

Abstract
Background
Clear-cell sarcoma (CCSA) is an orphan malignancy, characterized by a specific t(12;22) translocation, leading to rearrangement of the EWSR1 gene and overexpression of MET. We prospectively investigated the efficacy and safety of the tyrosine kinase inhibitor crizotinib in patients with advanced or metastatic CCSA.
Patients and methods
Patients with CCSA received oral crizotinib 250 mg twice daily. Primary end point was objective response rate (ORR), secondary end points included duration of response, disease control rate (DCR), progression-free survival (PFS), progression-free rate (PFR), overall survival (OS), OS rate and safety. The study design focused on MET+ disease with documented rearrangement of the EWSR1 gene by fluorescence in situ hybridization.
Results
Among 43 consenting patients with the local diagnosis of CCSA, 36 had centrally confirmed CCSA, 28 of whom were eligible, treated and assessable. Twenty-six out of the 28 patients had MET+ disease, of whom one achieved a confirmed partial response and 17 had stable disease (SD) (ORR 3.8%, 95% confidence interval: 0.1–19.6). Further efficacy end points in MET+ CCSA were DCR: 69.2% (48.2% to 85.7%), median PFS: 131 days (49–235), median OS: 277 days (232–442). The 3-, 6-, 12- and 24-month PFR was 53.8% (34.6–73.0), 26.9% (9.8–43.9), 7.7% (1.3–21.7) and 7.7% (1.3–21.7), respectively. Among two assessable MET− patients, one had stable disease and one had progression. The most common treatment-related adverse events were nausea [18/34 (52.9%)], fatigue [17/34 (50.0%)], vomiting [12/34 (35.3%)], diarrhoea [11/34 (32.4%)], constipation [9/34 (26.5%)] and blurred vision [7/34 (20.6%)].
Conclusions
The PFS with crizotinib in MET+ CCSA is similar to results achieved first-line in non-selected metastatic soft tissue sarcomas with single-agent doxorubicin. The PFS is similar to results achieved with pazopanib in previously treated sarcoma patients.
Clinical trial number
EORTC 90101, EudraCT number 2011-001988-52, NCT01524926.
Keywords: clear-cell sarcoma, MET gene, EWSR1 gene rearrangement, tyrosine kinase inhibitor, crizotinib
Introduction
Clear-cell sarcoma (CCSA) is a rare aggressive tumour that primarily affects adolescents and young adults, typically involves deep soft tissue of the lower extremities and tends to occur near tendons, fascias and aponeuroses [1–5]. CCSA is associated with a high rate of local recurrence, distant metastasis and lymph node involvement, which is uncommon in other types of soft tissue sarcoma (STS) [3–5]. The five-year OS is around 50%–67%, but this decreases to 20% in patients with metastatic disease [3, 6, 7]. CCSA tends to be resistant to conventional chemotherapy and to radiation therapy [3, 6].
CCSA is characterized by the t(12;22)(q13;q12) translocation resulting in the Ewing sarcoma breakpoint region 1/activating transcription factor-1 (EWSR1/ATF1) fusion gene, presenting in >90% of cases (supplementary Material, Introduction and Figures S1 and S2, available at Annals of Oncology online, provide details on the EWSR1/ATF1 fusion gene and the MET signalling pathway) [3, 8, 4]. Fluorescence in situ hybridization (FISH) is used to detect these fusions and to establish the diagnosis of CCSA (supplementary Figure S3, available at Annals of Oncology online). Davies et al. confirmed MET expression requires EWS-ATF1 expression, and the viability and motility of CCSA is dependent on the HGF/MET axis signalling [3]. MET inhibition may offer an indirect target for the treatment of CCSA [3]. In studies using specific inhibitors of MET signalling, MET activity was found to be essential for proliferation and survival of CCSA cell lines and xenograft models [3, 9, 10].
Based on MET involvement in CCSA and the absence of a standard treatment for patients with advanced disease, strong rationale exists to explore therapies that target the MET tyrosine kinase receptor, such as crizotinib (Xalkori®, PF-02341066, Pfizer Inc.) [8, 10]. MET engagement results in the activation of multiple downstream signalling pathways, including phosphatidylinositol 3 kinase (PI3K)/AKT, RAS/MAPK pathways etc. [3, 9, 11]. While several active targets in CCSA exist for future investigations, our study focused on the MET receptor as a target for crizotinib inhibition. Crizotinib, a small molecule TKI, inhibits MET, anaplastic lymphoma kinase (ALK) and ROS proto-oncogene 1 receptor tyrosine kinase (ROS1) [12–16]. It interferes with pathways by competitively inhibiting adenosine triphosphate from binding to the receptor, thereby preventing phosphorylation [12–16]. This blocks the downstream cascade of events, inhibiting the growth and survival of MET dependent cells [12–16]. Crizotinib is approved for first-line treatment of patients with metastatic non-small-cell lung cancer (NSCLC) whose tumours are either ALK- or ROS1-positive, 250 mg twice daily is the recommended oral adult dose [17].
