

Abstract
Trichloroethylene (TCE) and tetrachloroethylene (PCE) are ubiquitous environmental contaminants and occupational health hazards. Recent health assessments of these agents identified several critical data gaps, including lack of comparative analysis of their effects. This study examined liver and kidney effects of TCE and PCE in a dose-response study design. Equimolar doses of TCE (24, 80, 240, and 800 mg/kg) or PCE (30, 100, 300, and 1000 mg/kg) were administered by gavage in aqueous vehicle to male B6C3F1/J mice. Tissues were collected 24 h after exposure. Trichloroacetic acid (TCA), a major oxidative metabolite of both compounds, was measured and RNA sequencing was performed. PCE had a stronger effect on liver and kidney transcriptomes, as well as greater concentrations of TCA. Most dose-responsive pathways were common among chemicals/tissues, with the strongest effect on peroxisomal β-oxidation. Effects on liver and kidney mitochondria-related pathways were notably unique to PCE. We performed dose-response modeling of the transcriptomic data and compared the resulting points of departure (PODs) to those for apical endpoints derived from long-term studies with these chemicals in rats, mice, and humans, converting to human equivalent doses using tissue-specific dosimetry models. Tissue-specific acute transcriptional effects of TCE and PCE occurred at human equivalent doses comparable to those for apical effects. These data are relevant for human health assessments of TCE and PCE as they provide data for dose-response analysis of the toxicity mechanisms. Additionally, they provide further evidence that transcriptomic data can be useful surrogates for in vivo PODs, especially when toxicokinetic differences are taken into account.
Keywords: volatile organic compounds, agents, toxicogenomics, methods, dose-response, risk assessment, kidney, systems toxicology, liver, systems toxicology
Trichloroethylene (TCE) and tetrachloroethylene (perchloroethylene; PCE) are high-production volume chlorinated olefin solvents with many industrial and consumer applications, including metal degreasing and use as chemical feedstocks in synthesis of chlorofluorocarbons (Guha et al., 2012). Historically, the most well-known uses of these agents were vapor degreasing and dry-cleaning, although the use of TCE in dry-cleaning is now prohibited and the use of PCE is also being phased out (Cichocki et al., 2016). Due to their widespread use, high-production volume, and the challenges of remediating polluted sites, both TCE and PCE are ubiquitous environmental contaminants of air, soil, and drinking and ground water (IARC, 2014). TCE is the most frequently found groundwater pollutant at National Priority List sites in the United States (Fay and Mumtaz, 1996), and cocontamination with TCE and PCE is common (Fay and Mumtaz, 1996; Pohl et al., 2008). The high frequency of exposure to TCE and PCE has been indicated by the National Health and Nutrition Examination Survey; both chemicals are commonly detected in blood and/or exhaled air samples in the general population (Jia et al., 2012).
The cancer and/or noncancer toxicity of TCE and PCE has recently been evaluated by multiple state, federal, and international agencies, including the United States Environmental Protection Agency (U.S. EPA) (2011a,b), the International Agency for Research on Cancer (IARC, 2014), the National Toxicology Program (2015), and the California Environmental Protection Agency Office of Environmental Health Hazard Assessment Air Toxics Hot Spots Program (California Environmental Protection Agency, 2016). Both TCE and PCE are among the first 10 chemicals evaluated for potential risks to human health and the environment under the Frank R. Lautenberg Chemical Safety for the 21st Century Act (U.S. EPA, 2017). TCE is classified as “carcinogenic to humans” by both U.S. EPA (Chiu et al., 2013) and IARC (Guha et al., 2012), while PCE is classified as “likely to be carcinogenic to humans” by U.S. EPA (Guyton et al., 2014) and as “probably carcinogenic to humans (Group 2A)” by IARC (Guha et al., 2012). Although the mechanisms of TCE- or PCE-induced cancer or noncancer effects have been subjects of intense debate, bioactivation of either parent chemical to relatively reactive metabolites is accepted as a critical precursor step for subsequent toxicity (Cichocki et al., 2016).
The toxicity of TCE has been more widely studied than that of PCE (Cichocki et al., 2016; Lash et al., 2002; Rusyn et al., 2014). Due to structural similarities of these chemicals, the effects of PCE are often presumed to be similar to those of TCE. However, considerable qualitative and quantitative differences in metabolism exist between TCE and PCE (IARC, 2014; U.S. EPA, 2011a,b). For example, PCE is metabolized more slowly than TCE, and it is thought that their oxidative metabolism may be mediated by different cytochrome P450s (Cichocki et al., 2016, 2017a). Further, relative amounts and chemical reactivity of major reactive metabolites and precursor intermediates differ between these 2 compounds, trichloroacetic acid (TCA) and trichloroethanol (TCOH) are major oxidative metabolites of TCE, while PCE yields only TCA.
Few studies have directly compared the target organ metabolism and toxicity of both TCE and PCE; here, we address the critical gap in our knowledge of the relative potency for liver and kidney effects of these 2 common environmental toxicants. We conducted a comparative dose-response analysis between TCE and PCE in male B6C3F1 mice. We exposed animals to equal molar doses of TCE or PCE and evaluated formation of TCA, a major oxidative metabolite of TCE and PCE, as well as dose-response effects on gene expression as a function of external dose or internal dose of TCA in each organ. We then performed benchmark dose (BMD) modeling of the transcriptomic data (Thomas et al., 2007, 2013) and compared it with apical endpoint-derived points of departure (PODs) from long-term studies with these chlorinated solvents.
MATERIALS AND METHODS
Chemicals
TCE and PCE were acquired from Sigma Aldrich (St Louis, Missouri). All other reagents were obtained from commercially available sources and were of chemical-grade or higher.
Animals
Adult (5–6 weeks of age) male B6C3F1/J mice were obtained from the Jackson Laboratory (Bar Harbor, Maine). All mice were housed in polycarbonate cages on Sani-Chips (P.J. Murphy Forest Products, Montville, New Jersey) irradiated hardwood bedding. Animals were fed National Toxicology Program (NTP)-2000 (Zeigler Brothers, Gardners, Pennsylvania) wafer diet and water ad libitum on a 12 h light-dark cycle. All studies were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Study design
After a week-long acclimatization period, mice were intragastrically administered a single dose of TCE, PCE, or vehicle (5% Alkamuls El-620 in saline, 5 ml/kg). All dosing occurred between the hours of 07:00 and 09:00. In addition to vehicle-only treated animals (0 dose), the following doses were used for TCE (24, 80, 240, and 800 mg/kg) or PCE (30, 100, 300, and 1000 mg/kg). These approximate to 0.22, 0.67, 2.0, and 6.0 mmol/kg for each of TCE and PCE. Six mice were exposed to vehicle, while the other groups had 3 mice each. Dose ranges were selected based on previous studies showing that these amounts are well-tolerated in acute and subchronic studies, as well as corresponding to the dose range used in both 90-day and 2-year mouse studies (Buben and O’Flaherty, 1985; Cichocki et al., 2017a,b; National Toxicology Program, 1977, 1990; Philip et al., 2007; Yoo et al., 2015c). Necropsy was performed 24 h following exposure. Mice were anesthetized (pentobarbital, 50 mg/kg ip) and killed by exsanguination through the vena cava. Organs were rinsed in phosphate-buffered saline, blotted dry, and weights were recorded. A small section of the liver left and median lobes and kidney were placed in formalin. The remaining tissues were snap-frozen in liquid nitrogen and stored at −80°C until processing.
Quantification of TCA
TCA is a major oxidative and hepatotoxic metabolite of both TCE and PCE (Cichocki et al., 2016). TCA was measured in liver and kidney according to the method of (Cichocki et al., 2017b), which was modified from U.S. EPA method 815-B-03-002. Briefly, aqueous liver and kidney homogenates were spiked with 10 nmol of internal standard (aqueous 2-bromobutyric acid) and heated at 60°C for 2 h in the presence of methanolic sulfuric acid (10%, vol:vol) to generate the respective methyl esters. After liquid-liquid extraction, the derivatives were analyzed via gas chromatography-mass spectrometry. Eight-point calibration curves were generated by spiking known amounts of TCA into tissue homogenates from naïve mice. TCA content in experimental samples was determined using the peak area ratio of TCA to internal standard and extrapolation from the calibration curve. The assay was confirmed to be linear up to 1000 nmol/g tissue. The lower limit of quantitation was 4 nmol/g tissue, as determined by using a signal-to-noise ratio of > 3.
