

Abstract
Background
Pseudohypoparathyroidism (PHP) is a rare disease whose phenotypic features are rather difficult to identify in some cases. Thus, although these patients may present with the Albright’s hereditary osteodystrophy (AHO) phenotype, which is characterized by small stature, obesity with a rounded face, subcutaneous ossifications, mental retardation and brachydactyly, its manifestations are somewhat variable. Indeed, some of them present with a complete phenotype, whereas others show only subtle manifestations. In addition, the features of the AHO phenotype are not specific to it and a similar phenotype is also commonly observed in other syndromes. Brachydactyly type E (BDE) is the most specific and objective feature of the AHO phenotype, and several genes have been associated with syndromic BDE in the past few years. Moreover, these syndromes have a skeletal and endocrinological phenotype that overlaps with AHO/PHP. In light of the above, we have developed an algorithm to aid in genetic testing of patients with clinical features of AHO but with no causative molecular defect at the GNAS locus. Starting with the feature of brachydactyly, this algorithm allows the differential diagnosis to be broadened and, with the addition of other clinical features, can guide genetic testing.
Methods
We reviewed our series of patients (n = 23) with a clinical diagnosis of AHO and with brachydactyly type E or similar pattern, who were negative for GNAS anomalies, and classify them according to the diagnosis algorithm to finally propose and analyse the most probable gene(s) in each case.
Results
A review of the clinical data for our series of patients, and subsequent analysis of the candidate gene(s), allowed detection of the underlying molecular defect in 12 out of 23 patients: five patients harboured a mutation in PRKAR1A, one in PDE4D, four in TRPS1 and two in PTHLH.
Conclusions
This study confirmed that the screening of other genes implicated in syndromes with BDE and AHO or a similar phenotype is very helpful for establishing a correct genetic diagnosis for those patients who have been misdiagnosed with “AHO-like phenotype” with an unknown genetic cause, and also for better describing the characteristic and differential features of these less common syndromes.
Electronic supplementary material
The online version of this article (10.1186/s12881-018-0530-z) contains supplementary material, which is available to authorized users.
Keywords: Brachydactyly, Pseudohypoparathyroidism, Albright’s hereditary osteodystrophy, Hormone resistance, Short stature
Background
Albright’s hereditary osteodystrophy (AHO) is a unique phenotype classically associated with pseudohypoparathyroidism (PHP) [1, 2]. This phenotype was initially described by Albright et al. as a constellation of signs, including short stature, obesity with a rounded face, subcutaneous ossifications, mental retardation and brachydactyly. Parathyroid hormone (PTH) resistance was also originally included as a feature of the AHO phenotype as these authors noticed a reduced calcaemic and phosphaturic response to injected bovine parathyroid extract in such patients with normal renal function [1]. However, more patients with this phenotype but lacking hormone resistance were described in 1952, thus this disease was termed pseudopseudohypoparathyroidism (PPHP). Consequently, this type of hormonal resistance was included as a non-obligatory manifestation of AHO [3]. Many years later, genetic and/or epigenetic alterations at the guanine nucleotide-binding protein, alpha-stimulating (Gsα) locus (GNAS) were identified as the cause of this condition in about 70% of patients with a clinical diagnosis of PHP/PPHP [4], or iPPSD2 (inactivating PTH/PTHrP signalling disorder) and iPPSD3 according to the new proposed classification [5].
Despite this high detection rate of GNAS molecular defects, some patients with a clinical suspicion of PHP/PPHP still lack a confirmed molecular diagnosis, possibly due to the variability of the manifestations in terms of both number and severity, especially in cases in which there is no family history ([5–7] and personal data). In addition, the features of the AHO phenotype are not exclusive to PHP/PPHP. For example, AHO-like syndrome, or brachydactyly and mental retardation syndrome (BDMR, OMIM#600430), as its name indicates, includes a group of patients who show several features of AHO (BDE and mental retardation being the most notable) but with normal Gsα levels and with no endocrine abnormality [8]. These patients frequently carry deletions at the 2q37 chromosome or mutations in the gene coding for histone deacetylase 4 (HDAC4), which is found at this locus [8, 9]. Similarly, the biochemical alterations (hypocalcaemia and hyperphosphatemia) observed in PHP are also present in other syndromes associated with calcium homeostasis, such as hypoparathyroidism [10]. PTH resistance and brachydactyly (but in a more severe form) are also present in acrodysostosis with multihormonal resistance ACRDYS1 (or iPPSD4, OMIM#101800) [11–13], which is associated with mutations in the gene coding for the cAMP-dependent protein kinase type 1 regulatory subunit protein (PRKAR1A) [14]. Another type of acrodysostosis, which lacks hormone resistance (ACRDYS2 or iPPSD5, OMIM#6146139), is caused by mutations in the gene coding for phosphodiesterase 4D (PDE4D) [15, 16].
Considering recent publications in which a significant number of patients were clinically misdiagnosed as PHP when they actually had other syndromes [17], our goal was to validate our diagnostic algorithm starting with the brachydactyly feature to guide candidate gene testing in patients with features of AHO who do not carry genetic or epigenetic alterations at the GNAS locus.
Methods
Patients
The current series involved 23 out of a total of 149 patients referred to the Molecular (Epi)Genetics Laboratory at OSI Araba University Hospital for molecular diagnosis with a clinical suspicion of AHO phenotype with or without PTH resistance and the presence of brachydactyly type E or a similar pattern. (Epi)genetic alterations at the GNAS locus had been previously ruled out as described [18].
The clinical details of the whole series studied are summarized in Table 1. Some of these patients had already been reported, as indicated in the Table 1. The clinical history of the patients, including hand(s) (Additional files 1, 2 and 3: Figures S1-S3) and feet radiographs and clinical photos (if available), was requested from the physicians who referred the samples for genetic study.
Table 1.