The European Organization for Research and Treatment of Cancer (EORTC) initiated a multinational, multi-tumour, prospective phase II clinical trial (EORTC 90101 ‘CREATE’) to evaluate the efficacy and safety of crizotinib in patients with advanced tumours characterized by MET and/or ALK alterations. CREATE included ALK or MET driven tumour types in 6 disease-specific cohorts. We present the independent CCSA cohort results here.
Methods
Study design
This was a multicentre, biomarker-driven, single agent, non-randomized, open-label, two-stage phase II trial, assessing the activity and safety of crizotinib in patients with locally advanced or metastatic CCSA (EORTC 90101,ClinicalTrials.gov:NCT01524926). The patient population was divided into MET positive (MET+; presence of EWSR1 gene rearrangement) and MET negative (MET−; absence of EWSR1 gene rearrangement) sub-cohorts, which were analysed separately. Investigators were blinded to the centrally assessed MET status.
Ethics approval was obtained by competent committee(s) and according to national legislation. The study was conducted in accordance with: the Declaration of Helsinki; laws and regulations of each participating country/institution; and the International Conference on Harmonisation-Good Clinical Practice.
Patient enrolment
Patient enrolment was based on a single informed consent per patient but followed a multi-step procedure. Step 1 prerequisites for registration were a local diagnosis of advanced and/or metastatic CCSA deemed incurable by conventional surgery, radiotherapy or systemic therapy, the availability of a formalin-fixed paraffin embedded tumour-containing tissue block from primary tumour and/or metastatic site, and written informed consent for collection of the tissue and all other trial-specific procedures.
The requisite criteria for step 2 included receipt of the tissue block by a central biorepository (BioRep, Milan, Italy) with presence of tumour in the shipped material and confirmation of the correct diagnosis of CCSA by central pathology.
Screened patients were treated after completion of step 1 and 2, provided all eligibility criteria were met. There were no limitations in terms of previous systemic or local treatments for CCSA only prior exposure to crizotinib or other specific MET-inhibiting agents was not allowed. Details on the patient selection are described in the study protocol (http://www.eortc.be/services/doc/protocols/1v10.0.pdf).
Treatment, safety and efficacy assessment
Eligible patients with centrally confirmed diagnosis of CCSA were treated with oral crizotinib at a starting dose of 250 mg twice daily. One treatment cycle was defined as 21 days in duration. Treatment continued until progression, unacceptable toxicity, or patient refusal. Dosing instructions were in line with the standard use of crizotinib in the labelled NSCLC indication. Dose and schedule modifications were defined in the protocol.
Safety information was collected at baseline, day 15 of cycle 1 and 2, and at the end of every cycle applying the Common Terminology Criteria for Adverse Events [CTCAE] version 4.0. Tumour assessments were carried out every other cycle by the local investigator according to RECIST 1.1 on the basis of computer tomography or magnetic resonance imaging. Digital images were collected and objective responses were centrally reviewed.
Assessment of MET alterations
Patients were attributed to the MET+ sub-cohort based on the presence of the EWSR1 gene rearrangement. This was assessed by FISH with the commercially available dual colour break-apart rearrangement probe Vysis® LSI®EWSR1(22q12) (Abbott Molecular). At least 15% of tumour cells had to show a rearrangement for a case to be considered MET+. This threshold was established by validation of the specific FISH probe in the specific indication in the laboratory performing the evaluation, as well as running this test in clinical routine. EWSR1 rearrangement is a molecular hallmark of CCSA, so a very low rate of MET− cases was expected.
Outcomes
The main objective was to study the activity of crizotinib in MET+ CCSA patients. The primary end point was ORR per RECIST 1.1 with response confirmation, assessed by the local investigator. This end point was chosen based on the response pattern seen with crizotinib in the labelled indication of NSCLC and in the absence of reliable reference data on PFS or PFR in CCSA. Secondary end points included: duration of response, DCR, PFS, PFR, OS, OSR, safety and correlative/translational research end points. The DCR was defined as the percentage of patients achieving either a complete (CR) or partial response (PR) or SD.
Statistical analysis
The statistical design was conceptually focused on cases with centrally documented EWSR1 fusion (MET+ sub-cohort). It was decided that showing an ORR >10% (null hypothesis) in CCSA MET+ patients, a rare population, resistant to chemotherapy and radiotherapy, would be promising for future research. Therefore a Simon's optimal two-stage design [18] was implemented with the aim of excluding an ORR ≤10% under the alternative assumption that 30% ORR can be achieved with crizotinib. The type I error and power were set at 10%. In stage 1, if at least two out of the first 12 eligible and assessable CCSA MET+ patients achieved a confirmed RECIST PR or CR, a maximum of 35 patients were to be enrolled. In stage 2, if less than 6 out of the 35 eligible and assessable patients responded, the treatment was declared ineffective. If at least 6 out of the 35 patients responded, further study of crizotinib in CCSA was warranted.