Gene expression profiling
Total RNA was extracted from liver or kidney samples using miRNEasy kits (Qiagen, Valencia, California). RNA integrity was determined using Bioanalyzer (Agilent, Santa Clara, California). cDNA libraries were constructed using TruSeq Total RNA kits (Illumina, San Diego, California) following the manufacturer’s instructions. Library quality was assessed on a Bioanalyzer and sequenced on an Illumina HiSeq-2000 (Illumina). Sample multiplexing was performed prior to sequencing so that, in liver, for each chemical the control and highest concentration samples were sequenced one per lane for the highest number of mapped reads (approximately 130 million). The remaining samples were combined using 3 per lane and had approximately 45 million reads (Supplementary Figure 1). The rationale for the larger number of reads in the control and high-concentration conditions was to maximize the power to detect differential isoform usage due to treatment. For kidney, samples were analyzed using 3 per lane for all doses, with mean ± SD of mapped reads was 49.1 ± 4.4 million. Gene expression data are available from Gene Expression Omnibus (GSE100296).
Transcriptomics data analysis
Data preprocessing was performed following a standard CASAVA v.1.8.2 pipeline, providing FASTQ output. Overall expression was quantified using a TopHat2 mapping pipeline, with a secondary analysis and exon-specific calling using tools made available as part of the IsoDOT software (Sun et al., 2015). For primary expression calling, each read was aligned to the mm10 mouse reference sequence using TopHat v2.0.10 (Trapnell et al., 2009). A maximum of one match per read was used in the alignments, and the mouse genome mm10 RefSeq annotations downloaded from University of California-San Cruz (UCSC) Genomes were used as a guide for the alignments. The resulting .bam files were sorted and converted to .sam for use with HTSeq (Anders et al., 2015) with the options for no-stranded sequencing and intersection-nonempty, which calls the best (or longest) match to a gene as the gene being counted when there was more than one gene overlapped by a given read. A set of customized Perl scripts were written to aggregate the counts results into a single file with 24,602 genes, and to add the ENSEMBL identification numbers (IDs) associated with the RefSeq ID, where possible. The mapping between RefSeq ID and ENSEMBL ID was also obtained from UCSC Genomes browser files.
Analysis of per-gene differential expression analysis was performed using edgeR v3.0 (Nikolayeva and Robinson, 2014; Robinson et al., 2010), using log TCE or PCE administered dose, or liver or kidney TCA concentration as continuous predictors. The edgeR analyses were considered primary and are the basis of differential expression comparisons shown. We also performed secondary analyses as follows that are not shown as they yielded similar results. Specifically, for each gene a negative binomial model (Anders et al., 2013) was fit using R glm.nb in the R MASS package, with model log(count) ∼ log(concentration + 0.5) + log(library size) and Wald-statistics and P values using the summary function for the concentration regression coefficient. In addition, we computed rank (Spearman) correlation of expression (normalized to library size/total read count) versus chemical concentration, designed to provide robust results relatively insensitive to the precise concentration-response relationship.
Methods for testing of differential isoform usage are less mature than for differential total expression and can involve complex inference over potential isoforms (Katz et al., 2010). As articulated in (Anders et al., 2012), exon “usage” (the proportion of reads mapping to individual exons within a gene) can provide clear and powerful evidence of differential isoform abundance without the need for indirect isoform inference. To provide a simplified and readily interpretable approach, we directly tested association of chemical concentration to proportional “usage” of each exon as follows. For a single gene g with mg exons observed in the experiment, we denote the read count for the ith exon in sample j as cij, total read count for the gene , and proportional usage . Exons with an average of fewer than 10 reads per sample were considered as having insufficient precision and were collapsed into a single artificial exon. For each i, we computed a P value Pi using linear regression for the usage vector ui. versus rank of dose, weighted by the number of mapped reads per exon. For each gene, the exon-specific P values were corrected using Simes’ approach, which has been advocated as a robust correction useful for testing families of hypotheses (Peterson et al., 2016). The Simes’ P values were then further corrected for false discovery rate (FDR) control (Benjamini and Hochberg, 1995) to obtain q values per-gene. Estimates of the proportion of true null hypotheses were based on π0 estimation (the proportion of true nulls) from the R qvalue package. To maximize the chance of informative discoveries, q < 0.15 was considered significant, which ensuring the proportion of true discoveries was a useful 85% or above. As few genes were significant for differential isoform usage, pathway analysis of differential exon usage was performed using the liberal criterion of a gene list consisting of gene-level P values < 0.05, not corrected for multiple comparisons.
Pathway analysis
Gene expression levels and phenotype data were subjected to pathway analysis for evidence of significance enrichment. The DAVID/EASE (Huang et al., 2009) online tool was used to perform enrichment analysis of the differentially expressed genes for each of TCE and PCE, with FDR q < 0.05 considered statistically significant. Bioconductor was used to assign genes to the Gene Ontology (GO) domains (molecular function, cellular component, and biological process) and Kyoto Encyclopedia of Genes and Genomes (KEGG), for a total of 5015 GO and KEGG pathways tested with each pathway having at least 5 genes. For each GO and KEGG domain, the pathway P values were adjusted by Benjamini-Hochberg FDR (Benjamini and Hochberg, 1995) to obtain q values, with q < 0.05 considered statistically significant.
BMD analysis of the transcriptomic data
The BMDExpress software (Yang et al., 2007) was used to evaluate BMD differences between chemicals, with analysis organized around pathways provided by the Molecular Signatures Database (Subramanian et al., 2005) using dose values in mmol/kg.
Comparison of transcriptomic benchmark doses with apical data
Previous studies have suggested that transcriptomic PODs correlated with those for apical endpoints, and that therefore transcriptional BMD values have the potential to serve as POD for quantitative risk assessment (Thomas et al., 2011). We therefore compared transcriptomic BMDs with apical POD used in U.S. EPA’s Toxicological Reviews for TCE and PCE (U.S. EPA, 2011a,b). Specifically, U.S. EPA comprehensively reviewed the available literature and selected specific endpoints and studies for deriving PODs to support both noncancer reference doses and cancer slope factors. For noncancer, the PODs included no-observed-adverse-effect levels (NOAELs) and benchmark dose lower confidence limits(BMDLs) for a variety of kidney and liver effects. For cancer, we included only BMDLs for 10% responses for liver and kidney cancer to maintain comparability (some cancer PODs used 1% or 5% response). Because apical endpoint PODs were derived from a range of species, including mice, rats, and humans, each with differing toxicokinetics, we standardized all dose units to human equivalent doses (HEDs) using the most up-to-date multispecies physiologically based pharmacokinetic (PBPK) models (Chiu and Ginsberg, 2011; Chiu et al., 2009). Specifically, HEDs were based on the following dose metrics: area under the curve of PCE in blood for PCE kidney, liver oxidative metabolism for PCE and TCE liver, and glutathione (GSH) conjugation for TCE kidney. For PCE, maximum likelihood estimates of each internal dose metric from Chiu and Ginsberg (2011) were used. For TCE, median estimates of each internal dose metric from Chiu et al. (2009) were used. An additional reason for this standardization is that margins of exposure can be readily computed and compared based on HED. For each chemical (TCE or PCE) and tissue (liver or kidney), the apical endpoint HEDs were compared with median transcriptional BMDL values.
RESULTS
TCA is a major metabolite of both TCE and PCE, and it was used here as a metabolic parameter to conduct direct comparison between TCE and PCE 24 h after treatment (Figure 1). In liver, amounts of TCA were quantifiable at doses above 80 mg/kg (0.67 mmol/kg) for TCE and 30 mg/kg (0.22 mmol/kg) for PCE (Figs. 1A and 1B). In kidney, TCA was quantifiable at doses above 240 mg/kg (2 mmol/kg) for TCE and 300 mg/kg (2 mmol/kg) for PCE (Figs. 1C and 1D). At equimolar doses, more TCA was found in both liver and kidney tissue from PCE-exposed mice compared with TCE-exposed groups (2-way rank ANOVA P = 0.014 for liver, P = 1.3 × 10−4 for kidney). It is also notable that concentrations of TCA in liver were about 3-fold greater than those in kidney for both TCE and PCE treatments. Even though all correlations between the dose and TCA concentration in liver and kidney were significant, the correlations were stronger overall for PCE as compared with TCE.
Figure 1.

Liver (A and B) and kidney (C and D) levels of trichloroacetic acid (TCA) measured in male B6C3F1 mice 24 h following oral gavage with equimolar doses of trichloroethylene (TCE, top row) or tetrachloroethylene (PCE, bottom row) in aqueous vehicle (5% Alkamuls EL-620 in saline). Spearman (ρ) correlation coefficients and corresponding significance (P) values are shown for each dose-response relationship.
