [DOI] [PubMed] [Google Scholar]
- 17.Reja R, Vinayachandran V, Ghosh S, Pugh BF. Molecular mechanisms of ribosomal protein gene coregulation. Genes Dev. 2015;29:1942–1954. doi: 10.1101/gad.268896.115. Nucleosome positioning gates the TSS at ribosomal protein genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krietenstein N, et al. Genomic nucleosome organization reconstituted with pure proteins. Cell. 2016;167:709–721.e12. doi: 10.1016/j.cell.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark DJ. Nucleosome positioning, nucleosome spacing and the nucleosome code. J Biomol Struct Dyn. 2010;27:781–793. doi: 10.1080/073911010010524945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Ma H, Pugh BF. Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res. 2011;21:875–884. doi: 10.1101/gr.117465.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 23.Cairns BR. Chromatin remodeling complexes: strength in diversity, precision through specialization. Curr Opin Genet Dev. 2005;15:185–190. doi: 10.1016/j.gde.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Gkikopoulos T, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yen K, Vinayachandran V, Pugh BF. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013;154:1246–1256. doi: 10.1016/j.cell.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurard-Levin ZA, Quivy JP, Almouzni G. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem. 2014;83:487–517. doi: 10.1146/annurev-biochem-060713-035536. [DOI] [PubMed] [Google Scholar]
- 28.Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell. 2001;7:21–30. doi: 10.1016/s1097-2765(01)00151-4. [DOI] [PubMed] [Google Scholar]
- 29.Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010;24:748–753. doi: 10.1101/gad.1913210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13:373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skene PJ, Henikoff S. Histone variants in pluripotency and disease. Development. 2013;140:2513–2524. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- 33.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 34.Talbert PB, Henikoff S. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol. 2017;18:115–126. doi: 10.1038/nrm.2016.148. [DOI] [PubMed] [Google Scholar]
- 35.Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2016.166. http://dx.doi.org/10.1038/nrm.2016.166. [DOI] [PubMed]
- 36.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplan N, et al. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat Struct Mol Biol. 2010;17:918–920. doi: 10.1038/nsmb0810-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hughes AL, Rando OJ. Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys. 2014;43:41–63. doi: 10.1146/annurev-biophys-051013-023114. [DOI] [PubMed] [Google Scholar]
- 40.Mardis ER. ChIP-seq: welcome to the new frontier. Nat Methods. 2007;4:613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- 41.O’Geen H, Echipare L, Farnham PJ. Using ChIP-seq technology to generate high-resolution profiles of histone modifications. Methods Mol Biol. 2011;791:265–286. doi: 10.1007/978-1-61779-316-5_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet. 2014;15:709–721. doi: 10.1038/nrg3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsompana M, Buck MJ. Chromatin accessibility: a window into the genome. Epigenetics Chromatin. 2014;7:33. doi: 10.1186/1756-8935-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou X, Blocker AW, Airoldi EM, O’Shea EK. A computational approach to map nucleosome positions and alternative chromatin states with base pair resolution. eLife. 2016;5:e16970. doi: 10.7554/eLife.16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Annunziato AT. Split decision: what happens to nucleosomes during DNA replication? J Biol Chem. 2005;280:12065–12068. doi: 10.1074/jbc.R400039200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, et al. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 49.Wade JT, Hall DB, Struhl K. The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature. 2004;432:1054–1058. doi: 10.1038/nature03175. [DOI] [PubMed] [Google Scholar]
- 50.Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nat Genet. 2006;38:1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- 51.Segal E, Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat Rev Genet. 2009;10:443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, et al. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16:847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Albert I, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 55.Tosi A, et al. Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell. 2013;154:1207–1219. doi: 10.1016/j.cell.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science. 2013;340:195–199. doi: 10.1126/science.1229758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang F, Ranjan A, Wei D, Wu C. Comment on “A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme”. Science. 2016;353:358. doi: 10.1126/science.aad5921. [DOI] [PubMed] [Google Scholar]