Clinical description of patients studied (patient data as provided by the clinician at the reference centre)
| PATIENT | Age at consultation | Age of genetic diagnosis | Sex | Elevated PTH | Ca/P | Vitamin 25(OH)D | BD | MR | Height (cm) | BMI | Facial dimorphisms | Skeletal dysplasia | Advanced bone age | Dental defects | Other features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PHP01 (P9 [30]) |
7y | 12y6m | M | Yes (and TSH) | N | ND | Severe and generalized | Behavioural disorder | −1,5SD | + 1,3SD | broad face with widely spaced eyes | maxillonasal hypoplasia, severe hypoplasia of the skull, thickened calvarium, increased size of the jaw with severe malocclusion | No | ND | – |
| PHP02 (P8 [30]) |
6y6m | 8y6m | F | Yes | P↑ | ND | Severe and generalized | No | -2.5SD | 0.2SD | broad face with widely spaced eyes,maxillonasal hypoplasia | severe hypoplasia of the skull, thickened calvarium | No | Yes | pigmented skin spots |
| PHP03 (P14 [30]) |
3y | 3y10m | F | Yes (and TSH) | N | N | Severe and generalized | No | -1.8SD | 0.9SD | broad face with widely spaced eyes | maxillonasal hypoplasia, severe hypoplasia of the skull, thickened calvarium, dysplasia of both hips | yes | ND | – |
| PHP04 | 13y | 18y | F | Yes | N | ND | Severe and generalized (no X-ray) |
No | 135 cm (-3SD) | ND | Flat round face | genu valgum, Madelung deformity, exostosis in the knee (9y) | ND | ND | osteoporosis |
| PHP05 | – | 3y9m | F | Yes (and TSH) | ND | Low levels | Severe and generalized | ND | 45 cm (−2.7 SD) | + 1.8 SD | broad nasal root | – | Yes (5-6y) | ND | short neck, café-au-lait spots |
| PHP06 [31] |
42y | 45y | F | No (after Vit. D treatment) (and FSH |
N | Low levels at first | Severe and generalized | severe | 139 cm (− 4 SD) | >2SD | broad face with flattening of nasal ridge, facial dysostosis, spaced eyes | maxillonasal hypoplasia | – | ND | short neck, hyperinsulinism |
| PHP07 (P1 [33]) |
31y | 40y | F | Yes (and low GH) | N | N | MT: III-V outcarving cones of MP & TP |
learning difficulties (no test) | 141.5 cm (−4 SD) | 42.7 kg/m2 (> > 2SD) | round face, thin upper lip and prominent lower lip, pear-shaped nose | stubby fingers and toes | – | tooth hypoplasia | sparse hair, polyarthrosis, arthralgias of both hips and knees |
| PHP08 (P2 [33]) |
– | 11y | F | N | N | N | MT: II-V outcarving cones of MP & BP | N | 139.8 cm (-1SD) | 25.6 kg/m2 (+ 1.96 SD) | long flat philtrum, and thin upper lip, pear-shaped nose, protruding ears | – | ND | ND | sparse hair, laterally sparse eyebrows, type 2 diabetes |
| PHP09 | – | 32y | F | No | N | ND | Generalized shortening, severe outcarving of the epiphyses | ND | 152 cm (-2SD) | ND | thin upper lip, long philtrum, pear-shaped nose, sparse, eyebrows, prominent forehead | – | – | ND | Sparse hair |
| PHP09-D | 5m | 9m | F | No | N | ND | NA | ND | −2.8 SD | −2.4SD | thin upper lip, long philtrum, rounded nose, sparse eyebrows, prominent forehead, separated eyes | ND | ND | ND | Sparse hair |
| PHP10 | – | 33y | F | No | N | ND | Generalized shortening, stubby fingers (no X-ray received) |
ND | ND | ND | thin upper lip, long philtrum, pear-shaped nose | ND | ND | ND | Sparse hair |
| PHP10-S | 1y7m | M | No | N | ND | NA | ND | Growth failure | Growth failure | thin upper lip, long philtrum, rounded nose, sparse eyebrows, low-set ears | ND | ND | ND | Sparse hair, strabismus | |
| PHP10-D | 6y | F | No | N | ND | Yes (no X-ray) | ND | ND | ND | thin upper lip, long philtrum, rounded nose, sparse eyebrows, anteverted ears | ND | ND | ND | Sparse hair, strabismus | |
| PHP11 (P3 [34]) |
11.5y | 12y | F | N | N | N | MT: IV | N | −1 SD | N | No | – | Yes (13.5 years) (final height below target height) |
No | Advanced bone age |
| PHP12 [35] |
10y | 12y | F | No | N | ND | MT: II-V TP: I & III |
No | 148.7 cm (−0.4SD) | 1.7 SD | round face, long philtrum | – | Yes (12y) | No | short neck, descended and widely separated nipples |
| PHP13 | 28y | – | F | No | N | ND | Severe (especially IV & V MT) (no X-ray received) |
Yes | 139 cm (-4SD) | p3-p10 | small saddle nose, prominent forehead, epicanthal folds, upward slanting palpebral fissures, low and dysplastic ears | maxillonasal hypoplasia with severe prognathism | No | micrognatia | fine and sparse hair, parse eyebrows, café-au-lait spots, severe myopia |
| PHP14 | 10y | – | F | No | N | ND | MT: V (no X-ray received) |
ND | 130.6 cm (-1SD) | 85th | ND | clinodactyly, cone-shaped phalangeal epiphyses | ND | ND | – |
| PHP15 | 17y | – | F | No | N | ND | MT: IV (no X-ray received) |
No | p45 | p75-p90 | round face, facial asymmetry | – | ND | ND | hypothyroidism |
| PHP16 | 16y | – | M | No (after Vit. D treatment) |
N (after Vit. D treatment) |
Low levels at first | MT: IV & V (no X-ray received) |
ND | ND | (Obesity) | – | – | – | Dental malformations | acanthosis nigricans, short neck, hyperinsulinism |
| PHP17 | 9y7m | – | F | No | N | ND | MP: II-V at least. (BDA1?) | ND | −2,2SD | + 2,7SD | prominent forehead, depressed nasal root | rhizomelia | No | ND | increased subarachnoid space, ventriculomegaly, bilateral frontotemporal cortical atrophy, ectopic neurohypophysis |
| PHP18 | 9y11m | – | M | No | N | N | Stubby digits MT: III-IV at least |
No | 117 cm (−3,38SD) | N | flattening of nasal ridge | stocky build, hip hypoplasia, horizontal acetabulum, varum deformity, shortened tibia and femur, decreased interpedicular distance, scoliosis, bone dysplasias | ND | ND | – |
| PHP19 | 65y | – | F | No | N | N | MT: IV | No | (SS) | ND | big nose, thin upper lip | – | – | ND | – |
| PHP20 | 15y5m | – | M | No | N | N | MT: IV & V | No | 143,5 cm (−3,9SD) | + 4,22SD | ND | – | ND | ND | delayed puberty |
| PHP21 | 10y | – | F | N (TSH mildly increased) | N | ND | MT: IV; TP: I | No | p30 | N | prominent forehead, periorbital hyperpigmentation, long palpebral fissure, deep philtrum, thick eyebrows | – | ND | ND | – |
| PHP22 | 13y | – | F | N | N | ND | MP: II & V (BDA4?) | No | p50 | + 1.5SD | N | Bilateral cubitus valgus, short forearms, exostosis in both tibia, dorsolumbar hyperkyphosis in D12-L1 | ND | N | bicornuate uterus, short neck, wide thorax |
| PHP23 | 8y1m | – | F | No | N | N | MT: IV & V (mild) and clinodactyly of V | Mild | +3SD | > + 3SD | round face, thin upper lip, pear-shaped nose, sparse, arched eyebrows | – | ND | ND | sparse hair, epilepsy |
P patient, S son, D daughter, PTHr PTH resistance, Vit. D vitamin D, Ca calcemia, P phosphatemia, BD brachydactyly, MR mental retardation, N normal, NA not assessable due to short age, ND no data, MT metacarpal, TP telophalanx, MP mesophalanx, BP basophalanx, X-ray radiography, SD standard deviation, SS short stature
Candidate gene approach
The patients’ clinical features were reviewed to classify them according to the brachydactyly pattern and other clinical features, in accordance with the diagnostic algorithm proposed by us previously [19] and updated to include the most recent findings (Figure 1). The most probable candidate gene(s) were studied in each patient (Additional file 4: Table S1).