The focus of this study was on the dose-dependent transcriptional effects of TCE and PCE in liver and kidney. Transcriptional profiling was conducted by RNA-sequencing and logistic dose-response modeling was applied to all expressed transcripts in both tissues to determine the genes and pathways responsive to these treatments. Representative dose-response effects on the individual transcripts are shown in Figure 2. For example, expression of Cyp4a14 was strongly induced by both TCE and PCE in liver and in kidney, and significant correlation with administered dose was observed. At the same time, expression of Alox15 in liver was induced in a dose-dependent manner only by TCE, while no effect of either TCE or PCE was observed in the kidney. A complete matrix of dose-response analyses of all transcripts as compared with the administered dose of TCE or PCE, as well as to the liver and kidney levels of TCA, is provided in Supplementary Tables 1 and 2, respectively.
Figure 2.

Dose-dependent effects of TCE or PCE on mouse liver (A and B) and kidney (C and D) gene expression. Plots of logistic dose-response modeling of normalized gene expression values for each animal, with individual data shown as red (for TCE) and green (for PCE) dots and respective curve fits colored accordingly are shown. Representative genes are shown. A and C, Cyp4a14 is significantly upregulated by both TCE and PCE in both liver and kidney. B and D, Alox15 is upregulated by TCE in liver only but not PCE.
Next, we sought to determine the degree of the overall effect on dose-dependent transcriptional changes between TCE and PCE (Figure 3). Scatter plots show transcripts that were significantly positively or negatively correlated with the administered dose of TCE or PCE in liver (Figure 3A) and kidney (Figure 3D). The same comparisons based on the levels of TCA produced in each tissue after exposure to either TCE or PCE are shown in Supplementary Figure 2. Overall, each chemical elicited both unique and shared responses in these tissues (genes are labeled red for TCE and green for PCE). Dose-dependent effects on the transcriptome were highly correlated between the administered dose and the tissue level of TCA (Figs. 3B, 3C, 3E, and 3F); however, correlations were much greater for PCE, as compared with TCE. This result suggests close correspondence of differential expression results between the levels of TCA and the administered dose. Therefore, pathway analyses were conducted to elucidate the molecular networks perturbed by TCE and/or PCE in a dose-dependent manner.
Figure 3.

Correlation analysis of dose-response in gene expression in mouse liver (A–C) and kidney (D–F) following treatment with TCE or PCE. Plotted are −Log10(P values) for dose-responsive differential gene expression analysis, right and top of axes origin are genes with positive association, left and bottom of axes origin are genes with negative association. Dots are individual genes and the color scheme is as follows: red (genes that are significant with false discovery rate (FDR) q < 0.05 in TCE but not PCE), green (q < 0.05 in PCE but not TCE), blue (significant with q < 0.05 for both TCE and PCE), and black (not significant for both TCE and PCE). A, Genes significantly correlated with the administered dose of either PCE or TCE. B, Genes significantly correlated with both TCE dose and liver TCA levels. C, Genes significantly correlated with both PCE dose and liver TCA levels. Panels D–F are same as A–C but for kidney. Pearson (r) correlation coefficients are shown for each plot.
Pathway analysis of the chemical-specific or shared dose-responsive transcripts revealed that pathways associated with Peroxisome proliferator-activated receptor (PPAR) signaling and oxidation/reduction were largely identical between TCE and PCE and significantly positively correlated with the administered dose in the liver (Table 1, Supplementary Tables 3–6); no shared pathways were significantly negatively correlated. Positive dose-response transcriptional effects of PCE were largely related to fatty acid metabolism and membrane-associated transport (primarily ABC transporter family). Effects on the mitochondria and nucleotide metabolism pathways were negatively correlated with the administered dose of PCE. No pathways were significant and different from those also affected by PCE for positive correlation with TCE dose; acute-phase response was a significant pathway negatively correlated with TCE dose. In the kidney, nearly identical effects were observed for both chemicals (Table 2, Supplementary Tables 7–9), except that mitochondrial respiration was upregulated by PCE in a dose-dependent manner. In addition, PCE affected gluconeogenesis and retinol metabolism in the kidney.
Table 1.
Pathway Analysis of Dose-Responsive Genes in Mouse Liver Following Treatment With TCE or PCE as Illustrated in Figure 3A
| Category | Term | Count | pValue | qValuea |
|---|---|---|---|---|
| Pathways significantly (q < 0.05) positively correlated with both TCE and PCE doseb | ||||
| KEGG_PATHWAY | mmu03320:PPAR signaling pathway | 9 | 1.6E-10 | 6.5E-09 |
| GOTERM_BP_FAT | GO:0055114 ∼ oxidation reduction | 9 | 6.5E-04 | 9.8E-02 |
| Pathways significantly (q < 0.05) negatively correlated with both TCE and PCE dose | ||||
| None | ||||
| Pathways significantly (q < 0.05) positively correlated only with PCE dosec | ||||
| GOTERM_CC_FAT | GO:0005777 ∼ peroxisome | 12 | 9.1E-09 | 1.5E-06 |
| GOTERM_BP_FAT | GO:0006631 ∼ fatty acid metabolic process | 14 | 1.1E-07 | 1.1E-04 |
| KEGG_PATHWAY | mmu02010:ABC transporters | 6 | 1.4E-04 | 1.3E-02 |
| Pathways significantly (q < 0.05) negatively correlated only with PCE dosed | ||||
| GOTERM_CC_FAT | GO:0005739 ∼ mitochondrion | 50 | 7.0E-11 | 1.9E-08 |
| GOTERM_CC_FAT | GO:0070469 ∼ respiratory chain | 8 | 3.2E-05 | 8.4E-04 |
| KEGG_PATHWAY | mmu00240:Pyrimidine metabolism | 8 | 1.7E-03 | 4.2E-02 |
| GOTERM_CC_FAT | GO:0031974 ∼ membrane-enclosed lumen | 30 | 1.6E-03 | 3.5E-02 |
| Pathways significantly (q < 0.05) positively correlated only with TCE dose | ||||
| None | ||||
| Pathway significantly (q < 0.05) negatively correlated only with TCE dosee | ||||
| GOTERM_BP_FAT | GO:0006953 ∼ acute-phase response | 4 | 2.2E-05 | 6.2E-03 |
Representative pathways for each annotation cluster are shown, complete lists of significant pathways and clusters are included in Supplementary Tables 3–6.
Benjamini-Hochberg-corrected P value.
Full list of the significant pathways is in Supplementary Table 3.
Full list of the significant pathways is in Supplementary Table 4.
Full list of the significant pathways is in Supplementary Table 5.
Full list of the significant pathways is in Supplementary Table 6.
Table 2.
Pathway Analysis of Dose-Responsive Genes in Mouse Kidney Following Treatment With TCE or PCE as Illustrated in Figure 3D
| Category | Term | Count | pValue | qValuea |
|---|---|---|---|---|
| Pathways significantly (q < 0.05) positively correlated with both TCE and PCE doseb | ||||
| KEGG_PATHWAY | mmu03320:PPAR signaling pathway | 6 | 7.9E-08 | 2.6E-06 |
| UP_KEYWORDS | Oxidoreductase | 4 | 4.0E-03 | 4.7E-02 |
| Pathways significantly (q < 0.05) negatively correlated with both TCE and PCE dose | ||||
| None | ||||
| Pathways significantly (q < 0.05) positively correlated only with PCE dosec | ||||
| GOTERM_CC_DIRECT | GO:0005739 ∼ mitochondrion | 37 | 2.7E-07 | 4.3E-05 |
| GOTERM_CC_FAT | GO:0005777 ∼ peroxisome | 10 | 1.7E-06 | 1.4E-04 |
| GOTERM_BP_DIRECT | GO:0006631 ∼ fatty acid metabolic process | 12 | 1.3E-07 | 4.4E-05 |
| GOTERM_BP_DIRECT | GO:0032869 ∼ cellular response to insulin stimulus | 8 | 1.2E-05 | 2.0E-03 |
| GOTERM_BP_DIRECT | GO:0006412 ∼ translation | 18 | 9.4E-08 | 9.2E-05 |
| KEGG_PATHWAY | mmu00830:Retinol metabolism | 8 | 1.0E-04 | 1.4E-02 |
| Pathway significantly (q < 0.05) negatively correlated only with PCE dosed | ||||
| KEGG_PATHWAY | mmu00982:Drug metabolism—cytochrome P450 | 5 | 2.6E-04 | 1.7E-02 |
| Pathways significantly (q < 0.05) positively correlated only with TCE dose | ||||
| None | ||||
| Pathways significantly (q < 0.05) negatively correlated only with TCE dose | ||||
| None | ||||
Representative pathways for each annotation cluster are shown, complete lists of significant pathways and clusters are included in Supplementary Tables 7–9.
Benjamini-Hochberg-corrected P value.
Full list of the significant pathways is in Supplementary Table 7.
Full list of the significant pathways is in Supplementary Table 8.
Full list of the significant pathways is in Supplementary Table 9.
