

Abstract
Background. Pendrin, the chloride/bicarbonate exchanger of β-intercalated cells of the renal connecting tubule and the collecting duct, plays a key role in NaCl reabsorption by the distal nephron. Therefore, pendrin may be important for the control of extracellular fluid volume and blood pressure.
Methods. Here, we have used a genetic mouse model in which the expression of pendrin can be switched-on in vivo by the administration of doxycycline. Pendrin can also be rapidly removed when doxycycline administration is discontinued. Therefore, our genetic strategy allows us to test selectively the acute effects of loss of pendrin function.
Results. We show that acute loss of pendrin leads to a significant decrease of blood pressure. In addition, acute ablation of pendrin did not alter significantly the acid-base status or blood K+ concentration.
Conclusion. By using a transgenic mouse model, avoiding off-target effects related to pharmacological compounds, this study suggests that pendrin could be a novel target to treat hypertension.
Keywords: chloride, diuretics, hypertension, intercalated cells, pendrin
INTRODUCTION
Pendrin (Slc26a4) is an anion exchanger expressed in the kidney, thyroid and inner ear. Pendrin can mediate the uptake of chloride anion against either intracellular bicarbonate or iodide. Inactivating mutations of the SLC26A4 gene in humans lead to Pendred syndrome, an autosomal recessive disease characterized by sensorineural deafness associated with hypothyroidism and goiter [1–3]. In the kidney, pendrin participates in NaCl
uptake by epithelial cells of the distal nephron [4]. Pendrin is regulated in response to changes in chloride balance [5, 6], and is important for Cl− conservation [7, 8]. As a consequence, pendrin has been proposed to be one of the key elements that controls renal regulation of extracellular fluid volume and blood pressure. Accordingly, genetic ablation of pendrin has been shown to impair renal adaptation to a low-salt diet, leading to hypotension in this setting [8]. Conversely, mice with pendrin disruption are protected against mineralocorticoid-induced hypertension [9]. The mechanism by which ablation of pendrin lowers blood pressure is not completely clear. Indeed, pendrin is required for proper renal conservation of chloride, and hence of NaCl, the main determinant of vascular volume. However, pendrin has also been shown to control the activity of the sodium channel ENaC [10, 11], or the release of catecholamines by adrenal glands [12], two mechanisms by which pendrin inhibition can potentially affect blood pressure significantly. The observation, however, that transgenic mice overexpressing pendrin in intercalated cells are prone to develop salt-sensitive hypertension rather favors the possibility that the impact of pendrin on blood pressure is primarily through its role in renal chloride reabsorption.
All the aforementioned observations have led several groups to propose that pendrin might be a novel target to treat human hypertension [13–15]. However, the direct demonstration that pendrin inhibition is a good strategy to lower blood pressure, and hence, treat hypertension, has not been provided yet. Therefore, we used a conditional transgenic mouse model that allows the control of pendrin expression by a doxycycline-inducible system to test the effects of pendrin suppression on blood pressure in vivo.
MATERIALS AND METHODS
Animals
All animals were used in accordance with the French and European guidelines for the use of laboratory animals, in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All procedures and protocols were reviewed and approved by the ethic committee of Paris Descartes University and the French Ministry of Research (Protocol N°14-061). In this study, we used the binary transgenic mouse model developed initially by Choi et al. [16] in which pendrin expression can be induced with doxycycline. Briefly, the model requires the presence of two transgenes. The ‘effector’ transgene [Tg[E]; Tg(RP23-265L9/rtTA2S-M2/NeoR)1Ajg] allows the expression of the reverse tetracycline-controlled transactivator (rtTA) to be under the control of the mouse Slc26a4 promoter and cis-regulatory elements. The ‘responder transgene’ [Tg[R]; Tg(AcGFP/TRE/Slc26a4)2Ajg] carries the full-length mouse Slc26a4 cDNA under the control of the tetracycline operator TetO (Figure 1). Both transgenes are maintained on the Slc26a4Δ/Δ background initially developed by Everett et al. [17] so that the responder transgene is the only source of functional pendrin. This model has been characterized previously and proven to provide tight and inducible expression of pendrin in the inner ear [16, 18, 19]. To induce Slc26a4 expression, mice were administrated drinking water containing 0.2 g doxycycline hyclate (Sigma-Aldrich) with 5 g sucrose per 100 mL of deionized water [16].
FIGURE 1.