MET− patients served as a non-randomized, non-historical and treated internal control. The entry of ‘all comers’ independent of their MET status was allowed, to avoid delay of treatment of patients in need of an active intervention and to provide reference data for both subsets. The entry of MET− cases was considered ethical due to the lack of validated treatment alternatives for this disease.
The stopping rules and activity end points details are provided in supplementary Material/Methodology, available at Annals of Oncology online. Analyses were carried out using SAS version9.4 (SAS Institute, Cary).
Results
Patient disposition, reference pathology, clinical screening and enrolment
Between 28 January 2013 and 1 December 2014, 16 sites in 8 European countries recruited 43 patients with local diagnosis of CCSA. Only 36/43 (84%) patients had a confirmed CCSA according to reference pathology were eligible for screening and potential treatment.
The seven non-confirmed, non-eligible cases included two cases of melanoma, three non-specified malignancies and one case with insufficient material for reference pathology. One patient withdrew from study before central review. These patients were not treated.
A total of 34 of the 36 patients with centrally confirmed CCSA were enrolled in the study (step 3) and started treatment with crizotinib. The two remaining patients withdrew consent. A total of 28 eligible patients (26 MET+ and 2 MET−) with confirmed CCSA who started treatment with crizotinib were assessable for the primary end point (supplementary Figure S4, available at Annals of Oncology online, provides the CONSORT-like patient disposition).
As expected, the number of patients with MET− disease was very low (two patients eligible and assessable). As the trial was conceptually focused on EWSR1 rearranged cases, and due to the low sample size of MET− disease, only some key results obtained in the latter cases will be presented. Supplementary Figure S4, available at Annals of Oncology online, depicts the trial profile.
Genetic analysis and molecular epidemiology
FISH testing was completed according to protocol by the academic laboratory within a median time of 4 days (range 1–15) after receipt of technically useful, unstained slides from the central biorepository.
Among 36 patients with centrally confirmed CCSA diagnosis, 32 (88.9%) had documented EWSR1 rearrangement (MET+ cases). Only three patients (8.3%) had no detectable EWSR1. FISH testing could not be carried out in the remaining patient due to technical failure. Supplementary Table S1, available at Annals of Oncology online, shows an overview of the molecular characteristics.
Due to rapid accrual of patients, a delay in reporting clinical efficacy results to EORTC, and in the light of the lack of treatment alternatives for this highly resistant malignancy, we over-recruited the CCSA cohort of EORTC 90101. We recruited more than the 12 MET+ CCSA patients required to complete stage 1, but less than 35 eligible and assessable patients for completing stage 2 according to protocol. Recruitment to both the MET+ and MET− CCSA sub-cohorts was suspended on 5 February 2015, after having analysed the ORR in the first 12 eligible and assessable MET+ cases.
Patient characteristics
Among the total group with confirmed diagnosis of CCSA, 31 patients with MET+ disease were treated, two patients with MET− CCSA entered the treatment phase and one patient with unknown MET status (MET?) received crizotinib.
Table 1 shows the characteristics of these 34 treated patients. Their median age was 44 years, 35.3% (12/34) had an ECOG PS of 1 and the majority [91.2% (31/34)] had undergone prior surgery. Only 26.5% (9/34) had received prior systemic therapy, illustrating the lack of treatment options outside of clinical trials for such patients.
Table 1.
Key patient characteristics
|
MET status |
||||
|---|---|---|---|---|
| MET + (N =31) | MET − (N=2) | MET? (N=1) | Total (N=34) | |
| N (%) | N (%) | N (%) | N (%) | |
| Age (years) | ||||
| Median | 44.0 | 42.5 | 40.0 | 44.0 |
| Range | 18.0–64.0 | 34.0–51.0 | 40.0–40.0 | 18.0–64.0 |
| Eastern Cooperative Oncology Group performance status | ||||
| 0 | 15 (48.4) | 2 | 0 | 17 (50.0) |
| 1 | 12 (35.3) | 0 | 0 | 12 (35.3) |
| 2 | 4 (12.9) | 0 | 1 | 5 (14.7) |
| Sex | ||||
| Male | 19 (61.3) | 2 | 0 | 21 (61.8) |
| Female | 12 (38.7) | 0 | 1 | 13 (38.2) |
| Any previous surgery for CCSA | 28 (90.3) | 2 | 1 | 31 (91.2) |
| Any prior systemic anticancer therapy | 9 (29.0) | 0 | 0 | 9 (26.5) |
| Chemotherapy | 9 (29.0) | 0 | 0 | 9 (26.5) |
| Tyrosine kinase inhibitor | 1 | 0 | 0 | 1 |
| Immunotherapy | 1 | 0 | 0 | 1 |
Crizotinib treatment
As of 2 March 2017, with a median follow-up of 281 days (range 43–933), 2.9% (1/34) of patients were still receiving treatment. Only 38.2% (13/34) of treated patients required dose reductions or dose modifications (Table 2). The total treatment duration with crizotinib ranged from 3 to 849+ days. Reasons for treatment discontinuation are shown in Table 2.