Fig. 1.
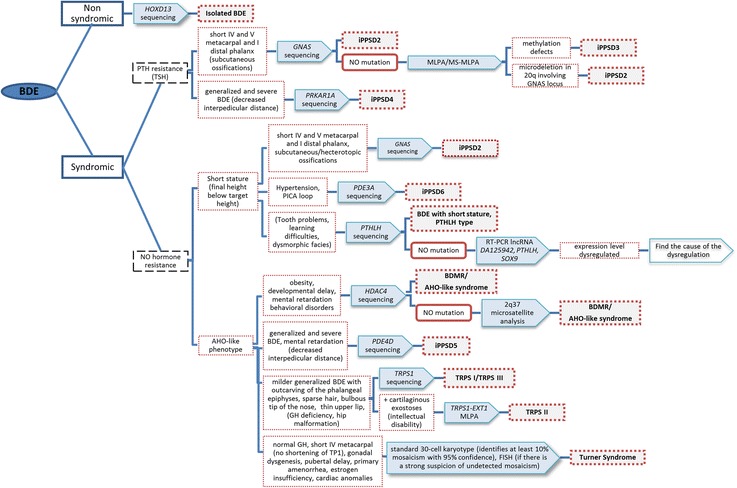
Updated clinical algorithm used during this project. Features in parentheses indicate that they are not obligatory features of the syndromes. BDE: brachydactyly type E; MLPA: multiplex ligation-dependent probe amplification; MS-MLPA: methylation specific-MLPA; iPPSD: inactivating PTH/PTHrP Signaling Disorder
The following disorders (all of which present BDE or similar types) were considered: (i) iPPSD4 [14] and iPPSD5 [15, 16]; (ii) hypertension with brachydactyly syndrome or iPPSD6 (HTNB, OMIM#112410), in which the responsible gene, phosphodiesterase 3A (PDE3A), has been identified very recently [20]; (iii) tricho-rhino-phalangeal syndrome type I and III (TRPS-I, OMIM#190350; TRPS-III, OMIM#190351), caused by mutations in the TRPS1 gene [21] or the more severe form, type II (OMIM#150230), which is a contiguous gene syndrome on 8q24.1 involving loss-of-function copies of the TRPS1 and EXT1 genes [22]; (iv) BDMR; (v) brachydactyly type E with short stature, PTHLH type (OMIM#613382), caused by mutations in the gene coding for parathyroid hormone-related protein (PTHLH) [23–25]; and (vi) isolated BDE in which the HOXD13 gene has been implicated [26, 27].
We should mention at this point that, although Turner syndrome chromosomal disorder (frequently, 45,X) is a relatively well-known entity, patients also show BDE [28] and short stature, which could give rise to some misdiagnoses. However, none of our patients presented clinical features compatible with Turner syndrome.
Mutational analysis of candidate genes
Genomic DNA was extracted from peripheral blood mononuclear cells using the QIAamp DNA Mini Kit (QIAGEN, Düren, Germany) according to the manufacturer’s instructions.
The DNA obtained was amplified by PCR for PRKAR1A (ref: NM_002734), PDE4D (ref: NM_001104631), TRPS1 (ref: NM_014112), HDAC4 (ref: NM_006037), PTHLH (ref: NM_198965.1) and/or HOXD13 (ref: NM_000523) coding exons and exon–intron junction, using specific primers (primers available on request). Direct Sanger sequencing was carried out using standard methods and an ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA). The reference sequences mentioned in parentheses were employed for mutation description according to the HGVS nomenclature.
Gene dosage analyses by multiplex ligation-dependent probe amplification (MLPA)
Gene dosage analyses were carried out using the SALSA MLPA P228-B1 TRPS1-EXT1, P179-B1 Limb− 1 and P264 B1 Human Telomere 9 probemix (MRC-Holland, Amsterdam, The Netherlands), when no alterations were identified by direct sequencing of the TRPS1 gene, HOXD13 gene or HDAC4 gene, respectively.
Microsatellite analysis
2q37 deletions were studied by microsatellite analysis as reported previously [29].
Results
Following the proposed candidate gene approach guided by the aforementioned diagnostic algorithm, 12 out of 23 patients were diagnosed genetically.
Mutations in PRKAR1A in the current series: iPPSD4
Six patients with PTH resistance and severe, generalized brachydactyly, both of which are typical characteristics of iPPSD4 (classically called ACRDYS1), were identified. Accordingly, the PRKAR1A gene was sequenced. Four of these patients harboured the recurrent c.1101C>T/p.Arg368* mutation (PHP01 and PHP02 already reported [30], and PHP04 and PHP05) and a fifth one carried the c.854A>G/p.Gln285Arg mutation (PHP03, reported [30]). The functional impact of this substitution was assessed experimentally and it was found to produce a similar impairment to the recurrent mutation, i.e., defects in PKA activation characterized by a reduced sensitivity to cAMP [30]. Parental studies suggested that all mutations were de novo. The remaining patient had no mutation in PRKAR1A.