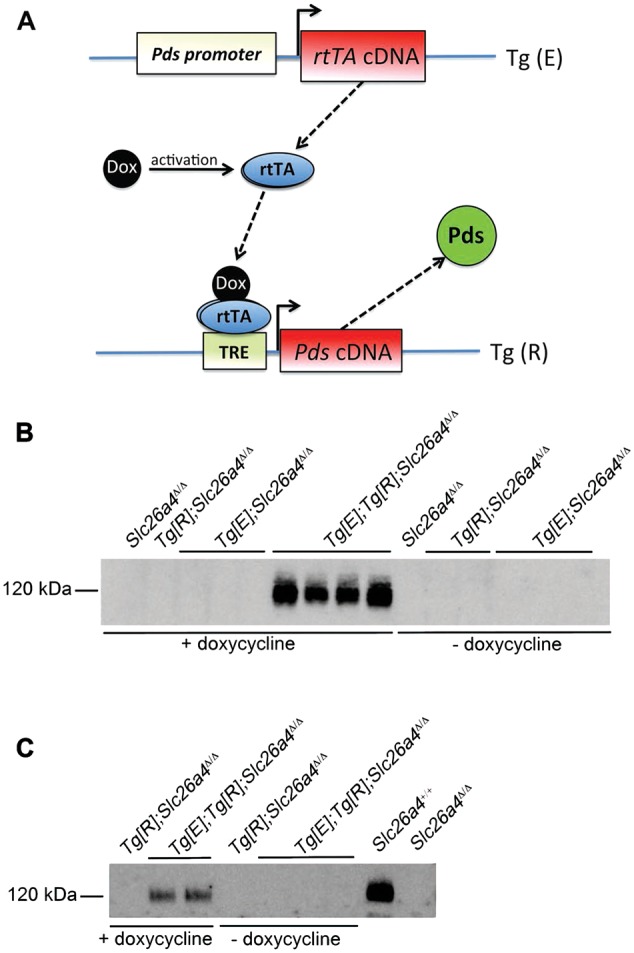
Schematic representation of the transgenes and its validation. (A) The transgenes are crossed onto a Slc26a4Δ/Δ background so the transgene Tg[R] is the only source of pendrin. When mice are administrated with doxycycline, rtTA expression is driven by the pendrin promoter and cis-regulatory elements in mice that normally express pendrin (i.e. the non-β-intercalated cells in the kidney), and then pendrin mRNA expression stops when doxycycline is withdrawn. Pds, pendrin; rtTA, reverse tetracyclin-dependent transactivator; Dox, doxycycline hyclate; TRE, tetracyclin responsive element. (B and C) Analyses of mouse pendrin expression by immunoblots of membrane fractions from Slc26a4+/+, Slc26a4Δ/Δ mice Tg[R];Slc26a4Δ/Δ, Tg[E];Slc26a4Δ/Δ and Tg[E];Tg[R];Slc26a4Δ/Δ. For the samples labeled ‘ + doxycycline’ tissue was extracted from the renal cortex doxycycline-administered mice for 2–3 weeks in the drinking water. For ‘−doxycycline’ tissue was extracted 5 days after doxycycline administration. Immunoblots were probed with the anti-mouse pendrin antibody.
Generation and genotyping of transgenic mice
Mice on a Slc26a4Δ/Δ background and harboring one allele (heterozygous) of the transgene Tg[E] were bred with mice also on a Slc26a4Δ/Δ background and harboring one copy (heterozygous) of the transgene Tg[R] to produce Slc26a4Δ/Δ TgR+/− mice to produce Tg[R];Tg[E];Slc26a4Δ/Δ. For all the different studies, only sex- and age-matched Tg[R];Tg[E];Slc26a4Δ/Δ animals of 8–12 weeks of age were used. Controls included littermates (hence, on a Slc26a4Δ/Δ background) in which either the Tg[E] (Tg[R];Slc26a4Δ/Δ) or the Tg[R] (Tg[E];Slc26a4Δ/Δ) was absent. Other controls included wild-type 129sv mice because the original Slc26a4Δ/Δ strain has been backcrossed for more than five generations on this background.
The genotype of the resulting offspring was determined by Polymerase chain reaction (PCR) using the three following primers as originally described by Everett et al. [17]:
mPds-ex7s: 5′-TGCCGATTTCATCGCTGG-3′
mPds-ex8as: 5′-GCATTGTAGTTCTTTTCCAAGTTGG-3′
mPds-KO: 5′-GGGTGCGGAGAAAGAGGTAATG-3′
The first two primers allowed the amplification of two bands of 287 and 168 bp, corresponding to the Tg[E] and Tg[R] transgenes, respectively. The first and third primers amplified a band of 243 bp, corresponding to the Slc26a4Δ allele. In addition, the presence of the Tg[E] was confirmed using primers Tg(E).2F.115 (5′-TCCAGCCTCTCTTAAAGTGC-3′) and Tg(E).2R.116 (5′-CCGTCGACATTTAGGTGACA-3′), which yielded a 445-bp fragment, as described by Choi et al. [16].