Table 2.
Study treatment
|
MET status |
||||
|---|---|---|---|---|
| MET + (N=31) | MET − (N=2) | MET? (N=1) | Total (N=34) | |
| N (%) | N (%) | N (%) | N (%) | |
| Treatment, dose intensity and dose adjustments | ||||
| Relative dose intensity (%) | ||||
| Median | 100.0 | 97.2 | 100.0 | 100.0 |
| Range | 78.2–104.0 | 93.6–100.7 | 100.0–100.0 | 78.2–104.0 |
| Number of patients with at least one treatment modification | 12 (38.7.0) | 1 | 0 | 13 (38.2) |
| Reduction to dose level-1 (200 mg twice daily) | 3 (9.7) | 1 | 0 | 4 (11.8) |
| Reduction to dose level-2 (250 mg once daily) | 3 (9.7) | 0 | 0 | 3 (8.8) |
| Other dose level modification | – | – | – | – |
| Interruption of treatment | 10 (32.3) | 0 | 0 | 10 (29.4) |
| Treatment duration | ||||
| Duration of treatment (weeks) | ||||
| Median | 12.0 | 14.9 | 2.3 | 11.5 |
| Range | 0.4–121.3 | 9.4–20.3 | 2.3–2.3 | 0.4–121.3 |
| Number of cycles | ||||
| Median | 4 | 5.5 | 1 | 4 |
| Range | 1–40 | 4–7 | 1–1 | 1–40 |
| Reasons for treatment discontinuation | ||||
| Treatment status | ||||
| Ongoing | 1 | 0 | 0 | 1 |
| Stopped | 30 (96.8) | 2 | 1 | 33 (97.1) |
| Major reason for protocol treatment discontinuation | ||||
| Progression of CCSA | 20 (66.7) | 2 | 0 | 22 (66.7) |
| Toxicity | 2 | 0 | 0 | 2 |
| Fatigue grade 3, nausea, emesis | 1 | |||
| Grade 2 hepatic toxicity | 1 | |||
| Patient's decision | 2 | 0 | 0 | 2 |
| Symptomatic deterioration without radiological evidence of progression of CCSA | 4 (13.3) | 0 | 1 | 5 (15.2) |
| Other | 2 | 0 | 0 | 2 |
| Patient died at home with general deterioration of clinical condition | 1 | |||
| Investigator decision | 1 | |||
Activity of crizotinib in MET+ CCSA
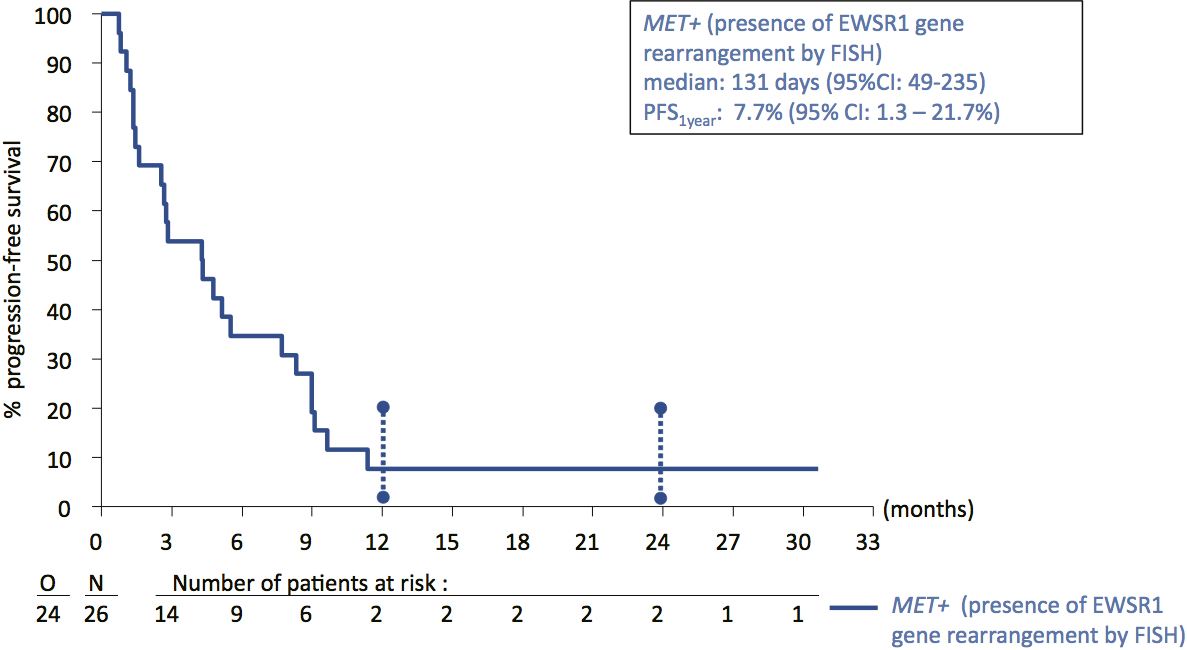
The primary end point was the ORR as assessed by the local investigator, with response confirmation. An objective PR was observed in 1/26 MET+ patients [3.8% ORR; 95% confidence interval (CI): 0.1% to 19.6%]. The primary end point of the trial was not met. The duration of response in the responding patient was 851+ days; the patient is still on active treatment at the data cut-off, having received 40+ cycles of treatment. RECIST SD was observed in 17/26 MET+ patients (65.4%). Disease progression was seen in eight patients (30.8%). Disease control was achieved in 18/26 MET+ patients (DCR; 69.2%, 95% CI: 48.2–85.7). The median PFS was 131 days (95% CI: 49–235; supplementary Figure S5, available at Annals of Oncology online). The 3, 6, 12 and 24 month PFR was 53.8% (34.6–73.0), 26.9% (9.8–43.9), 7.7% (1.3–21.7) and 