A comparison of tissue-specific transcriptional effects of TCE and PCE was also explored based on the administered dose of TCE or PCE (Figure 4) or the levels of TCA in liver and kidney (Supplementary Figure 3). From these comparisons, it is evident that TCE had a much weaker overall dose-response effect on the transcriptome in both liver and kidney, with few genes representing a common signature. PCE, on the other hand, elicited a much stronger dose-response effect on transcription with many transcripts exhibiting strong dose-response effects between liver and kidney. Even though there were differences in the overall magnitude of the effect on the transcriptome, shared pathways between both organs and chemicals were related to peroxisomal fatty acid metabolism (Table 3, Supplementary Tables 10 and 11).
Figure 4.

Correlation analysis of gene expression responses between mouse liver and kidney following treatment with TCE (A) or PCE (B). Plotted are −log10(P values) for dose-responsive differential gene expression analysis (compared with TCE or PCE dose). Colors and directionality in effects are same as in the legend to Figure 3. Pearson (r) correlation coefficients are shown for each correlation.
Table 3.
Pathway Analysis of Concordant Dose-Responsive Genes in Mouse Liver and Kidney Following Treatment With TCE or PCE as Illustrated in Figure 4
| Category | Term | Count | pValue | qValuea |
|---|---|---|---|---|
| Pathway significantly (q < 0.05) positively correlated with TCE dose in both liver and kidney (Figure 4A)b | ||||
| KEGG_PATHWAY | mmu03320:PPAR signaling pathway | 5 | 3.7E-07 | 5.5E-06 |
| Pathways significantly (q < 0.05) negatively correlated TCE dose in both liver and kidney | ||||
| None | ||||
| Pathways significantly (q < 0.05) positively correlated with PCE dose in both liver and kidney (Figure 4B)c | ||||
| GOTERM_CC_DIRECT | GO:0005777 ∼ peroxisome | 7 | 3.4E-07 | 2.2E-05 |
| GOTERM_BP_DIRECT | GO:0006637 ∼ acyl-CoA metabolic process | 5 | 6.2E-07 | 1.9E-04 |
| Pathways significantly (q < 0.05) negatively correlated PCE dose in both liver and kidney | ||||
| None | ||||
Representative pathways for each annotation cluster are shown, complete lists of significant pathways and clusters are included in Supplementary Tables 10 and 11.
Benjamini-Hochberg-corrected P value.
Full list of the significant pathways is in Supplementary Table 10.
Full list of the significant pathways is in Supplementary Table 11.
Although transcript- and pathway-based analysis detailed above was focused on the significance of the dose-response relationships, we also examined the magnitude of the transcriptional effects of TCE and PCE in each tissue by analyzing fold-change in the expression of dose-responsive transcripts at the highest dose (Figure 5). In the liver, 54 transcripts were significantly affected by both TCE (800 mg/kg or 6 mmol/kg) and PCE (1000 mg/kg or 6 mmol/kg); only 16 transcripts were significantly perturbed in the kidney. Interestingly, expression of these transcripts in the liver was up- or downregulated to nearly identical extent by both TCE and PCE (Figure 5A, slope approximately 1). In the kidney, while the correlation was significant, PCE treatment elicited a somewhat stronger induction of the common transcripts (Figure 5D, slope > 1). Fewer genes were significantly up- or downregulated by exposure to the highest dose of TCE in both liver (n = 97, Figure 5B) and kidney (n = 12, Figure 5E), the range of effects was up to 30-fold for upregulated transcripts and even greater for a few downregulated transcripts. The effects of the highest tested dose of PCE were much more prominent with 545 transcripts significantly affected in the liver (Figure 5C) and 290 in the kidney (Figure 5F). Although the number of transcripts affected by PCE was greater, the fold-change in expression was largely not over 10- to 15-fold. Interestingly, more transcripts were down- than upregulated in response to PCE in the liver but the opposite was true in the kidney. Pathway analysis of the transcripts in each category shown in Figure 5 (Table 4, Supplementary Tables 13–17) showed that shared pathways in both liver and kidney were related to peroxisomal fatty acid oxidation. Distinctively, PCE had a strong effect on the mitochondria-related pathways in both liver and kidney.
Figure 5.

Analysis of the transcriptional effects of TCE (800 mg/kg or 6 mmol/kg) and PCE (1000 mg/kg or 6 mmol/kg) in mouse liver (A–C) and kidney (D–F). Shown are genes that were significantly (FDR q < 0.05) up- or downregulated by treatment with either chemical. A and D, Genes that were significant for both TCE and PCE in liver (n = 54, only genes with official gene symbols counted) or kidney (n = 16, only genes with official gene symbols counted). Maximum fold-change in gene expression, converted to log2 values, is plotted for each gene. Regression analysis slope, correlation coefficients and significance are shown. Select genes are highlighted. B and C, Histograms of log2 maximum fold-change values for genes that are significant in liver either for TCE (n = 97) or PCE (n = 545). E and F, Histograms of log2 maximum fold-change values for genes that are significant in kidney either for TCE (n = 12) or PCE (n = 290). Lists of genes shown in each panel are provided as Supplementary Table 12.
Table 4.
Pathway Analysis of Gene Expression Following Treatment With TCE (800 mg/kg or 6 mmol/kg) or PCE (1000 mg/kg or 6 mmol/kg) as Illustrated in Figure 5
| Category | Term | Count | pValue | qValuea |
|---|---|---|---|---|
| Pathways significantly (q < 0.05) affected in liver by both TCE and PCE (Figure 5A)b | ||||
| KEGG_PATHWAY | mmu03320:PPAR signaling pathway | 11 | 3.0E-13 | 2.0E-11 |
| UP_KEYWORDS | Oxidoreductase | 11 | 1.6E-06 | 8.8E-05 |
| GOTERM_CC_DIRECT | GO:0005789 ∼ endoplasmic reticulum membrane | 9 | 3.9E-04 | 1.6E-02 |
| Pathway significantly (q < 0.05) affected in liver only by TCE (Figure 5B)c | ||||
| GOTERM_CC_DIRECT | GO:0005615 ∼ extracellular space | 19 | 7.4E-05 | 8.5E-03 |
| Pathways significantly (q < 0.05) affected in liver only by PCE (Figure 5C)d | ||||
| GOTERM_CC_DIRECT | GO:0005739 ∼ mitochondrion | 80 | 1.2E-08 | 2.3E-06 |
| GOTERM_BP_DIRECT | GO:0006629 ∼ lipid metabolic process | 33 | 1.7E-07 | 1.1E-04 |
| GOTERM_CC_DIRECT | GO:0005789 ∼ endoplasmic reticulum membrane | 33 | 4.8E-04 | 2.6E-02 |
| GOTERM_CC_DIRECT | GO:0005777 ∼ peroxisome | 15 | 3.1E-06 | 4.0E-04 |
| KEGG_PATHWAY | mmu02010:ABC transporters | 9 | 3.7E-05 | 4.3E-03 |
| Pathways significantly (q < 0.05) affected in kidney by both TCE and PCE (Figure 5D)e | ||||
| KEGG_PATHWAY | mmu03320:PPAR signaling pathway | 7 | 4.8E-09 | 2.0E-07 |
| KEGG_PATHWAY | mmu00071:Fatty acid degradation | 4 | 1.0E-04 | 1.5E-03 |
| Pathways significantly (q < 0.05) affected in kidney only by TCE (Figure 5E) | ||||
| None | ||||
| Pathways significantly (q < 0.05) affected in kidney only by PCE (Figure 5F)f | ||||
| GOTERM_BP_DIRECT | GO:0055114 ∼ oxidation-reduction process | 27 | 1.2E-06 | 7.5E-04 |
| GOTERM_BP_DIRECT | GO:0006631 ∼ fatty acid metabolic process | 13 | 1.2E-06 | 1.5E-03 |
| KEGG_PATHWAY | mmu00982:Drug metabolism-cytochrome P450 | 10 | 1.5E-06 | 8.2E-05 |
| GOTERM_CC_DIRECT | GO:0005777 ∼ peroxisome | 10 | 6.4E-05 | 6.5E-03 |
| GOTERM_CC_DIRECT | GO:0005743 ∼ mitochondrial inner membrane | 16 | 2.2E-04 | 1.5E-02 |
Representative pathways for each annotation cluster are shown, complete lists of significant pathways and clusters are included in Supplementary Tables 13–17.
Benjamini-Hochberg-corrected P value.
Full list of the significant pathways is in Supplementary Table 13.
Full list of the significant pathways is in Supplementary Table 14.
Full list of the significant pathways is in Supplementary Table 15.
Full list of the significant pathways is in Supplementary Table 16.
Full list of the significant pathways is in Supplementary Table 17.