Mutations in PDE4D in the current series: iPPSD5
In the patient (PHP06) with no mutation in PRKAR1A, the other gene, PDE4D, associated with iPPSD5 (formerly named ACRDYS2) was sequenced. Initially this patient presented elevated PTH (PTH = 107 pg/ml, normal range: 10–65; 25-OH vitamin D = 13.9 ng/ml, normal range 20–100), which is why iPPSD4 was suspected. However, after vitamin D treatment her PTH level normalized [31] (it was probably secondary to vitamin D deficiency) [32]. A novel heterozygous mutation (c.934C>G/p.Leu312Val) was identified in PDE4D. Functional studies of this mutation confirmed its pathogenicity [31].
Mutations in TRPS1 in the current series: TRPS-I
Four patients with features suggestive of TRPS-I (i.e. short stature, severe BDE with outcarving of the phalangeal epiphyses, sparse hair, bulbous tip of the nose or pear shaped nose, long philtrum, thin upper lip, as described in table 1) were positive for TRPS1 gene mutation: two patients (PHP08, previously described [33]; and PHP09) carried the recurrent c.2762G >C/p.Arg921Gln mutation and the remaining two patients each showed a different mutation: c.2830delA/p.Arg944Glyfs*3 in one case (PHP07, previously described [33]) and c.3159_3160delAAinsT/p.Lys1053Asnfs* (PHP10) in the other. Although neither of these has been described previously in the literature, the cosegregation in other family members (Additional file 4: Table S1) and frameshift characteristics (i.e., they would lead to truncated proteins if translated) suggested that they were possibly pathogenic.
Mutations in PTHLH in the current series: BDE with short stature PTHLH type
The BDE PTHLH type was suspected in two patients with BDE and advanced bone maturation for their chronological age. Indeed, two different novel mutations (c.101+ 3delAAGT, in PHP11 [34] and c.166C>T/p.Arg56*, in PHP12 [35]) were identified in the PTHLH gene in these patients. The characteristics of these mutations (frameshift and nonsense, respectively) suggested that they were causative of the pathology.
Discussion
PHP includes a heterogenic group of rare disorders associated with the AHO phenotype [1, 2]. Except for subcutaneous ossifications, the features of AHO are rather nonspecific as they also appear in other disorders, such as AHO-like syndrome [8] or acrodysostosis [11–13]. Less frequently, misdiagnosis with other entities has been observed because of the presence of BDE combined with short stature and obesity, which are also typically associated with other dysmorphic features and sometimes also with hormonal imbalances [33–37]. In this constellation of features, obesity or overweight and short stature could act as confusing factors as both are nonspecific [5, 33]. In addition, although obesity, intellectual disability, and resistance to several hormones are still extensively related to AHO, they may not be directly associated with genetic defects in GNAS [5].
The discovery of new genes implicated in syndromes with a phenotype similar to AHO, as well as other molecular mechanisms causative of iPPSD2 (classically named PHP/PPHP) [38], has been very helpful for establishing a correct genetic diagnosis for patients diagnosed with an “AHO-like phenotype” of unknown genetic cause [11, 14, 17, 33–35, 39], as well as for better describing the characteristic features of these less common syndromes. In ours and previously reported experiences [19, 40], BDE is the most specific and objective feature. For this reason, it was used as the inclusion criterion in this study and as a starting point to classify the aforementioned disorders in the previously proposed diagnostic algorithm [19]. It is well known that BDE was initially described as a variable shortening of the metacarpals/metatarsals with a more or less normal length of the phalanges [41].
As a result of the clinical re-evaluation of this series of patients, half of them (12/23) could be genetically diagnosed (supporting the importance of a good clinical examination, and the need of multidisciplinary approaches in the follow-up of these patients) and new knowledge acquired regarding these pathologies and the characteristic features detailed below.
We also analysed the features observed in our iPPSD4 (5/12) and iPPSD5 (1/12) patients and other cases described in the literature and noticed that the skeletal dysmorphisms (broad face, widely spaced eyes, maxillonasal hypoplasia, severe and generalized brachydactyly in hands/feet, severe short stature, cone-shaped epiphyses with early epiphyseal fusion, and advanced bone age [17, 30]) are very similar in both groups, although the facial dysmorphisms are often more severe in iPPSD5 [17, 30, 42]. Decreased interpedicular distance and mental retardation also appear to be more specific for iPPSD5 [6, 17, 30] since iPPSD4 patients show only behavioural disorders [30]. Finally, hormone resistance, which was initially used as a main differential characteristic to classify the patients with acrodysostosis, seems not to be as specific as initially appeared because more exceptions are found as more patients are reported (PTH resistance was recorded in 76% of iPPSD4 and 27% of iPPSD5 cases in the last review of Elli et al. [17]). All the iPPSD4 patients in our series exhibited PTH resistance, and although the iPPSD5 patient (PHP06) initially presented elevated PTH levels, PTH normalized after correcting the vitamin D deficiency, which is consistent with secondary hyperparathyroidism [32]. It is noteworthy that in contrast with the rest of the syndromes reflected in the algorithm, in which brachydactyly is usually not marked until the age of 6 years [39, 40], in acrodysostosis (both iPPSD4 and iPPSD5) the shortening and cone-shaped epiphyses are manifested during early childhood [16, 30].
Given the presence of brachydactyly and short stature, TRPS could be confused with the AHO phenotype, especially so if obesity (or overweight) and/or PTH resistance [37] and another hormone imbalance (GH deficiency has also been reported in some TRPS cases [43–47]) appear in the clinical profile, as shown in our two previously published cases [33] reviewed here. Keeping in mind all the identified cases in our series of patients, and comparing with those reported previously, in our opinion the most characteristic and illustrative features of TRPS syndrome are: bulbous tip of the nose (or pear-shaped nose), thin upper lip, involvement of the phalanges in the brachydactyly pattern and the typical outcarving of the phalangeal epiphyses, and sparse, slowly growing scalp hair.