7.7% (1.3–21.7), respectively. The median OS was 277 days (95% CI: 232–442), and the OSR was 36.1% (95% CI: 18.2% to 54.3%) at 1 year and 9.4% (95% CI: 1.7% to 25.3%) at 2 years (supplementary Figure S6, available at Annals of Oncology online). Figure 1A illustrates the maximum shrinkage of target lesions during treatment. Half of the MET+ CCSA cases had a measurable reduction of target lesions. Figure 1B summarizes the clinical course of all treated patients.
Figure 1.

(A) Maximum shrinkage of target lesions (per protocol) according to local investigator’s assessment. (B) Clinical course of patients in the CCSA MET+ and MET− sub-cohorts.
Activity of crizotinib in MET−/MET? CCSA
None of the MET− patients had a response: one had SD followed PD at day 143 and one had PD at day 66. Key efficacy data for the total study population are summarized in Table 3.
Table 3.
Response assessment and efficacy summary, according to investigator assessment
| MET status | |||
|---|---|---|---|
| MET + (N =26) | MET − (N =2) | Total (N =28) | |
| N (%) | N (%) | N (%) | |
| Best response | |||
| Complete response | – | – | – |
| Partial response | 1 | 0 | 1 |
| Stable disease | 17 (65.4) | 1 | 18 (64.3) |
| Progressive disease | 8 (30.8) | 1 | 9 (32.1) |
| Objective response rate | 3.8% | 0% | 3.6% |
| (95% CI ) | (0.1% to 19.6%) | – | (0.1% to 18.3%) |
| Disease control rate | 69.2% | 50% | 67.9% |
| (95% CI) | (48.2% to 85.7%) | (1.3% to 98.7%) | (47.7% to 84.1%) |
| Progression-free survival | |||
| Alive with no evidence of disease | 2 | 0 | 2 |
| Progression of CCSA or died | 24 (92.3) | 2 | 26 (92.9) |
| Median in days | 131 | 96.5 | 128.5 |
| (95% CI) | (49–235) | (66–127) | (66–168) |
| 1-Year progression-free survival rate | 7.7% | 0 | 7.1% |
| (95% CI) | (1.3% to 21.7%) | (1.3% to 20.4%) | |
| 2-Year progression-free survival rate | 7.7% | n.a. | 7.1% |
| (95% CI) | (1.3% to 21.7%) | (1.3% to 20.4%) | |
| Survival status | |||
| Alive | 4 (15.4) | 1 | 5 (17.9) |
| Dead | 22 (84.6) | 1 | 23 (82.1) |
| Reason of death : | |||
| Progression of CCSA | 18 (69.2) | 1 | 19 (67.9) |
| Toxicity and progression of CCSA | 1 | 0 | 1 |
| Other reason | 1 | 0 | 1 |
| Unknown | 2 | 0 | 2 |
| Median in days | 277 | n.a. | 286 |
| (95% CI) | (232–442) | (232–442) | |
| 1-Year survival rate | 36.1% | 100% | 40.8% |
| (95% CI) | (18.2% to 54.3%) | (100% to 100%) | (22.6% to 58.3%) |
| 2-Year survival rate | 9.4% | 50.0% | 10.6% |
| (95% CI) | (1.7% to 25.3%) | (0.6% to 91.0%) | (2.1% to 27.4%) |
CI, confidence interval.