- 58.Vassileva I, Yanakieva I, Peycheva M, Gospodinov A, Anachkova B. The mammalian INO80 chromatin remodeling complex is required for replication stress recovery. Nucleic Acids Res. 2014;42:9074–9086. doi: 10.1093/nar/gku605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peterson CL, Almouzni G. Nucleosome dynamics as modular systems that integrate DNA damage and repair. Cold Spring Harb Perspect Biol. 2013;5:a012658. doi: 10.1101/cshperspect.a012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Green CM, Almouzni G. When repair meets chromatin. First in series on chromatin dynamics EMBO Rep. 2002;3:28–33. doi: 10.1093/embo-reports/kvf005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dion MF, et al. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–1408. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- 62.Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ray-Gallet D, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 64.Kraushaar DC, et al. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013;14:R121. doi: 10.1186/gb-2013-14-10-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yildirim O, et al. A system for genome-wide histone variant dynamics in ES cells reveals dynamic MacroH2A2 replacement at promoters. PLoS Genet. 2014;10:e1004515. doi: 10.1371/journal.pgen.1004515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svensson JP, et al. A nucleosome turnover map reveals that the stability of histone H4 Lys20 methylation depends on histone recycling in transcribed chromatin. Genome Res. 2015;25:872–883. doi: 10.1101/gr.188870.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keppler A, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 68.Deaton AM, et al. Enhancer regions show high histone H3.3 turnover that changes during differentiation. eLife. 2016;5:e15316. doi: 10.7554/eLife.15316. Demonstration of a role for histone turnover in enhancers during differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belotserkovskaya R, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 70.Jansen A, Verstrepen KJ. Nucleosome positioning in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2011;75:301–320. doi: 10.1128/MMBR.00046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell. 2000;103:423–433. doi: 10.1016/s0092-8674(00)00134-3. [DOI] [PubMed] [Google Scholar]
- 72.Hughes AL, Jin Y, Rando OJ, Struhl K. A functional evolutionary approach to identify determinants of nucleosome positioning: a unifying model for establishing the genome-wide pattern. Mol Cell. 2012;48:5–15. doi: 10.1016/j.molcel.2012.07.003. By placing DNA from one yeast species into another, nucleosome organization and TSS are found to track together, indicating their mutual influence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, et al. Evidence against a genomic code for nucleosome positioning. Reply to “Nucleosome sequence preferences influence in vivo nucleosome organization”. Nat Struct Mol Biol. 2010;17:920–923. doi: 10.1038/nsmb0810-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ioshikhes I, Hosid S, Pugh BF. Variety of genomic DNA patterns for nucleosome positioning. Genome Res. 2011;21:1863–1871. doi: 10.1101/gr.116228.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fenouil R, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de Dieuleveult M, et al. Genome-wide nucleosome specificity and function of chromatin remodelers in ES cells. Nature. 2016;530:113–116. doi: 10.1038/nature16505. Genomic analysis of chromatin remodeller interactions with specific nucleosome positions involved in regulating transcription programmes in embryonic stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter? Dev Biol. 2010;339:258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137:445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 80.de la Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet. 2006;7:461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 81.Mitra D, Parnell EJ, Landon JW, Yu Y, Stillman DJ. SWI/SNF binding to the HO promoter requires histone acetylation and stimulates TATA-binding protein recruitment. Mol Cell Biol. 2006;26:4095–4110. doi: 10.1128/MCB.01849-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roberts CW, Orkin SH. The SWI/SNF complex — chromatin and cancer. Nat Rev Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 83.Ryan MP, Jones R, Morse RH. SWI-SNF complex participation in transcriptional activation at a step subsequent to activator binding. Mol Cell Biol. 1998;18:1774–1782. doi: 10.1128/mcb.18.4.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dutta A, et al. Swi/Snf dynamics on stress-responsive genes is governed by competitive bromodomain interactions. Genes Dev. 2014;28:2314–2330. doi: 10.1101/gad.243584.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de La Serna IL, et al. Mammalian SWI-SNF complexes contribute to activation of the hsp70 gene. Mol Cell Biol. 2000;20:2839–2851. doi: 10.1128/mcb.20.8.2839-2851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tolstorukov MY, et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci USA. 2013;110:10165–10170. doi: 10.1073/pnas.1302209110. SWI/SNF remodelling at promoters is crucial for proper gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cairns BR, et al. RSC, an essential, abundant chromatin-remodeling complex. Cell. 1996;87:1249–1260. doi: 10.1016/s0092-8674(00)81820-6. [DOI] [PubMed] [Google Scholar]