Alterations which lead to the haploinsufficiency in PTHLH, the gene coding for parathyroid hormone related protein (PTHrP), have been identified as a cause of autosomal-dominant BDE in 11 families, two of them within our series [23, 24, 34, 35, 48, 49]. Although initially named as “BDE with short stature, PTHLH type” (OMIM#613382), because it is almost always associated with short stature [23–25], we have observed in both PTHLH patients in our series (PHP11 [34] and PHP12 [35]) that this short stature may not manifest until middle or late childhood. In both these cases, the patients had normal stature for their age but advanced bone age. Consequently, they experienced early epiphyseal closure, an early halt to growth, and their predicted final height is estimated to be below their target height. Thus, both the progenitors’ final height and bone age should be taken into account when determining whether patients show a height in the lower range of normality.
Overall, our use of a diagnostic algorithm in the current study has helped to determine the genetic cause in 12/23 patients with BDE who were clinically misdiagnosed as PHP/PPHP. Similarly to the 12 cases solved, the remaining cases were also classified and studied using the candidate gene approach guided by the proposed algorithm. However, we did not find any genetic alterations in the candidate genes studied, possibly due to some limitations of the study, such as (i) analysis of putative deletions at PTHLH is lacking; (ii) hand X-rays are missing for four patients, therefore it is difficult to propose any other potential diagnosis, (iii) although a large number of genes have been identified as the cause of BDE in recent years, the genetic cause of some BDE cases remains unknown [50].
Conclusions
We conclude that use of the presented algorithm in patients with idiopathic BDE is helpful for establishing a correct genetic diagnosis for those patients who have been misdiagnosed as PHP/PPHP. [5]
Additional files
Figure S1. Hand X-rays for patients with acrodysostosis, caused by mutation at either PRKAR1A (PHP02, panel A) or PDE4D (PHP06, panel B) They presented severe shortening of all hand bones with cone-shaped epiphysis (rows). (TIFF 1641 kb)
Figure S2. Hand X-rays for a mother (PHP09, panel A) and her daughter (PHP09-D, panel B) with tricho-rhino-phalangeal syndrome caused by the same mutation in TRPS1. The mother’s hands showed severe bilateral shortening of the bone with the characteristic outcarving of the phalangeal epiphysis (row). However her daughter was too young to manifest this brachydactyly and outcarving. (TIFF 3376 kb)
Figure S3. Hand X-rays for patients without genetic diagnosis: (A) Patient PHP18 exhibits stubby digits and shortening of at least metacarpals (MT) III-IV; (B) Patient PHP19’s hands show bilateral shortening of MT IV; (C) Patient PHP20 presents shortening of MT IV and V; (D) Patient PHP21’s hand reveals bilateral shortening of MT IV and first telophalanx; (E) Patient PHP22’s hands present bilateral shortening of II and V mesophalanges (similar to BDA4); (F) Patient PHP23 presents mild shortening of MT IV and V and clinodactyly of the V digit. (TIFF 3217 kb)
Table S1. Brief summary of the candidate genes analysed for each patient and the results. (DOCX 21 kb)
Acknowledgements
We thank the patients and parents who participated in the study.
Members of the Spanish Network for Imprinting Disorders include the following institutions: Complejo Hospitalario de Navarra, Navarra (Anda E., Berrade S., Ramos-Arroyo M. A., and Rodríguez Erdozain R.); Complejo Hospitalario de Toledo, Toledo (Vicente A.); Consorcio Hospital General de Valencia, Valencia (Rodriguez-Lopez R.); Corporacio de Salut del Maresme i la Selva, Barcelona (Moreno A.); Corporació Sanitària Parc Tauli, Barcelona (Guitart M.); Hospital Clínico San Carlos, Madrid (Oancea-Ionescu R. and Perez Rodriguez O.); Hospital da Barbanza, A Coruña (Molinos Castro S.); Hospital Ernest Lluch Martin en Calatayud, Zaragoza (Meriño E.); Hospital General de Castellón, Castellón (Salvador-Sanchis J. L.); Hospital General Universitario de Alicante, Alicante (López Mondejar P. and Zapico M.); Hospital General Universitario de Ciudad Real, Ciudad Real (Palomo E. and Rozas Moreno P.); Hospital General Universitario de Elda, Alicante (Aleixandre-Blanquer F.); Hospital Infantil Universitario Niño Jesús, Madrid (Argente Oliver J., Martos G., Pozo J. and Rubio-Cabezas O.); Hospital Sant Joan de Déu, Barcelona (Bilbao Gasso L., Marti G., Martorell L., Cardona R., Suarez L. and Zambudio Sert S.); Hospital Universitari de Girona Doctor Josep Trueta, Girona (Obon M.); Hospital Universitari Doctor Pese, Valencia (Sanchis Calvo A.); Hospital Universitari i Politecnic La Fe de Valencia, Valencia (Moreno Macian F.); Hospital Universitario 12 de Octubre, Madrid (Cruz-Rojo J., Garzon Lorenzo L., and Sanchez del Pozo J.); Hospital Universitario Central de Asturias, Asturias (Riaño I.); Hospital Universitario de La Princesa, Madrid (Lahera Vargas M.); Hospital Universitario Fundación Jiménez Díaz, Madrid (Blanco-Kelly F., Lorda Sanchez M. I., Soriano Guillen L. and Tahsin Swafiri S.); Hospital Universitario Infanta Sofía, Madrid (Azriel A.); Hospital Universitario La Paz, Madrid (Lecumberri B. and Moreno J. C.); Hospital Universitario Ntra. Sra. de Candelaria, Tenerife (Garcia Nieto V.); Hospital Universitario Príncipe de Asturias, Madrid (Garcia Diaz J.); Hospital Universitario Puerta del Mar, Cádiz (Marin Iglesias R.); Hospital Universitario Quironsalud Madrid, Madrid (Laura Fernandez A.); Hospital Universitario Santa Cristina, Madrid (Martin Fuentes M.); Hospital Universitario Vall D’Hebron, Barcelona (Casteras A. and Clemente Leon M.); Hospital Universitario Virgen de la Arrixaca, Murcia (Ballesta-Martinez M. and Sanchez Soler M. J.); Hospital Universitario Virgen del Rocío, Sevilla (Gonzalez Meneses A.); Hospital Virgen de la Salud, Toledo (Lopez Lopez J.); OSI Bilbao-Basurto, Bizkaia (Garcia Barcina M. J.); OSI Ezkerraldea-Enkarterri-Cruces, Bizkaia (Gener B and Llano I.) and Parc de Salut Mar, Barcelona (Bonet Alcaina M.).