Immunofluorescence studies and immunoblot analyses
Procedures for immunofluorescence studies or immunoblot analyses have been previously detailed extensively elsewhere [20]. Briefly, kidney sections were labeled with a rabbit polyclonal anti-pendrin antibody [20] (diluted 1:200), a guinea pig anti-AE1 [21] (diluted 1:5000), a goat anti-AQP2 (diluted 1:500) and a chicken anti-Atp6v1e1, which detects the V H+-ATPase [22] (diluted 1:500), using standard techniques as previously described in detail [23]. For western blot analyses, 10–15 μg of the membrane-enriched protein fraction was separated on reducing 7.5% polyacrylamide-SDS gels. Protein loading was assessed on gels run in parallel and stained with Coomassie blue [5]. Blots were probed with a primary rabbit polyclonal anti-pendrin antibody [12, 24] a kind gift from Susan Wall (Emory University, Atlanta, GA, USA). Proteins were detected by chemiluminescence (ECL kit, Amersham Biosciences).
Biochemical and hormonal measurements
Blood gases analyses were performed by retro-orbitary punction on awake animals and pH, PCO2, PO2, [Na+], [K+], hematocrit and [Cl−] were measured with an ABL 77 pH/blood gas analyzer (Radiometer). Blood bicarbonate concentration was calculated by the auto-analyzer from the measured values using the Henderson–Hasselbach equation.
Measurements of blood pressure by radiotelemetry
Age-matched (2–3 months old) male mice were first treated for at least 15 days with doxycycline to induce strong renal pendrin expression. Since doxycycline-inducing pendrin expression is a genetic induced process, occurring progressively, blood pressure was not recorded during this phase. After anesthesia with pentobarbital (50 mg/kg) by intraperitoneal (IP) injection, telemetric devices were implanted according to the standard protocols 1 week before the doxycycline suspension. Briefly, catheters were inserted into the right femoral artery and attached to a radiotransmitter (Physio Tel HD-X11; Data Sciences International) located on the back. Mice were allowed to recover for 2–3 days at which time they were placed in cages (Hatteras Instruments) and permitted to acclimate to the cages for an additional 5 days. Blood pressure was continuously recorded using a telemetry receiver (Physio Tel DSI receiver; Data Sciences International).
RESULTS
Doxycycline induces specific expression of pendrin in renal β-intercalated cells of Tg[R];Tg[E];Slc26a4Δ/Δ mice
We first verified whether the strategy used by Choi et al. (Figure 1A and [16]) to control pendrin expression in the inner ear of pendrin KO mice with doxycycline also works in the kidney. Kidneys from Slc26a4+/+, Tg[E];Tg[R];Slc26a4Δ/Δ, Tg[R];Slc26a4Δ/Δ and Tg[E];Slc26a4Δ/Δ mice were harvested after 2–3 weeks of doxycycline administration. In some mice, doxycycline was administrated for 2–3 weeks and then withdrawn for 5 days to check whether pendrin expression driven by the Tet-on system is reversible. Kidney cortices were dissected manually and pendrin expression was assessed by western blot. Figure 1B and C shows that the anti-pendrin antibody interacted with a polypeptide of ∼120 kDa on a renal cortical membrane fraction isolated from Tg[E];Tg[R];Slc26a4Δ/Δ mice pre-treated for 2–3 weeks with doxycycline. The same signal was observed in Slc26a4+/+ mice. Importantly, this band was completely absent from the renal cortex isolated from Slc26a4Δ/Δ mice, confirming that our antibody is specific for pendrin protein. No pendrin immunoreactivity was detected in the renal cortex of Tg[E];Tg[R];Slc26a4Δ/Δ mice after 5 days of doxycycline withdrawal, confirming that our system allows a temporal control of pendrin expression. Importantly, no signal was detected in the kidney from ‘non-responder’ controls (Tg[R];Slc26a4Δ/Δ), which lack the ‘effector’ transgene Tg[E], and hence, cannot restore pendrin expression with doxycycline administration.