- 89.Parnell TJ, Huff JT, Cairns BR. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008;27:100–110. doi: 10.1038/sj.emboj.7601946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kubik S, et al. Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol Cell. 2015;60:422–434. doi: 10.1016/j.molcel.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 91.Lorch Y, Maier-Davis B, Kornberg RD. Role of DNA sequence in chromatin remodeling and the formation of nucleosome-free regions. Genes Dev. 2014;28:2492–2497. doi: 10.1101/gad.250704.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Boer CG, Hughes TR. Poly-dA:dT tracts form an in vivo nucleosomal turnstile. PLoS ONE. 2014;9:e110479. doi: 10.1371/journal.pone.0110479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618:30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kari V, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016;17:1609–1623. doi: 10.15252/embr.201642352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Torigoe SE, Patel A, Khuong MT, Bowman GD, Kadonaga JT. ATP-dependent chromatin assembly is functionally distinct from chromatin remodeling. eLife. 2013;2:e00863. doi: 10.7554/eLife.00863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nodelman IM, et al. The Chd1 chromatin remodeler can sense both entry and exit sides of the nucleosome. Nucleic Acids Res. 2016;44:7580–7591. doi: 10.1093/nar/gkw406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McKnight JN, Tsukiyama T, Bowman GD. Sequence-targeted nucleosome sliding in vivo by a hybrid Chd1 chromatin remodeler. Genome Res. 2016;26:693–704. doi: 10.1101/gr.199919.115. Provides evidence for nucleosome positioning against a fixed barrier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee JS, et al. Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev. 2012;26:914–919. doi: 10.1101/gad.186841.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park D, Shivram H, Iyer VR. Chd1 co-localizes with early transcription elongation factors independently of H3K36 methylation and releases stalled RNA polymerase II at introns. Epigenetics Chromatin. 2014;7:32. doi: 10.1186/1756-8935-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
- 101.Dang W, Bartholomew B. Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol Cell Biol. 2007;27:8306–8317. doi: 10.1128/MCB.01351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hota SK, et al. Nucleosome mobilization by ISW2 requires the concerted action of the ATPase and SLIDE domains. Nat Struct Mol Biol. 2013;20:222–229. doi: 10.1038/nsmb.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Corona DF, Tamkun JW. Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochim Biophys Acta. 2004;1677:113–119. doi: 10.1016/j.bbaexp.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 104.Aydin OZ, Vermeulen W, Lans H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014;13:3016–3025. doi: 10.4161/15384101.2014.956551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Langst G, Becker PB. Nucleosome mobilization and positioning by ISWI-containing chromatin-remodeling factors. J Cell Sci. 2001;114:2561–2568. doi: 10.1242/jcs.114.14.2561. [DOI] [PubMed] [Google Scholar]
- 107.Hopfner KP, Gerhold CB, Lakomek K, Wollmann P. Swi2/Snf2 remodelers: hybrid views on hybrid molecular machines. Curr Opin Struct Biol. 2012;22:225–233. doi: 10.1016/j.sbi.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deuring R, et al. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol Cell. 2000;5:355–365. doi: 10.1016/s1097-2765(00)80430-x. [DOI] [PubMed] [Google Scholar]
- 109.Strohner R, et al. NoRC — a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001;20:4892–4900. doi: 10.1093/emboj/20.17.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mellor J, Morillon A. ISWI complexes in Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1677:100–112. doi: 10.1016/j.bbaexp.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 111.Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li M, et al. Dynamic regulation of transcription factors by nucleosome remodeling. eLife. 2015;4:e06249. doi: 10.7554/eLife.06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 114.Weintraub H, Palter K, Van Lente F. Histones H2a, H2b, H3, and H4 form a tetrameric complex in solutions of high salt. Cell. 1975;6:85–110. doi: 10.1016/0092-8674(75)90077-x. [DOI] [PubMed] [Google Scholar]
- 115.Weintraub H, Worcel A, Alberts B. A model for chromatin based upon two symmetrically paired half-nucleosomes. Cell. 1976;9:409–417. doi: 10.1016/0092-8674(76)90085-4. [DOI] [PubMed] [Google Scholar]