An additional advantage of RNA-sequencing approach for gene expression analyses is the ability to examine the expression of the individual exons. The role of alternative splicing as a potential mechanism of toxicity has not been widely examined; therefore, we “over-sequenced” vehicle- and highest dose-treated samples from liver (Supplementary Figure 1). Greater sequencing depth allowed us to examine TCE and PCE effects on expression changes of the individual exons, as illustrated by the example of TCE effect in the liver (Figure 6). No genes demonstrated evidence for the differential exon usage in response to treatment at q < 0.05. Using a relatively liberal criterion of FDR q < 0.2, 14 genes showed significant differential isoform evidence as a function of TCE dose, and 121 genes as a function of PCE dose. Even though the findings were not highly significant, using the q value approach (Storey and Tibshirani, 2003) one can estimate the overall proportion of genes with differential isoform usage, even if not individually significant. Using this approach, a total of 9.0% and 21.0% of genes were estimated to have differential exon usage in liver for treatment with TCE and PCE, respectively. For kidney, no genes showed differential exon usage for TCE, and 2 genes for PCE at q < 0.2, and the corresponding overall estimates of genes with differential isoform usage was 2.7% and 1.1%.
Figure 6.
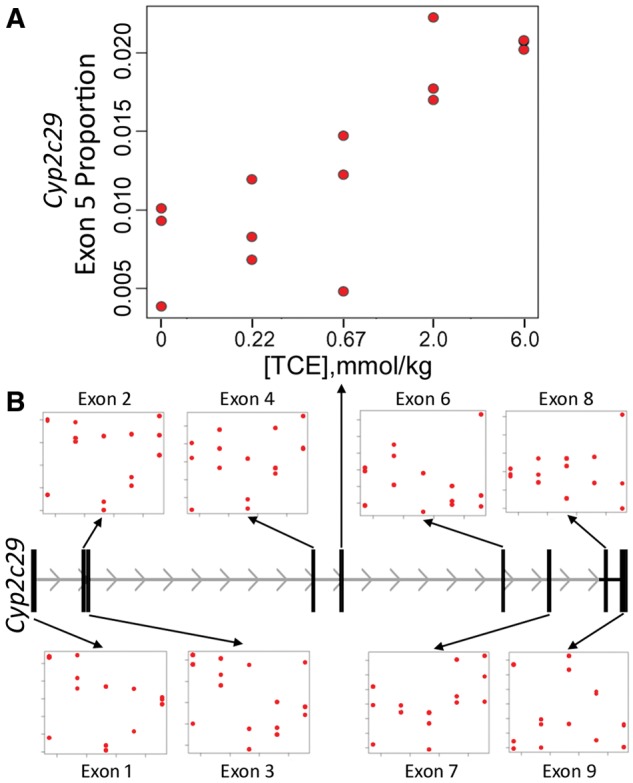
Association of Cyp2c29 exon “usage” proportion with TCE dose (P = 2.1 × 10−5, q value = 0.18). The result is largely driven by reads mapping to exon 5, for which the exon-specific usage was highly significant (P = 2.6 × 10−6) while for other exons it was not significant.
Several cytochrome P450 genes were among those with differential isoform evidence (q < 0.2) in the liver, including Cyp2c29, Cyp2c67, and Cyp2j6. The most significant gene was Cyp2c29, with unadjusted P = 2.31 × 10−5 and false discovery q = 0.18. This gene serves as an interesting illustration as it was selected in this analysis even though overall differential expression for this gene was not significant for either TCE or PCE (P > 0.6). Figure 6 shows that the exon-usage result is largely driven by relative expression in exon 5 increasing with TCE dose.
For the differential exon-usage results in liver, DAVID/EASE pathway analyses were performed for 340 genes with P < 0.01 for TCE and 507 genes with P < 0.01 for PCE. The functional annotation clustering for TCE pathways with enrichment q < 0.1 included metabolism of xenobiotics by cytochrome P450 (Table 5, Supplementary Table 18). Significant pathways for PCE included nucleoside binding, membrane-enclosed lumen and chaperone. Using the same approach and criteria, no pathways were significant for either chemical in the kidney.
Table 5.
Differential Exon-Usage Pathways in Liver in Response to Treatment With TCE
| Category | Term | Count | pValue | qValuea |
|---|---|---|---|---|
| Pathways significantly (q < 0.05) associated with differential usage of exons after PCE treatmentb | ||||
| GOTERM_MF_FAT | GO:0001882 ∼ nucleoside binding | 65 | 9.2E-05 | 4.7E-02 |
| GOTERM_CC_FAT | GO:0031974 ∼ membrane-enclosed lumen | 50 | 3.5E-04 | 4.0E-02 |
| SP_PIR_KEYWORDS | Chaperone | 13 | 5.1E-04 | 4.3E-02 |
| Pathways significantly (q < 0.05) associated with differential usage of exons after TCE treatmentb | ||||
| KEGG_PATHWAY | mmu00980:Metabolism of xenobiotics by cytochrome P450 | 8 | 3.2E-04 | 1.8E-02 |
Representative pathways for each annotation cluster are shown, complete lists of significant pathways and clusters are included in Supplementary Table 18.
Benjamini-Hochberg-corrected P value.
Full list of the significant pathways is in Supplementary Table 18.
Additionally, we sought to compare transcriptomics-derived dose-response effects between TCE and PCE and between the liver and kidney. Using transcriptomics data from this study, we derived transcriptional POD values for individual genes (BMDs and BMDLs) and then aggregated them into pathways (eg, median BMDs and BMDLs) in response to treatment with TCE or PCE in liver and kidney (Supplementary Table 19). Figure 7A shows POD values for pathways in liver and kidney that were significantly perturbed (with respect to dose-response) by TCE or PCE. Overall, liver transcriptional POD was lower than those for kidney, with PCE effects in the liver effected at the lowest doses. A number of common pathways were found (highlighted in Supplementary Table 19), with PPARα-mediated signaling being shared across both chemicals and tissues.
Figure 7.

Dose-response analysis of the transcriptional and apical effects of TCE and PCE in mouse liver and kidney. A, Point of departure (POD) (median BMDL) for pathways (Kyoto Encyclopedia of Genes and Genomes [KEGG] or Reactome) that were significantly perturbed (Fisher’s exact 2-tailed P < 0.05) by treatment with TCE (red) or PCE (green) in mouse liver (circles) and kidney (squares). Open symbols are KEGG pathways and closed symbols are Reactome pathways. A complete list of pathways and associated dose-response PODs are included as Supplementary Table 19. Select common pathways are labeled. B, Comparison of the apical POD (noncancer endpoints are black vertical bars, cancer endpoints are blue vertical bars) and transcriptomic POD ranges for all pathways (red box and whisker plots showing distribution of median pathway BMDLs) and selected pathways (individual red vertical lines showing individual median pathway BMDLs) after treatment with TCE or PCE. Complete details of the apical endpoint types, studies from which they were derived, and PODs for the pathways are available in Supplementary Tables 19 and 20. To enable direct comparison of the apical and transcriptional PODs to human exposure, all doses were converted to human equivalent doses using the following metrics: liver oxidative metabolism (for TCE and PCE Liver), GSH conjugation metabolism (for TCE Kidney), and PCE area under the curve (for PCE Kidney). C, Relationship between transcriptional and apical PODs converted to human equivalent doses. Symbols and error bars are the geometric means and ranges, respectively, of the median transcriptional BMDLs plotted against the corresponding geometric means and range of the apical PODs, from each panel in (B). Symbol shapes and colors represent different treatment (PCE or TCE), target tissue (kidney or liver), and type of apical POD (noncancer or cancer) as shown in the legend of panel C. Blue dotted lines are ± 1 order of magnitude deviation from perfect correspondence.
Finally, as described in the Materials and Methods section, we compared transcriptional BMDLs in this acute exposure study in the mouse with traditional apical PODs for noncancer and cancer endpoints in the same tissues from subchronic chronic mouse or rat studies, or epidemiological human data (Supplementary Table 20). The apical endpoint POD were those previously used by U.S. EPA (2011a,b) to derive toxicity values. Because the PODs were derived from several species, for this comparison, we converted both types of PODs to HEDs using multispecies PBPK models, also described in Materials and Methods, with the results shown in Figure 7B. For PCE, the transcriptional POD tended to be somewhat less sensitive than the apical PODs, with the exception of the apical POD for kidney cancer. For TCE, the transcriptional POD covered the same range. Overall, transcriptional POD were correlated well with apical endpoints PODs, as shown by the plots of the geometric means and ranges in POD in apical and transcriptional data for each chemical and tissue (Figure 7C). For both PCE and TCE, the median transcriptional BMDLs were generally within 10-fold of the apical PODs for the corresponding tissue.