Funding
The study was supported by funding from a research project grant (PI13/00467) from the Instituto de Salud Carlos III (Carlos III Institute of Health) of the Ministry of Economy and Competitiveness (Spain), co-financed by the European Regional Development Fund, and the Department of Health of the Basque Government (GV2014111017). AP is partly supported by the University of the Basque Country (Ref: 48198). GPN is partly supported by the I3SNS Program of the Spanish Ministry of Health (CP03/0064; SIVI 1395/09).
Availability of data and materials
Data and materials are available upon request.
Abbreviations
- ACRDYS1
Acrodysostosis type 1with multihormonal resistance
- ACRDYS2
Acrodysostosis type 2 without hormone resistance
- AHO
Albright’s hereditary osteodystrophy
- BDE
Brachydactyly type E
- BDMR
Brachydactyly and mental retardation syndrome
- GH
Growth hormone
- GNAS
Gene coding alpha subunit of the stimulatory guanine nucleotide-binding protein
- Gsα
Gs protein alpha subunit
- HDAC4
Gene coding for histone deacetylase 4
- HOXD13
Gene coding for homeobox D13
- HTNB
Hypertension with brachydactyly syndrome
- iPPSD
inactivating PTH/PTHrP signalling disorder
- PDE3A
Gene coding for phosphodiesterase 3A
- PDE4D
Gene coding for phosphodiesterase 4D
- PHP
Pseudohypoparathyroidism
- PKA
Protein kinase type 1A
- PPHP
Pseudopseudohypoparathyroidism
- PRKAR1A
Gene coding for the cAMP-dependent protein kinase type 1 regulatory subunit
- PTH
Parathyroid hormone
- PTHLH
Gene coding for parathyroid hormone-related protein
- PTHrP
Parathyroid hormone related protein
- TRPS
Tricho-rhino-phalangeal syndrome
- TRPS1
Gene coding for zinc finger transcription factor TRPS1
Authors’ contributions
GPN designed the project. AP and IG participated in the molecular analysis of the syndromes. The members of the Spanish Network for Imprinting Disorders participated in the recruitment, clinical description of the patients and the clinical discussion. AP and GPN designed and wrote the first draft. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the committee concerned. This project was approved by the Basque Clinical Research Ethics Committee (CEIC-E: 2010–021; PI2013124). Patients were informed about this study and informed written consent was obtained from all patients (or legal guardians for minors) and relatives included in the study.
Consent for publication
Written consent to publish clinical data was obtained.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12881-018-0530-z) contains supplementary material, which is available to authorized users.
Contributor Information
Arrate Pereda, Email: arrate.peredaaguirre@osakidetza.eus.
Intza Garin, Email: intza.garinelkoro@osakidetza.eus.
Guiomar Perez de Nanclares, Phone: +34945007000, Email: gnanclares@osakidetza.eus.
Spanish Network for Imprinting Disorders:
E. Anda, S. Berrade, M. A. Ramos-Arroyo, R. Rodríguez Erdozain, A. Vicente, R. Rodriguez-Lopez, A. Moreno, M. Guitart, R. Oancea-Ionescu, O. Perez Rodriguez, S. Molinos Castro, E. Meriño, J. L. Salvador-Sanchis, P. López Mondejar, M. Zapico, E. Palomo, P. Rozas Moreno, F. Aleixandre-Blanquer, J. Argente Oliver, G. Martos, J. Pozo, O. Rubio-Cabezas, L. Bilbao Gasso, G. Marti, L. Martorell, R. Cardona, L. Suarez, S. Zambudio Sert, M. Obon, A. Sanchis Calvo, F. Moreno Macian, J. Cruz-Rojo, L. Garzon Lorenzo, J. Sanchez del Pozo, I. Riaño, M. Lahera Vargas, F. Blanco-Kelly, M. I. Lorda Sanchez, L. Soriano Guillen, S. Tahsin Swafiri, A. Azriel, B. Lecumberri, J. C. Moreno, V. Garcia Nieto, J. Garcia Diaz, R. Marin Iglesias, M. Martin Fuentes, A. Casteras, M. Clemente Leon, M. Ballesta-Martinez, M. J. Sanchez Soler, A. Gonzalez Meneses, J. Lopez Lopez, M. J. Garcia Barcina, B. Gener, I. Llano, and M. Bonet Alcaina
References
- 1.Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidsm- an example of “Seabright syndrome”. Endocrinology. 1942;30:922–932. [Google Scholar]
- 2.Fitch N, Opitz JM, Herrmann J. Albright’s hereditary osteodystrophy: a review. Am J Med Genet. 1982;11:11–29. doi: 10.1002/ajmg.1320110104. [DOI] [PubMed] [Google Scholar]
- 3.Albright F, Forbes AP, Henneman PH. Pseudo-pseudohypoparathyroidism. TransAssoc Am Physicians. 1952;65:337–350. [PubMed] [Google Scholar]
- 4.Mantovani G, Linglart A, Garin I, Silve C, Elli FM, de Nanclares GP. Clinical utility gene card for: pseudohypoparathyroidism. Eur J Hum Genet. 2013;21 [DOI] [PMC free article] [PubMed]
- 5.Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016;175:P1–17. doi: 10.1530/EJE-16-0107. [DOI] [PubMed] [Google Scholar]
- 6.Silve C, Clauser E, Linglart A, et al. Horm Metab Res. 2012;44:749–758. doi: 10.1055/s-0032-1304252. [DOI] [PubMed] [Google Scholar]
- 7.Qu L, Zhang TT, Mu YM. [Clinical analysis of 15 cases of pseudohypoparathyroidism]. Nan.Fang Yi.Ke.Da.Xue.Xue.Bao. 2012;32:685–6. [PubMed]
- 8.Wilson LC, Leverton K, Oude Luttikhuis ME, Oley CA, Flint J, Wolstenholme J, et al. Brachydactyly and mental retardation: an Albright hereditary osteodystrophy-like syndrome localized to 2q37. Am J Hum Genet. 1995;56:400–407. [PMC free article] [PubMed] [Google Scholar]
- 9.Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, DR ML, et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioural problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012/08/07. 2012;26:517–22. [DOI] [PubMed]