Safety and toxicity
No new or unexpected safety signals were detected. The most common treatment-related adverse events occurring in ≥10% of the 34 patients who started crizotinib were nausea (52.9% of patients), fatigue (50.0%), vomiting (35.3%), diarrhoea (32.4%), constipation (26.5%) and blurred vision (20. 6%). The reported treatment-related grade 3/4 adverse events were nausea (two patients), fatigue (two), gastritis (one), Qt prolongation (one) and anorexia (one). Supplementary Tables S2 and S3, available at Annals of Oncology online, show adverse events occurring in ≥10% of treated patients.
Three deaths occurred on treatment or within 4 weeks of treatment-discontinuation, all deemed unrelated to crizotinib treatment. There was one possibly treatment-related death. This patient developed pneumonia, pulmonary embolism and pneumonitis during cycle 12 of treatment.
Discussion
The treatment of advanced, inoperable CCSA remains a challenge due to the lack of an established systemic treatment standard. Information from prospective clinical trials on the efficacy of systemic treatments for CCSA is limited. EORTC 90101 CREATE is likely the first, well powered study in this setting. The main objective of this phase II study was to assess the activity of crizotinib in this rare and chemotherapy-resistant, translocation-related sarcoma. The ORR was 3.8% (1/26 PR, 95% CI: 0.1% to 19.6%) and the primary end point of the trial was not met, as we did not observe at least two objective and radiologically confirmed responses among the first 12 eligible and assessable MET+ cases.
Multiple factors led to over-recruitment of patients beyond stage 1 of Simon’s optimal two-stage design. We saw rapid accrual of CCSA cases, with the majority of patients previously untreated, reflecting the high unmet medical need in this orphan malignancy. Investigators observed a large proportion of patients achieving early SD under treatment with crizotinib, and all these cases could theoretically still convert upon further exposure to an objective response. Furthermore, all responses had to be confirmed by a second scan, to be in line with RECIST 1.1. This led to a delay in reporting efficacy data to EORTC, as investigators had to wait until their patients came off study or had reached a confirmed PR. By that time we had exceeded the minimum sample size to assess the futility of crizotinib, without having reached the stage 2 sample size according to trial design. Due to the lack of treatment alternatives we accepted this over-recruitment and this also provided an opportunity to gain deeper insight into the natural course of this cancer.
Even though we found alterations leading to MET expression in 88.9% of our cases, which is in line with the literature, the inhibition of MET by crizotinib only translated in one, however, durable response. It is unclear why this patient had such exceptional response, but we hope that further tissue-based analysis will provide an explanation. We cannot exclude that this response was induced by effects other than MET inhibition, as crizotinib inhibits more than one target. Interestingly, 18/26 (69.2%, 95% CI: 48.2–85.7) of our cases achieved disease control for a median of 131 days. This suggests that PFS or PFR would have been better end points for assessing the efficacy of crizotinib in this disease. The response pattern of MET-driven malignancies to crizotinib might be different from the volumetric responses seen in ALK-driven NSCLC.
Although it was shown that the presence of EWSR1–ATF1 fusion protein, characteristic of CCSA, was required for MET expression and that MET inhibition significantly reduced CCSA cell growth in vitro [3] it is not yet known if MET expression and/or activation is present at the same level in all CCSA cases. It is possible that other factors (e.g. level of HGF expression) contributes to the role of MET in oncogenesis and therefore variable elements may influence the response to MET inhibitors. This hypothesis, along with the influence of MET-related pathways status, is currently being tested.
Based on a retrospective statistical analysis of multiple EORTC sarcoma trials, Van Glabbeke et al. [2] proposed reference values for potentially active agents in STS. For first-line therapy, she recommended a 6 month PFR of ≥30%–56% and for second-line therapy a 3 month PFR of ≥40% as an indicator of promising activity, while a 6 month PFR of ≤20% would suggest inactivity. In our CCSA MET+ group, the 3- and 6-month PFR was 53.8% (34.6–73.0) and 26.9% (9.8–43.9). In an exploratory analysis, we looked at the outcome achieved with crizotinib in pre-treated (N = 7/26) versus non-pre-treated (N = 19/26) patients with EWSR1 rearrangement. The first-line subset had a 3- and 6-month PFR of 52.6% (30.2–75.1) and 42.1% (19.9%–64.3%). The second-line subset had a 3- and 6-month PFR of 57.1% (20.5%–93.8%) and 14.3% (0.0%–40.2%). This post hoc analysis suggests that crizotinib is active in this setting, following Van Glabbeke’s criteria.
The approval and routine use of most drugs for the treatment of STS has been based on trials pooling various types of sarcoma together, which limits the interpretation of the efficacy of such agents in a given histological subtype. Our phase II trial was highly histotype-specific, involved reference pathology and genetic characterization of CCSA as one of the rarest and most treatment-resistant members of the STS family. In this context, it is noteworthy that the PFS seen with crizotinib in MET+ CCSA was similar to results achieved in non-selected patients with advanced STS treated with single-agent doxorubicin in first line (4.6 months, 95% CI: 2.9–5.6) [19], or with the oral angiogenesis inhibitor pazopanib in previously treated patients (4.6 months, 95% CI: 3.7–4.8) [2]. In our smaller, more exploratory study, crizotinib achieved a median PFS in CCSA with MET alterations of 4.4 months (95% CI: 1.6–7.8).