DISCUSSION
After decades of toxicological research on major environmental pollutants TCE and PCE, our knowledge remains incomplete concerning how these chemicals induce toxicity and whether their effects are as similar as their resemblance in structure and metabolite profile may suggest. TCE is a well-studied chemical; however, considerably less experimental and epidemiologic evidence is available for PCE, one of the most widely used chlorinated solvents. Furthermore, the database on the comparative toxicity of chlorinated solvents is relatively sparse and is limited to apical measures of toxicity (eg, LD50, serum liver enzyme levels, urinary proteins, etc.). Thus, this study was undertaken to narrow the knowledge gaps and to better inform risk management decisions by directly comparing adverse health effects of TCE and PCE through a comprehensive dose-response evaluation of transcriptomic phenotypes.
This study was not intended for comprehensive toxicokinetic evaluation of TCE and PCE across multiple tissues; still, the dose-response study design and the standardization of the administered doses provided important information on TCE and PCE metabolism to TCA in liver and kidney. TCA is a known toxicant, and liver carcinogen in rodents (Bull et al., 1990). Specifically, we found that exposure to equimolar doses of both chemicals results in greater amounts of TCA produced by PCE in both tissues and that concentrations of TCA in liver are about 3-fold greater than those in kidney for both TCE and PCE. These findings are consistent with the oxidative metabolism of TCE and PCE in both humans (Bernauer et al., 1996; Lash et al., 1999; Volkel et al., 1998) and rodents (Cichocki et al., 2017b; Lash et al., 2014; Lash and Parker, 2001; Yoo et al., 2015a,b) whereby there are 2 major oxidative metabolites for TCE (trichloroethanol and TCA), while TCA is the only major oxidative metabolite of PCE. It is noteworthy that previous studies seldom compared tissue levels of TCA between TCE and PCE in the same study and at the same range of doses; therefore, our work provides important direct comparison that should increase confidence in tissue dosimetry of a shared toxic oxidative metabolite of both chemicals. However, a limitation of our study is that it did not evaluate formation of glutathione conjugates, species that are thought to be key for kidney-specific effects of both TCE and PCE (Cichocki et al., 2016). Such an examination requires development of sensitive methods for detection of glutathione conjugates (Kim et al., 2009a; Luo et al., 2017) and a different study design as these molecules are cleared rapidly from the body (Kim et al., 2009b).
Dose-response analyses of transcriptional effects yield important clues on similarities and differences in tissue-specific effects of TCE and PCE. Our data suggest that the oxidative metabolism of TCE and PCE is mechanistically related to the observed alterations in gene expression in both liver and kidney. First, we observed that induction of peroxisomal fatty acid β-oxidation and associated pathways was among the most robust and dose-responsive effects of both TCE and PCE. Indeed, it is known that TCA, but not TCE, TCOH or PCE can activate mouse PPARa (Maloney and Waxman, 1999; Zhou and Waxman, 1998). Second, dose-responsive differential gene expression in liver and kidney was highly correlated between TCE or PCE external dose and internal TCA (liver or kidney) dose levels, further suggesting that TCA is not merely a surrogate for exposure.
Importantly, while PPARα-mediated signaling is a well-established effect of these chemicals in the rodent liver, the evidence in support of a role for this pathway as a mechanism of kidney toxicity for both TCE and PCE is deemed to be relatively modest (Cichocki et al., 2016). Thus, our study provides additional (Yoo et al., 2015b,c) strong evidence that not only is there robust concordance and dose-response in both liver and kidney but also that transcripts in PPARα-mediated pathways are induced by TCE and PCE to nearly the same extent. Other shared mechanisms of noncancer toxicity for TCE and PCE are cell proliferation, apoptosis, and oxidative stress; however, the time-point employed in this study, 24 h after treatment, makes interpreting transcriptional changes in these pathways challenging, as the acute effects of these agents may or may not be associated with apical chronic responses.
Transcriptional profiling data obtained in this study also offer important clues about metabolic pathways of TCE and PCE in both liver and kidney. First, a connection between PPARα activation and TCE metabolism has been recently demonstrated in mouse liver (Venkatratnam et al., 2017; Yoo et al., 2015c). The fact that PPARα activation is equally pronounced for liver and kidney, suggests that similar connections may exist in the kidney, and may also be true for PCE; however, these hypotheses need to be examined further. Second, we found that PCE induced ABC-family transporters in the liver but solute carrier (SLC)-family transporters in the kidney in a dose-dependent manner; TCE treatment had no dose-response effect on these pathways. Most of the data that exist on the role of transporters in the potential toxicity of chlorinated solvents has been from studies of TCE and the fate of its glutathione conjugates in the kidney (Lash, 2011; Tsirulnikov et al., 2010). Thus, our findings that PCE may have a more pronounced effects on transporters, and also in the liver, also suggests intriguing avenues for future research. Third, we found that alternative splicing of cytochrome P450 genes upon exposure to TCE may also effect its metabolism. Oxidative metabolism of TCE and PCE has been attributed primarily to CYP2E1 activity owing to their structural similarity to other CYP2E1 substrates (Lash and Parker, 2001), although experimental studies in knockout mice showed that it is not an exclusive pathway for TCE (Forkert et al., 2006; Kim and Ghanayem, 2006; Ramdhan et al., 2008). We found that several cytochrome P450 genes exhibited significant differential isoform usage upon exposure, these include Cyp2c29, Cyp2c67, and Cyp2j6. The most significant gene was Cyp2c29, where presence of exon 5 in the transcripts was positively associated with the dose of TCE. CYP2C family enzymes have been implicated in interindividual variability in metabolism of chlorinated solvents (Snawder and Lipscomb, 2000; Wang et al., 1996), and Cyp2c29 is a highly expressed hepatic enzyme in the mouse. Although no study examined the role of exon 5 in Cyp2c29 function, this exon does not have an amino acid that encodes across a splice junction and the loss of this exon is likely to result in shortening of the protein. In addition, several predicted transcript variants of Cyp2c29 gene that are missing exon 5 (XM_006526636 and XP_006526699) have been annotated based on RNA-seq alignments. Thus, further exploration of the alternative splicing as a potential mechanism of chemical effects on metabolism and toxicity is warranted (Nelson et al., 2004) and is now enabled by RNA-seq experimental data.
Equally informative are observed discordances in the transcriptional effect of TCE and PCE. Our data show that mitochondria-related transcriptional pathways are strongly affected, in dose-response manner, in both liver and kidney by PCE but not TCE. These results are concordant with observations made previously using liver and kidney in vitro models in rats and mice. Studies in rat liver preparations showed that PCE had a much stronger effect on uncoupling mitochondrial oxidative phosphorylation than TCE (Ogata and Hasegawa, 1981). Subsequent studies demonstrated that in rat liver submitochondrial particles, PCE increased the Michaelis constant (Km) and decreased the maximum velocity (Vmax) of cytochrome c reduction by NADH-cytochrome c reductase, an effect that suggests that PCE may impact the electron flow at the mitochondrial inner membrane (Miyazaki and Takano, 1983). Lash et al. (2002) showed that PCE can impair mitochondrial respiration in suspensions of isolated mitochondria from rat and mouse kidneys, and this effect was much more pronounced than that for TCE; however, this study did not find significant effects of PCE on the isolated liver mitochondria from rats or mice (Lash et al., 2002). Thus, our results obtained in vivo further add to this body of knowledge regarding the potential for PCE, but not TCE, to impair mitochondrial respiration in both liver and kidney.
Finally, we explored the utility of the transcriptomic data obtained from this acute single-dose study for dose-response assessment and derivation of POD values. Thomas and coworkers have demonstrated that pathway-based PODs based on gene expression data from short-term exposure studies are well-correlated with the PODs for apical endpoints in the same species and route of exposure derived from traditional 90-day and 2-year animal studies (Farmahin et al., 2017; Thomas et al., 2011, 2013). We found that transcriptional PODs in the mouse after an acute exposure correlated well with POD for apical endpoints from subchronic and chronic studies across multiple exposure routes and species, including humans—after correcting for toxicokinetics, differences were less than an order of magnitude. For PCE, the transcriptional POD tended to be somewhat less sensitive than the apical POD; for TCE, the transcriptional POD covered the same range, with the exception of the apical POD for kidney cancer. These difference may be related the dosimetry conversion to HEDs; the TCE PBPK model (Chiu et al., 2009, 2014) is based on a more robust toxicokinetic database than the PCE model (Chiu and Ginsberg, 2011). Thomas et al. (2011) used the median transcriptional BMD or BMDL for the most sensitive pathway in each tissue as a comparator for apical POD. They found that the transcriptional POD was more conservative, generally within one order of magnitude (Thomas et al., 2011). In our study, transcriptional PODs were also within 10-fold of apical PODs but the differences were in both directions as opposed to being consistently conservative. Additionally, whereas previous analyses compared transcriptional and apical endpoint PODs only in the same species and route of exposure, we used PBPK modeling to standardize PODs from multiple species and routes to human equivalent oral doses. For instance, many of the apical endpoint PODs we used were based on inhalation studies, and importantly, in one case, the apical endpoint POD was based on human data. Conversion to HEDs has the additional utility of being directly comparable to human exposure estimates and derivation of the margins of exposure. These results provide further evidence that transcriptomic data can be used as surrogates for in vivo PODs and suggest that this approach can be generalized across species and exposure routes when toxicokinetic differences are taken into account.