- 11.Ablow RC, Hsia YE, Brandt IK. Acrodysostosis coinciding with pseudohypoparathyroidism and pseudo pseudohypoparathyroidism. Am J Roentgenol. 1977;128:95–99. doi: 10.2214/ajr.128.1.95. [DOI] [PubMed] [Google Scholar]
- 12.Davies SJ, Hughes HE. Familial acrodysostosis: can it be distinguished from Albright’s hereditary osteodystrophy? Clin Dysmorphol. 1992;1:207–215. doi: 10.1097/00019605-199210000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Graham JM, Krakow D, Tolo VT, Smith AK, Lachman RS. Radiographic findings and Gs-alpha bioactivity studies and mutation screening in acrodysostosis indicate a different etiology from pseudohypoparathyroidism. Pediatr Radiol. 2001;31:2–9. doi: 10.1007/s002470000355. [DOI] [PubMed] [Google Scholar]
- 14.Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, et al. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med. 2011;364:2218–2226. doi: 10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 15.Lee H, Graham JM, Rimoin DL, Lachman RS, Krejci P, Tompson SW, et al. Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet. 2012;90:746–751. doi: 10.1016/j.ajhg.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michot C, Le Goff C, Goldenberg A, Abhyankar A, Klein C, Kinning E, et al. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012;90:740–745. doi: 10.1016/j.ajhg.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elli FM, Bordogna P, De Sanctis L, Giachero F, Verrua E, Segni M, et al. Screening of PRKAR1A and PDE4D in a large Italian series of patients clinically diagnosed with Albright hereditary Osteodystrophy and/or Pseudohypoparathyroidism. J Bone Miner Res. 2016;31:1215–1224. doi: 10.1002/jbmr.2785. [DOI] [PubMed] [Google Scholar]
- 18.De Nanclares GP, Fernández-Rebollo E, Santin I, García-Cuartero B, Gaztambide S, Menéndez E, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 2007;92:2370–2373. doi: 10.1210/jc.2006-2287. [DOI] [PubMed] [Google Scholar]
- 19.Pereda A, Garin I, Garcia-Barcina M, Gener B, Beristain E, Ibanez AM, et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J Rare Dis. 2013;8:141. doi: 10.1186/1750-1172-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maass PG, Aydin A, Luft FC, Schächterle C, Weise A, Stricker S, et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet. 2015;47:647–653. doi: 10.1038/ng.3302. [DOI] [PubMed] [Google Scholar]
- 21.Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet. 2000:71–4. [DOI] [PubMed]
- 22.Lüdecke H, Wagner MJ, Nardmann J, La Pillo B, Parrish JE, Willems PJ, et al. Molecular dissection of a contiguous gene syndrome: localization of the genes involved in the langer - giedion syndrome. Hum Mol Genet. 1995;4:31–36. doi: 10.1093/hmg/4.1.31. [DOI] [PubMed] [Google Scholar]
- 23.Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, et al. Deletion and point mutations of PTHLH cause Brachydactyly type E. Am J Hum Genet. 2010;86:434–439. doi: 10.1016/j.ajhg.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maass PG, Wirth J, Aydin A, Rump A, Stricker S, Tinschert S, et al. A cis-regulatory site downregulates PTHLH in translocation t(8;12)(q13;p11.2) and leads to Brachydactyly Type E. Hum Mol Genet. 2009/12/18. 2010;19:848–860. [DOI] [PMC free article] [PubMed]
- 25.Wang J, Wang Z, An Y, Wu C, Xu Y, Fu Q, et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clin Chim Acta. 2015;446:9–14. doi: 10.1016/j.cca.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Johnson D, Kan S-H, Oldridge M, Trembath RC, Roche P, Esnouf RM, et al. Missense mutations in the Homeodomain of HOXD13 are associated with Brachydactyly types D and E. Am J Hum Genet 2003;72:984–997. [DOI] [PMC free article] [PubMed]
- 27.Jamsheer A, Sowińska A, Kaczmarek L, Latos-Bieleńska A. Isolated brachydactyly type E caused by a HOXD13 nonsense mutation: a case report. BMC Med Genet. 2012;13:4. doi: 10.1186/1471-2350-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poznanski AK, Werder EA, Giedion A, Martin A, Shaw H. The pattern of shortening of the bones of the hand in PHP and PPHP--A comparison with brachydactyly E, turner syndrome, and acrodysostosis. Radiology. 1977;123:707–718. doi: 10.1148/123.3.707. [DOI] [PubMed] [Google Scholar]
- 29.Fernández-Rebollo E, Pérez O, Martinez-Bouzas C, Cotarelo-Pérez MC, Garin I, Ruibal JL, et al. Two cases of deletion 2q37 associated with segregation of an unbalanced translocation 2;21: Choanal atresia leading to misdiagnosis of CHARGE syndrome. Eur J Endocrinol. 2009;160:711–717. doi: 10.1530/EJE-08-0865. [DOI] [PubMed] [Google Scholar]
- 30.Linglart A, Fryssira H, Hiort O, Holterhus PM, Perez De Nanclares G, Argente J, et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab 2012;97:E2328–E2338. [DOI] [PubMed]
- 31.Briet C, Pereda A, Le Stunff C, Motte E, de Dios Garcia-Diaz J, de Nanclares GP, et al. Mutations causing acrodysostosis−2 facilitate activation of phosphodiesterase 4D3. Hum Mol Genet. 2017; [DOI] [PubMed]
- 32.Akin L, Kurtoǧlu S, Yildiz A, Akin MA, Kendirci M. Vitamin D deficiency rickets mimicking pseudohypoparathyroidism. JCRPE J Clin Res Pediatr Endocrinol. 2011/01/29. 2010;2:173–5. [DOI] [PMC free article] [PubMed]
- 33.Pereda A, Azriel S, Bonet M, Garin I, Gener B, Lecumberri B, et al. Pseudohypoparathyroidism vs. tricho-rhino-phalangeal syndrome: patient reclassification. J Pediatr Endocrinol Metab. 2014;27:1089–1094. [DOI] [PubMed]