Statistically, we focused on cases with EWSR1 positive CCSA as assessed by FISH, while allowing ‘all comers’ to enter the study, provided they had a centrally confirmed diagnosis. This served multiple strategic purposes: to avoid delay during study entry after consent; to give the laboratory sufficient time for FISH; to offer an experimental treatment to patients with an unmet medical need; to collect data on the molecular epidemiology of advanced CCSA; and to provide a reference for future trials in this rare cancer. The expected small sample size of the MET− sub-cohort of our trial population however precludes drawing definitive conclusions for this group.
Reference pathology was an important component of the trial. Our pathology review was completed within a median of 3 days (range 1–17) after receipt of unstained slides. It was based on a review of local pathology and FISH reports, central microscopy and additional stainings if required, and FISH testing in Leuven in all cases. Of note, only 36 of the 43 consenting patients had a centrally confirmed CCSA. This highlights once again the complexity of proper morphological characterization of rare malignancies, even in highly dedicated academic institutions, especially in tumour types that can mimick other diseases. The misclassification rate here was similar to that seen in other STS studies.
Another specific feature was the mandatory collection of a non-returnable, archived tissue block from all participating patients for research. Many investigators are hesitant to provide commercial trial sponsors with a tissue block for research, while EORTC as an academic non-for-profit organization has the capacity to function as a biobank and non-commercial facilitator of translational work. We were able to collect tissue from 42 consenting patients, including material from 36 individuals with centrally confirmed CCSA. This precious resource is now the basis for multiple ongoing and planned exploratory studies, which will lead to a better understanding of the CCSA biology and the identification of prognostic or predictive biomarkers and treatment strategies for this rare cancer.
Our study showed variable response in the MET+ cohort which suggests the presence of other factors in combination with EWSR1 rearrangement which might predict crizotinib’s efficacy. We are currently performing correlative studies using whole exome sequencing to evaluate the mutational profile and low-coverage whole genome sequencing to study copy number changes, which will be supplemented by research using tissue microarrays constructed from the tissue blocks, to better understand the molecular background of CCSA and individual cases’ sensitivity or resistance to crizotinib.
In our complex trial we demonstrated that EORTC can perform multi-tumour, precision-medicine phase II trials in rare cancers with collection of tissue blocks, real time reference pathology and molecular characterization. Given the inherent limitations of performing larger prospective trials in ultra-rare diseases, innovative trial methodology like the basket approach chosen in EORTC 90101, and new regulatory mechanisms are required to provide patients with orphan malignancies with potentially active drugs such as crizotinib.
The adverse events observed in this study were consistent with safety data for crizotinib in patients with NSCLC. No new adverse events were observed. Dose intensity was high and the incidence of dose modifications was relatively low.
This study illustrates once again the methodological limitations using response rate in early clinical trials in oncology. This end point had been chosen based on the impressive volumetric responses seen with crizotinib in the labelled indication of ALK+ NSCLC and due to the absence of reliable reference data on PFS or PFR in this rare type of sarcoma. In general, EORTC is recommending using time-related end points such as PFR during the early exploration of novel agents in STS [1], which provided the phase II rationale for at least two successful sarcoma registration trials during the past 5 years [2, 20].
We were able to demonstrate that crizotinib provided clinical benefit to patients with locally advanced or metastatic MET+ CCSA. This is noteworthy as this orphan malignancy is renowned for being profoundly resistant to conventional systemic agents. Response rate should not be the primary end point for future phase II trials with MET inhibiting agents for CCSA. DCR, PFS and/or PFR would be a more appropriate reflection of the therapeutic effects of treatments in this disease, where progression arrest might be more important than shrinkage of the tumour and its metastasis. Other MET inhibitors, such as small molecules (highly specific or multi-targeted) or monoclonal antibodies, either given as single agents or in combination with other drugs, could be studied in future CCSA trials.
Other local investigators
Germany: Prof. B. Kasper, MD.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
CREATE was sponsored by EORTC and supported by Pfizer. We thank the involved patients and their families for participating in this study. We thank the EORTC Headquarters team, investigators, nurses and other study staff for their contributions to this trial. PS funded editorial support, and J. O’Regan (Bingham Mayne and Smith Ltd.) provided editorial assistance with an early draft of the manuscript. This publication was supported by the EORTC Cancer Research Fund. TvC and JC helped with the review of radiology. SS is supported by the NIHR, University College London Hospitals Biomedical Research Centre. ML received a fellowship supported by Fonds Cancer (Brussels, Belgium). We also want to thank the members of Steering Committee for their contribution.