- 116.Zlatanova J, Bishop TC, Victor JM, Jackson V, van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 117.Black BE, Cleveland DW. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell. 2011;144:471–479. doi: 10.1016/j.cell.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 119.Hondele M, et al. Structural basis of histone H2A–H2B recognition by the essential chaperone FACT. Nature. 2013;499:111–114. doi: 10.1038/nature12242. [DOI] [PubMed] [Google Scholar]
- 120.Aguilar-Gurrieri C, et al. Structural evidence for Nap1-dependent H2A–H2B deposition and nucleosome assembly. EMBO J. 2016;35:1465–1482. doi: 10.15252/embj.201694105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15:1405–1421. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 123.Koerber RT, Rhee HS, Jiang C, Pugh BF. Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol Cell. 2009;35:889–902. doi: 10.1016/j.molcel.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Harada BT, et al. Stepwise nucleosome translocation by RSC remodeling complexes. eLife. 2016;5:e10051. doi: 10.7554/eLife.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dalal Y, Furuyama T, Vermaak D, Henikoff S. Structure, dynamics, and evolution of centromeric nucleosomes. Proc Natl Acad Sci USA. 2007;104:15974–15981. doi: 10.1073/pnas.0707648104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ramachandran S, Henikoff S. Nucleosome dynamics during chromatin remodeling in vivo. Nucleus. 2016;7:20–26. doi: 10.1080/19491034.2016.1149666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xi Y, Yao J, Chen R, Li W, He X. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011;21:718–724. doi: 10.1101/gr.117101.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Knight B, et al. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev. 2014;28:1695–1709. doi: 10.1101/gad.244434.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chereji RV, Ocampo J, Clark DJ. MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol Cell. 2017;65:565–577.e3. doi: 10.1016/j.molcel.2016.12.009. MNase-sensitive regions of the genome do not necessarily contain histones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fei J, et al. The prenucleosome, a stable conformational isomer of the nucleosome. Genes Dev. 2015;29:2563–2575. doi: 10.1101/gad.272633.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Henikoff S, et al. The budding yeast centromere DNA element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife. 2014;3:e01861. doi: 10.7554/eLife.01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Krassovsky K, Henikoff JG, Henikoff S. Tripartite organization of centromeric chromatin in budding yeast. Proc Natl Acad Sci USA. 2012;109:243–248. doi: 10.1073/pnas.1118898109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hasson D, et al. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat Struct Mol Biol. 2013;20:687–695. doi: 10.1038/nsmb.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Furuyama T, Codomo CA, Henikoff S. Reconstitution of hemisomes on budding yeast centromeric DNA. Nucleic Acids Res. 2013;41:5769–5783. doi: 10.1093/nar/gkt314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rhee HS, Pugh BF. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol. 2012 doi: 10.1002/0471142727.mb2124s100. http://dx.doi.org/10.1002/0471142727.mb2124s100. [DOI] [PMC free article] [PubMed]
- 136.Ramachandran S, Zentner GE, Henikoff S. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res. 2015;25:381–390. doi: 10.1101/gr.182618.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry. 2012;51:3302–3309. doi: 10.1021/bi300129b. [DOI] [PubMed] [Google Scholar]
- 138.Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20:2009–2017. doi: 10.1101/gad.1435706. [DOI] [PubMed] [Google Scholar]
- 139.Studitsky VM, Nizovtseva EV, Shaytan AK, Luse DS. Nucleosomal barrier to transcription: structural determinants and changes in chromatin structure. Biochem Mol Biol J. 2016;2:8. doi: 10.21767/2471-8084.100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Eaton ML, et al. Chromatin signatures of the Drosophila replication program. Genome Res. 2011;21:164–174. doi: 10.1101/gr.116038.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.MacAlpine DM, Almouzni G. Chromatin and DNA replication. Cold Spring Harb Perspect Biol. 2013;5:a010207. doi: 10.1101/cshperspect.a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stunkel W, Kober I, Seifart KH. A nucleosome positioned in the distal promoter region activates transcription of the human U6 gene. Mol Cell Biol. 1997;17:4397–4405. doi: 10.1128/mcb.17.8.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhao X, Pendergrast PS, Hernandez N. A positioned nucleosome on the human U6 promoter allows recruitment of SNAPc by the Oct-1 POU domain. Mol Cell. 2001;7:539–549. doi: 10.1016/s1097-2765(01)00201-5. [DOI] [PubMed] [Google Scholar]