In conclusion, we posit that comparative analyses of the molecular effects (eg, liver and kidney gene expression) elicited by highly similar chlorinated solvents (TCE and PCE) show both similarities and differences in the mechanisms of their toxicity in the mouse. Quantitative assessment of such differences and similarities through dose-response study designs yields important additional clues for interpretation of the concordance in the mechanisms of toxicity. Notably, comparisons with apical endpoints observed in cancer and noncancer studies suggest that transcriptional data, when used in combination with tissue-specific dosimetry, may be useful for conducting risk-based evaluations of chemical exposures.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge assistance from Dr Scott Auerbach (National Institute of Environmental Health Sciences) in interpreting BMDExpress results. The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of National Institutes of Health (NIH) or EPA. The authors express their gratitude to Dr Thomas McDonald (Texas A&M University) for assistance with analytical instrumentation.
FUNDING
J.A.C. was a recipient of a National Research Service Award through the National Institute of Environmental Health Sciences (F32 ES026005). This work was supported, in part, by a cooperative agreement (STAR RD83561202) from U.S. EPA to Texas A&M University.
REFERENCES
- Anders S., McCarthy D. J., Chen Y., Okoniewski M., Smyth G. K., Huber W., Robinson M. D. (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P. T., Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Reyes A., Huber W. (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate - A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 57, 289–300. [Google Scholar]
- Bernauer U., Birner G., Dekant W., Henschler D. (1996). Biotransformation of trichloroethene: Dose-dependent excretion of 2,2,2-trichloro-metabolites and mercapturic acids in rats and humans after inhalation. Arch. Toxicol. 70, 338–346. [DOI] [PubMed] [Google Scholar]
- Buben J. A., O’Flaherty E. J. (1985). Delineation of the role of metabolism in the hepatotoxicity of trichloroethylene and perchloroethylene: A dose-effect study. Toxicol. Appl. Pharmacol. 78, 105–122. [DOI] [PubMed] [Google Scholar]
- Bull R. J., Sanchez I. M., Nelson M. A., Larson J. L., Lansing A. J. (1990). Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology 63, 341–359. [DOI] [PubMed] [Google Scholar]
- California Environmental Protection Agency. (2016). Perchloroethylene Inhalation Cancer Unit Risk Factor Technical Support Document for Cancer Potency Factors, Appendix B, Public Review Draft (February, 2016) Air Toxics Hot Spots Program. https://oehha.ca.gov/media/downloads/air//061016srpreviewpceurf.pdf, Accessed June 1, 2017.
- Chiu W. A., Campbell J. L., Clewell H. J., Zhou Y. H., Wright F. A., Guyton K. Z., Rusyn I. (2014). Physiologically-based pharmacokinetic (PBPK) modeling of inter-strain variability in trichloroethylene metabolism in the mouse. Environ. Health Perspect. 122, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu W. A., Ginsberg G. L. (2011). Development and evaluation of a harmonized physiologically based pharmacokinetic (PBPK) model for perchloroethylene toxicokinetics in mice, rats, and humans. Toxicol. Appl. Pharmacol. 253, 203–234. [DOI] [PubMed] [Google Scholar]
- Chiu W. A., Jinot J., Scott C. S., Makris S. L., Cooper G. S., Dzubow R. C., Bale A. S., Evans M. V., Guyton K. Z., Keshava N., et al. (2013). Human health effects of trichloroethylene: Key findings and scientific issues. Environ. Health Perspect. 121, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu W. A., Okino M. S., Evans M. V. (2009). Characterizing uncertainty and population variability in the toxicokinetics of trichloroethylene and metabolites in mice, rats, and humans using an updated database, physiologically based pharmacokinetic (PBPK) model, and Bayesian approach. Toxicol. Appl. Pharmacol. 241, 36–60. [DOI] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Konganti K., Luo Y. S., McDonald T. J., Iwata Y., Chiu W. A., Threadgill D. W., Pogribny I. P., Rusyn I. (2017a). Impact of nonalcoholic fatty liver disease on toxicokinetics of tetrachloroethylene in mice. J. Pharmacol. Exp. Ther. 361, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Venkatratnam A., McDonald T. J., Knap A. H., Wade T., Sweet S., Chiu W. A., Threadgill D. W., Rusyn I. (2017b). Characterization of variability in toxicokinetics and toxicodynamics of tetrachloroethylene using the collaborative cross mouse population. Environ. Health Perspect. 125, 057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Guyton K. Z., Guha N., Chiu W. A., Rusyn I., Lash L. H. (2016). Target organ metabolism, toxicity, and mechanisms of trichloroethylene and perchloroethylene: Key similarities, differences, and data gaps. J. Pharmacol. Exp. Ther. 359, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmahin R., Williams A., Kuo B., Chepelev N. L., Thomas R. S., Barton-Maclaren T. S., Curran I. H., Nong A., Wade M. G., Yauk C. L. (2017). Recommended approaches in the application of toxicogenomics to derive points of departure for chemical risk assessment. Arch. Toxicol. 91, 2045–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay R. M., Mumtaz M. M. (1996). Development of a priority list of chemical mixtures occurring at 1188 hazardous waste sites, using the HazDat database. Food Chem. Toxicol. 34, 1163–1165. [DOI] [PubMed] [Google Scholar]
- Forkert P. G., Millen B., Lash L. H., Putt D. A., Ghanayem B. I. (2006). Pulmonary bronchiolar cytotoxicity and formation of dichloroacetyl lysine protein adducts in mice treated with trichloroethylene. J. Pharmacol. Exp. Ther. 316, 520–529. [DOI] [PubMed] [Google Scholar]
- Guha N., Loomis D., Grosse Y., Lauby-Secretan B., El Ghissassi F., Bouvard V., Benbrahim-Tallaa L., Baan R., Mattock H., Straif K., et al. (2012). Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 13, 1192–1193. [DOI] [PubMed] [Google Scholar]
- Guyton K. Z., Hogan K. A., Scott C. S., Cooper G. S., Bale A. S., Kopylev L., Barone S., Makris S. L., Glenn B., Subramaniam R. P., et al. (2014). Human health effects of tetrachloroethylene: Key findings and scientific issues. Environ. Health Perspect. 122, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. (2014). TRICHLOROETHYLENE, TETRAC-HLOROETHYLENE, AND SOME OTHER CHLORINATED AGENTS. IARC monographs on the evaluation of carcinogenic risks to humans, 106, 1. [PMC free article] [PubMed]
- Jia C., Yu X., Masiak W. (2012). Blood/air distribution of volatile organic compounds (VOCs) in a nationally representative sample. Sci. Total Environ. 419, 225–232. [DOI] [PubMed] [Google Scholar]
- Katz Y., Wang E. T., Airoldi E. M., Burge C. B. (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Ghanayem B. I. (2006). Comparative metabolism and disposition of trichloroethylene in Cyp2e1-/-and wild-type mice. Drug Metab. Dispos. 34, 2020–2027. [DOI] [PubMed] [Google Scholar]
- Kim S., Collins L. B., Boysen G., Swenberg J. A., Gold A., Ball L. M., Bradford B. U., Rusyn I. (2009a). Liquid chromatography electrospray ionization tandem mass spectrometry analysis method for simultaneous detection of trichloroacetic acid, dichloroacetic acid, S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine. Toxicology 262, 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Kim D., Pollack G. M., Collins L. B., Rusyn I. (2009b). Pharmacokinetic analysis of trichloroethylene metabolism in male B6C3F1 mice: Formation and disposition of trichloroacetic acid, dichloroacetic acid, S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine. Toxicol. Appl. Pharmacol. 238, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H. (2011). Renal membrane transport of glutathione in toxicology and disease. Vet. Pathol. 48, 408–419. [DOI] [PubMed] [Google Scholar]
- Lash L. H., Chiu W. A., Guyton K. Z., Rusyn I. (2014). Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity. Mutat. Res. Rev. Mutat. Res. 762, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H., Parker J. C. (2001). Hepatic and renal toxicities associated with perchloroethylene. Pharmacol. Rev. 53, 177–208. [PubMed] [Google Scholar]
- Lash L. H., Putt D. A., Brashear W. T., Abbas R., Parker J. C., Fisher J. W. (1999). Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J. Toxicol. Environ. Health A 56, 1–21. [DOI] [PubMed] [Google Scholar]