Funding
This work was done by EORTC and supported by Pfizer Inc. as an investigator-initiated trial (no grant number is applicable). EORTC was the legal sponsor. Pfizer Inc. provided the investigational agent and funding, but had no role in the study design, data collection, analysis, interpretation, writing of the report or decision to publish this report. The database is held by EORTC, and EORTC statisticians carried out the analysis.
Disclosure
PR and LHL received honoraria from Pfizer outside the scope of this study. JYB received research support and honoraria from Pfizer outside the scope of this study. VG received honoraria from Bristol Myers Squibb, Novartis, Pfizer; advisory board for Bristol Myers Squibb, Novartis, Pfizer, Roche; received travel grant from Bristol Myers Squibb, MSD, Novartis, Pfizer. PR received grants and personal fees from Novartis, received personal fees from Pfizer, Bayer, PharmaMar, Amgen, AstraZeneca, Clinigen, Lilly, Deciphera, outside the submitted work. JS received honoraria from Roche, Novartis, Swedish Orphan, Merck. WvdG received research support from Novartis, honoraria from Bayer. SB received honoraria from Pfizer for consulting and CME activities. PS, AW, SS, AA, FD, SR, MGL, RS, MDR, SM, ML, TR and LC have declared no conflicts of interest.
References
- 1. Van Glabbeke M, Verweij J, Judson I, Nielsen OS.. EORTC Soft Tissue and Bone Sarcoma Group. Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer 2002; 38: 543–549. [DOI] [PubMed] [Google Scholar]
- 2. van der Graaf WT, Blay JY, Chawla SP. et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012; 379: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 3. Davis IJ, McFadden AW, Zhang Y. et al. Identification of the receptor tyrosine kinase c-Met and its ligand, Hepatocyte Growth Factor, as therapeutic targets in clear cell. Sarcoma. Cancer Res 2010; 70: 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang WL, Mayordomo E, Zhang W. et al. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod Pathol 2009; 22: 1201–1209. [DOI] [PubMed] [Google Scholar]
- 5. Hocar O, Le Cesne A, Berissi S. et al. Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 52 cases. Dermatol Res Pract 2012; 984096. doi:10.1155/2012/984096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Speleman R, Sciot F, Clear cell sarcoma of soft tissue In Fletcher C, Unni K, Mertens F (eds), World Health Organization classification of tumours pathology and genetics of tumours of soft tissue and bone. Lyon, France: IARC Press, 2002, pp. 211–212. [Google Scholar]
- 7. Hisaoka M, Ishida T, Kuo TT. et al. Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 2008; 32: 452–460. [DOI] [PubMed] [Google Scholar]
- 8. Schaefer KL, Brachwitz K, Wai DH. et al. Expression profiling of t(12;22) positive clear cell sarcoma of soft tissue cell lines reveals characteristic up-regulation of potential new marker genes including ERBB3. Cancer Res 2004; 64: 3395–3405. [DOI] [PubMed] [Google Scholar]
- 9. McGill GG, Haq R, Nishimura EK, Fisher DE.. c-Met expression is regulated by Mitf in the melanocyte lineage. J Biol Chem 2006; 281: 10365–10373. [DOI] [PubMed] [Google Scholar]
- 10. Wagner AJ, Goldberg JM, Dubois SG. et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: results of a multicenter phase 2 trial. Cancer 2012; 118: 5894–5902. [DOI] [PubMed] [Google Scholar]
- 11. Meyer R, D'Alessandro LA, Kar S. et al. Heterogeneous kinetics of AKT signaling in individual cells are accounted for by variable protein concentration. Front Physiol 2012; 3: 451.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodig SJ, Shapiro GI.. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr Opin Investig Drugs 2010; 11: 1477–1490. [PubMed] [Google Scholar]
- 13. Sahu A, Prabhash K, Noronha V. et al. Crizotinib: a comprehensive review. South Asian J Cancer 2013; 2: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karachaliou N, Rosel R, Molina MA, Viteri S.. Predicting resistance by selection of signaling pathways. Transl Lung Cancer Res 2014; 3: 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stone A, Harrington K, Frakes M. et al. EGFR and c-Met inhibitors are effective in reducing tumorigenicity in cancer. J Carcinog Mutagen 2014; 5: 173. [Google Scholar]
- 16. Camidge DR, Ou S-HI, Shapiro G. et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC).2014 ASCO Annual Meeting. J Clin Oncol 2014; 32: 5s (suppl; abstr 8001). [Google Scholar]
- 17.Crizotinib Summary of Product Characteristics (SPC); http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002489/WC500134759.pdf (2 March 2015, date last accessed).
- 18. Simon R. Optimal two-stage designs for phase II clinical trials. Controlled Clinical Trials 1989; 10: 1–10. [DOI] [PubMed] [Google Scholar]
- 19. Judson I, Verweij J, Gelderblom H. et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 2014; 15: 415–423. [DOI] [PubMed] [Google Scholar]
- 20. Schöffski P, Chawla S, Maki RG. et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016; 387: 1629–1637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.