- 146.Tramantano M, et al. Constitutive turnover of histone H2A.Z at yeast promoters requires the preinitiation complex. eLife. 2016;5:e14243. doi: 10.7554/eLife.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brickner DG, et al. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5:e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grimaldi Y, Ferrari P, Strubin M. Independent RNA polymerase II preinitiation complex dynamics and nucleosome turnover at promoter sites in vivo. Genome Res. 2014;24:117–124. doi: 10.1101/gr.157792.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Duzdevich D, et al. The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell. 2015;58:483–494. doi: 10.1016/j.molcel.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bell SP, Labib K. Chromosome duplication in Saccharomyces cerevisiae. Genetics. 2016;203:1027–1067. doi: 10.1534/genetics.115.186452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Franco AA, Lam WM, Burgers PM, Kaufman PD. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005;19:1365–1375. doi: 10.1101/gad.1305005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang H, et al. A unique binding mode enables MCM2 to chaperone histones H3–H4 at replication forks. Nat Struct Mol Biol. 2015;22:618–626. doi: 10.1038/nsmb.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang J, et al. The histone chaperone FACT contributes to DNA replication-coupled nucleosome assembly. Cell Rep. 2016;14:1128–1141. doi: 10.1016/j.celrep.2015.12.096. [DOI] [PubMed] [Google Scholar]
- 155.Deniz O, Flores O, Aldea M, Soler-Lopez M, Orozco M. Nucleosome architecture throughout the cell cycle. Sci Rep. 2016;6:19729. doi: 10.1038/srep19729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bar-Ziv R, Voichek Y, Barkai N. Chromatin dynamics during DNA replication. Genome Res. 2016;26:1245–1256. doi: 10.1101/gr.201244.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Annunziato AT. The fork in the road: histone partitioning during DNA replication. Genes (Basel) 2015;6:353–371. doi: 10.3390/genes6020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Katan-Khaykovich Y, Struhl K. Splitting of H3–H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc Natl Acad Sci USA. 2011;108:1296–1301. doi: 10.1073/pnas.1018308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu M, et al. Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science. 2010;328:94–98. doi: 10.1126/science.1178994. [DOI] [PubMed] [Google Scholar]
- 160.Tran V, Lim C, Xie J, Chen X. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science. 2012;338:679–682. doi: 10.1126/science.1226028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nekrasov M, et al. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat Struct Mol Biol. 2012;19:1076–1083. doi: 10.1038/nsmb.2424. [DOI] [PubMed] [Google Scholar]
- 162.Greaves IK, Rangasamy D, Ridgway P, Tremethick DJ. H2A.Z contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci USA. 2007;104:525–530. doi: 10.1073/pnas.0607870104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dumesic PA, et al. Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell. 2015;160:204–218. doi: 10.1016/j.cell.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sirbu BM, Couch FB, Cortez D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc. 2012;7:594–605. doi: 10.1038/nprot.2012.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vasseur P, et al. Dynamics of nucleosome positioning maturation following genomic replication. Cell Rep. 2016;16:2651–2665. doi: 10.1016/j.celrep.2016.07.083. Nucleosomes return to their pre-replication positions in a transcriptionally linked manner after DNA replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ramachandran S, Henikoff S. Transcriptional regulators compete with nucleosomes post-replication. Cell. 2016;165:580–592. doi: 10.1016/j.cell.2016.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Aird D, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rizzo JM, Bard JE, Buck MJ. Standardized collection of MNase-seq experiments enables unbiased dataset comparisons. BMC Mol Biol. 2012;13:15. doi: 10.1186/1471-2199-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mieczkowski J, et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun. 2016;7:11485. doi: 10.1038/ncomms11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wal M, Pugh BF. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 2012;513:233–250. doi: 10.1016/B978-0-12-391938-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kelly TK, et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497–2506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ishii H, Kadonaga JT, Ren B. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci USA. 2015;112:E3457–E3465. doi: 10.1073/pnas.1424804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. Description of a rapid and facile method (assay for transposable-accessible chromatin (ATAC)–seq) for measuring chromatin accessibility on a genomic scale. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schep AN, et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25:1757–1770. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brogaard K, Xi L, Wang JP, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hsieh TH, et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 2015;162:108–119. doi: 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]