- Lash L. H., Qian W., Putt D. A., Hueni S. E., Elfarra A. A., Sicuri A. R., Parker J. C. (2002). Renal toxicity of perchloroethylene and S-(1,2,2-trichlorovinyl)glutathione in rats and mice: Sex- and species-dependent differences. Toxicol. Appl. Pharmacol. 179, 163–171. [DOI] [PubMed] [Google Scholar]
- Luo Y. S., Cichocki J. A., McDonald T. J., Rusyn I. (2017). Simultaneous detection of the tetrachloroethylene metabolites S-(1,2,2-trichlorovinyl) glutathione, S-(1,2,2-trichlorovinyl)-L-cysteine, and N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine in multiple mouse tissues via ultra-high performance liquid chromatography electrospray ionization tandem mass spectrometry. J. Toxicol. Environ. Health A 80, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney E. K., Waxman D. J. (1999). Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 161, 209–218. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y., Takano T. (1983). Impairment of mitochondrial electron transport by tetrachloroethylene. Toxicol. Lett. 18, 163–166. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program. (1977). Bioassay of tetrachloroethylene for possible carcinogenicity. Natl. Cancer Inst. Carcinog. Tech. Rep. Ser. 13, 1–83. [PubMed] [Google Scholar]
- National Toxicology Program. (1990). Carcinogenesis studies of trichloroethylene (without epichlorohydrin) (CAS No. 79-01-6) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 243, 1–174. [PubMed] [Google Scholar]
- National Toxicology Program, N. (2015). Report on Carcinogens, Monograph for Trichloroethylene https://ntp.niehs.nih.gov/ntp/roc/monographs/finaltce_508.pdf, Accessed June 1, 2017. [PubMed]
- Nelson D. R., Zeldin D. C., Hoffman S. M., Maltais L. J., Wain H. M., Nebert D. W. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18. [DOI] [PubMed] [Google Scholar]
- Nikolayeva O., Robinson M. D. (2014). edgeR for differential RNA-seq and ChIP-seq analysis: An application to stem cell biology. Methods Mol. Biol. 1150, 45–79. [DOI] [PubMed] [Google Scholar]
- Ogata M., Hasegawa T. (1981). Effects of chlorinated aliphatic hydrocarbons on mitochondrial oxidative phosphorylation in the rat with reference to the effects of chlorinated aromatic hydrocarbons. Ind. Health 19, 71–75. [DOI] [PubMed] [Google Scholar]
- Peterson C. B., Bogomolov M., Benjamini Y., Sabatti C. (2016). Many phenotypes without many false discoveries: Error controlling strategies for multitrait association studies. Genet. Epidemiol. 40, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip B. K., Mumtaz M. M., Latendresse J. R., Mehendale H. M. (2007). Impact of repeated exposure on toxicity of perchloroethylene in Swiss Webster mice. Toxicology 232, 1–14. [DOI] [PubMed] [Google Scholar]
- Pohl H. R., Tarkowski S., Buczynska A., Fay M., De Rosa C. T. (2008). Chemical exposures at hazardous waste sites: Experiences from the United States and Poland. Environ. Toxicol. Pharmacol. 25, 283–291. [DOI] [PubMed] [Google Scholar]
- Ramdhan D. H., Kamijima M., Yamada N., Ito Y., Yanagiba Y., Nakamura D., Okamura A., Ichihara G., Aoyama T., Gonzalez F. J., et al. (2008). Molecular mechanism of trichloroethylene-induced hepatotoxicity mediated by CYP2E1. Toxicol. Appl. Pharmacol. 231, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J., Smyth G. K. (2010). edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I., Chiu W. A., Lash L. H., Kromhout H., Hansen J., Guyton K. Z. (2014). Trichloroethylene: Mechanistic, epidemiologic and other supporting evidence of carcinogenic hazard. Pharmacol. Therap. 141, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snawder J. E., Lipscomb J. C. (2000). Interindividual variance of cytochrome P450 forms in human hepatic microsomes: Correlation of individual forms with xenobiotic metabolism and implications in risk assessment. Regul. Toxicol. Pharmacol. 32, 200–209. [DOI] [PubMed] [Google Scholar]
- Storey J. D., Tibshirani R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Liu Y., Crowley J. J., Chen T. H., Zhou H., Chu H., Huang S., Kuan P. F., Li Y., Miller D. R., et al. (2015). IsoDOT detects differential RNA-isoform expression/usage with respect to a categorical or continuous covariate with high sensitivity and specificity. J. Am. Stat. Assoc. 110, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. S., Allen B. C., Nong A., Yang L., Bermudez E., Clewell H. J. III, Andersen M. E. (2007). A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol. Sci. 98, 240–248. [DOI] [PubMed] [Google Scholar]
- Thomas R. S., Clewell H. J., Allen B. C., Wesselkamper S. C., Wang N. C., Lambert J. C., Hess-Wilson J. K., Zhao Q. J., Andersen M. E. (2011). Application of transcriptional benchmark dose values in quantitative cancer and noncancer risk assessment. Toxicol. Sci. 120, 194–205. [DOI] [PubMed] [Google Scholar]
- Thomas R. S., Wesselkamper S. C., Wang N. C., Zhao Q. J., Petersen D. D., Lambert J. C., Cote I., Yang L., Healy E., Black M. B., et al. (2013). Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol. Sci. 134, 180–194. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S. L. (2009). TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirulnikov K., Abuladze N., Koag M. C., Newman D., Scholz K., Bondar G., Zhu Q., Avliyakulov N. K., Dekant W., Faull K., et al. (2010). Transport of N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine, a metabolite of trichloroethylene, by mouse multidrug resistance associated protein 2 (Mrp2). Toxicol. Appl. Pharmacol. 244, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA. (2011a). Toxicological Review of Tetrachloroethylene (CAS No. 127-18-4): In Support of Summary Information on the Integrated Risk Information System (IRIS). U.S. Environmental Protection Agency, ed. EPA/635/R-08/011F.
- U.S. EPA. (2011b). Toxicological Review of Trichloroethylene (CAS No. 79-01-6): In Support of Summary Information on the Integrated Risk Information System (IRIS). National Center for Environmental Assessment, ed. EPA/635/R-09/011D.
- U.S. EPA. (2017). EPA Names First Chemicals for Review Under New TSCA Legislation. Available at: https://www.epa.gov/newsreleases/epa-names-first-chemicals-review-under-new-tsca-legislation. Accessed May 19, 2017.
- Venkatratnam A., Furuya S., Kosyk O., Gold A., Bodnar W., Konganti K., Threadgill D. W., Gillespie K. M., Aylor D. L., Wright F. A., et al. (2017). Collaborative cross mouse population enables refinements to characterization of the variability in toxicokinetics of trichloroethylene and provides genetic evidence for the role of PPAR pathway in its oxidative metabolism. Toxicol. Sci. 158, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkel W., Friedewald M., Lederer E., Pahler A., Parker J., Dekant W. (1998). Biotransformation of perchloroethene: Dose-dependent excretion of trichloroacetic acid, dichloroacetic acid, and N-acetyl-S-(trichlorovinyl)-L-cysteine in rats and humans after inhalation. Toxicol. Appl. Pharmacol. 153, 20–27. [DOI] [PubMed] [Google Scholar]
- Wang R. S., Nakajima T., Tsuruta H., Honma T. (1996). Effect of exposure to four organic solvents on hepatic cytochrome P450 isozymes in rat. Chem. Biol. Interact. 99, 239–252. [DOI] [PubMed] [Google Scholar]
- Yang L., Allen B. C., Thomas R. S. (2007). BMDExpress: A software tool for the benchmark dose analyses of genomic data. BMC Genomics 8, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. S., Bradford B. U., Kosyk O., Shymonyak S., Uehara T., Collins L. B., Bodnar W. M., Ball L. M., Gold A., Rusyn I. (2015a). Comparative analysis of the relationship between trichloroethylene metabolism and tissue-specific toxicity among inbred mouse strains: Liver effects. J. Toxicol. Environ. Health A 78, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. S., Bradford B. U., Kosyk O., Uehara T., Shymonyak S., Collins L. B., Bodnar W. M., Ball L. M., Gold A., Rusyn I. (2015b). Comparative analysis of the relationship between trichloroethylene metabolism and tissue-specific toxicity among inbred mouse strains: Kidney effects. J. Toxicol. Environ. Health A 78, 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. S., Cichocki J. A., Kim S., Venkatratnam A., Iwata Y., Kosyk O., Bodnar W., Sweet S., Knap A., Wade T., et al. (2015c). The contribution of peroxisome proliferator-activated receptor alpha to the relationship between toxicokinetics and toxicodynamics of trichloroethylene. Toxicol. Sci. 147, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. C., Waxman D. J. (1998). Activation of peroxisome proliferator-activated receptors by chlorinated hydrocarbons and endogenous steroids. Environ. Health Perspect. 106(Suppl. 4), 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.