- 34.Thomas-Teinturier C, Pereda A, Garin I, Diez-Lopez I, Linglart A, Silve C, et al. Report of two novel mutations in PTHLH associated with brachydactyly type E and literature review. Am J Med Genet Part A. 2016;170:734–742. doi: 10.1002/ajmg.a.37490. [DOI] [PubMed] [Google Scholar]
- 35.Pereda A, Garzon-Lorenzo L, Garin I, Cruz-Rojo J, Sanchez Del Pozo J, Perez de Nanclares G. The p.R56* mutation in PTHLH causes variable brachydactyly type E. Am J Med Genet A. 2017;173:816–819. doi: 10.1002/ajmg.a.38067. [DOI] [PubMed] [Google Scholar]
- 36.Van Der Werff Ten Bosch JJ. The syndrome of Brachymetacarpal dwarfism (“ pseudo-Pseudohypoparathyroidism ”) with and without Gonadal Dysgenesis. Lancet. 1959;273:69–71. doi: 10.1016/S0140-6736(59)91137-7. [DOI] [PubMed] [Google Scholar]
- 37.Böhles H, Ott R. Pseudohypohyperparathyroidism with the phenotype of the tricho-rhino-phalangeal syndrome. Klin Padiatr. 1983;195:117–120. doi: 10.1055/s-2008-1034053. [DOI] [PubMed] [Google Scholar]
- 38.Garin I, Elli FM, Linglart A, Silve C, De Sanctis L, Bordogna P, et al. Novel microdeletions affecting the GNAS locus in pseudohypoparathyroidism: characterization of the underlying mechanisms. J Clin Endocrinol Metab. 2015/01/17. 2015;100:E681–E687. [DOI] [PubMed]
- 39.Toka O, Maass PG, Aydin A, Toka H, Hübner N, Rüschendorf F, et al. Childhood hypertension in autosomal-dominant hypertension with brachydactyly. Hypertension. 2010;56:988–994. doi: 10.1161/HYPERTENSIONAHA.110.156620. [DOI] [PubMed] [Google Scholar]
- 40.Virágh K, Töke J, Sallai Á, Jakab Z, Rácz K, Tóth M. Gradual development of brachydactyly in pseudohypoparathyroidism. J Clin Endocrinol Metab. 2014/04/02. 2014;99:1945–1946. [DOI] [PubMed]
- 41.Temtamy SA, McKusick V. The genetics of hand malformations. Birth defects Orig Artic ser. New York: Alan R Liss, Inc.; 1978. [PubMed] [Google Scholar]
- 42.Silve C, Le-Stunff C, Motte E, Gunes Y, Linglart A, Clauser E. Acrodysostosis syndromes. Bonekey Rep Nature Publishing Group. 2012;1:225. doi: 10.1038/bonekey.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stagi S, Bindi G, Galluzzi F, Lapi E, Salti R, Chiarelli F. Partial growth hormone deficiency and changed bone quality and mass in type I trichorhinophalangeal syndrome. Am J Med Genet Part A. 2008:1598–604. [DOI] [PubMed]
- 44.Sohn YB, Ki C-S, Park SW, Cho S-Y, Ko A-R, Kwon M-J, et al. Clinical, biochemical, and genetic analysis of two korean patients with trichorhinophalangeal syndrome type I and growth hormone deficiency. Ann Clin Lab Sci. 2012;42:307–312. [PubMed] [Google Scholar]
- 45.Riedl S, Giedion A, Schweitzer K, Müllner-Eidenböck A, Grill F, Frisch H, et al. Pronounced short stature in a girl with tricho-rhino-phalangeal syndrome II (TRPS II, Langer-Giedion syndrome) and growth hormone deficiency. Am J Med Genet. 2004;131(A):200–203. doi: 10.1002/ajmg.a.30374. [DOI] [PubMed] [Google Scholar]
- 46.Shao C, Tian J, Hong SD, Xiao YC, Xu C, Cheng WL, et al. A novel mutation in TPRS1 gene caused tricho-rhino-phalangeal syndrome in a Chinese patient with severe osteoporosis. Chin Med J. 2011;124:1583–1585. [PubMed] [Google Scholar]
- 47.Macchiaiolo M, Mennini M, Digilio MC, Buonuomo PS, Lepri FR, Gnazzo M, et al. Thricho-rhino-phalangeal syndrome and severe osteoporosis: a rare association or a feature? An effective therapeutic approach with biphosphonates. Am J Med Genet Part A. 2014;164:760–763. doi: 10.1002/ajmg.a.36327. [DOI] [PubMed] [Google Scholar]
- 48.Gray MJ, van Kogelenberg M, Beddow R, Morgan T, Wordsworth P, Shears DJ, et al. A new acro-osteolysis syndrome caused by duplications including PTHLH. J Hum Genet. 2014;59:484–487. doi: 10.1038/jhg.2014.58. [DOI] [PubMed] [Google Scholar]
- 49.Jamsheer A, Sowińska-Seidler A, Olech EM, Socha M, Kozłowski K, Pyrkosz A, et al. Variable expressivity of the phenotype in two families with brachydactyly type E, craniofacial dysmorphism, short stature and delayed bone age caused by novel heterozygous mutations in the PTHLH gene. J Hum Genet. 2016:1–5. [DOI] [PubMed]
- 50.Silve C. A cup half-full or half-empty? When PTHrP levels matter. IBMS Bonekey. 2010;7:325–332. doi: 10.1138/20100465. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hand X-rays for patients with acrodysostosis, caused by mutation at either PRKAR1A (PHP02, panel A) or PDE4D (PHP06, panel B) They presented severe shortening of all hand bones with cone-shaped epiphysis (rows). (TIFF 1641 kb)
Figure S2. Hand X-rays for a mother (PHP09, panel A) and her daughter (PHP09-D, panel B) with tricho-rhino-phalangeal syndrome caused by the same mutation in TRPS1. The mother’s hands showed severe bilateral shortening of the bone with the characteristic outcarving of the phalangeal epiphysis (row). However her daughter was too young to manifest this brachydactyly and outcarving. (TIFF 3376 kb)
Figure S3. Hand X-rays for patients without genetic diagnosis: (A) Patient PHP18 exhibits stubby digits and shortening of at least metacarpals (MT) III-IV; (B) Patient PHP19’s hands show bilateral shortening of MT IV; (C) Patient PHP20 presents shortening of MT IV and V; (D) Patient PHP21’s hand reveals bilateral shortening of MT IV and first telophalanx; (E) Patient PHP22’s hands present bilateral shortening of II and V mesophalanges (similar to BDA4); (F) Patient PHP23 presents mild shortening of MT IV and V and clinodactyly of the V digit. (TIFF 3217 kb)
Table S1. Brief summary of the candidate genes analysed for each patient and the results. (DOCX 21 kb)
Data Availability Statement
Data and materials are available upon request.