Pendrin expression was next assessed by immunofluorescence on kidney sections (Figure 2). After 2 weeks of doxycycline administration, pendrin expression in the kidney of Tg[E];Tg[R];Slc26a4Δ/Δ mice was identical to that observed in Slc26a4+/+ mice. Pendrin was observed in a subset of epithelial cells restricted to the kidney cortex exhibiting typical features of β-intercalated cells. Importantly, no pendrin immunolabeling was detected in Tg[E];Tg[R];Slc26a4Δ/Δ mice after the withdrawal of doxycycline for 5 days. Also, no pendrin staining was detected in kidney sections from the Tg[E];Slc26a4Δ/Δ and Tg[R];Slc26a4Δ/Δ controls, treated or not with doxycycline. This confirms that the transgene responder (Tg[R]) is the only source of pendrin in our model and that this transgene is not leaky, i.e. does not express pendrin when Tg[E] is not present.
FIGURE 2.

Immunolocalization of pendrin on kidney sections from Slc26a4+/+, Tg[R];Slc26a4Δ/Δ, Tg[E];Slc26a4Δ/Δ and Tg[E];Tg[R];Slc26a4Δ/Δ mice. Anti-pendrin antibody stained a subset of renal epithelial cells in the renal cortex of Slc26a4+/+ mice. The same pattern of expression of pendrin was observed in Tg[E];Tg[R];Slc26a4Δ/Δ mice treated for at least 15 days with doxycycline. After 5 days of doxycycline withdrawal, pendrin expression was completely undetectable. As expected, neither Tg[R];Slc26a4Δ/Δ nor Tg[E];Slc26a4Δ/Δ mice expressed pendrin protein, either in the presence or absence of doxycycline treatment.
To determine more precisely which renal cell-type expresses pendrin upon doxycycline administration in Tg[E];Tg[R];Slc26a4Δ/Δ mice, we performed colocalization studies. Principal cells were stained for the water channel Aqp2, and β-intercalated cells were identified by an antibody specific for the anion exchanger Ae1. No pendrin expression was detected in the kidneys of Tg[E];Slc26a4Δ/Δ or Tg[R];Slc26a4Δ/Δ mice, whether or not they were administered doxycycline (Figure 3). By contrast, in the kidney of Tg[E];Tg[R];Slc26a4Δ/Δ mice treated with doxycycline, pendrin-positive cells are found exclusively in cortical renal tubules that also harbor Aqp2- and Ae1-positive cells, indicating that they are connecting tubules or cortical collecting duct (i.e. the renal tubules that normally express pendrin). Furthermore, in this particular subset of renal tubules, pendrin-positive cells were always negative for both Ae1 and Aqp2, indicating that they are neither β-intercalated nor principal cells and, thus, are β-intercalated cells. Pendrin was not detected in the kidneys of Tg[E];Tg[R];Slc26a4Δ/Δ mice after doxycycline withdrawal.
FIGURE 3.

Cellular composition of collecting duct from Tg[E];Tg[R];Slc26a4Δ/Δ, Tg[E];Slc26a4Δ/Δ or Tg[R]; Slc26a4Δ/Δ mice. Colocalization studies with anti-pendrin (red), anti-AE1 (blue) and anti-AQP2 (green) antibodies. Doxycycline administration for 2 weeks does not induce pendrin staining in Tg[R];Slc26a4Δ/Δ or Tg[E];Slc26a4Δ/Δ mice, while the expression of pendrin at the apical pole of a subset of cells in cortical renal tubules that are also labeled by Ae1- and Aqp2-specific antibodies, and thus are identified as connecting tubules or cortical collecting ducts, occurs in Tg[E];Tg[R];Slc26a4Δ/Δ mice. Pendrin staining is restricted to a subset of cells that are identified as β-intercalated cells because they are negative for Ae1 or for Aqp2 staining, and it disappears in mice in which doxycycline has been withdrawn for 5 days.
Effects of pendrin expression on plasma electrolytes
In the kidney, pendrin is a chloride/bicarbonate exchanger, which controls the excretion of base into urine in response to metabolic alkalosis or to a dietary alkali load. Accordingly, disruption of the Slc26a4 gene has been shown to impair the capability of the kidney to cope with an alkali load and can lead to the development of metabolic alkalosis [25]. Therefore, we tested the effects of the presence or absence of pendrin on blood pH, PCO2 and [HCO3−], as well as on blood [Na+], [K+] and [Cl−]. Figure 4 shows that Tg[E];Tg[R];Slc26a4Δ/Δ mice exhibited a slightly higher bicarbonate concentration accompanied by a converse mild increase in [Na+] and [Cl−] concentrations when pendrin expression is switched off by doxycycline withdrawal. These changes are consistent with the premise that pendrin preferentially mediates renal bicarbonate excretion [25] and renal reclamation of NaCl [20]. However, it should be noted that the changes following the deletion of pendrin expression were of very limited magnitude since changes in either [Na+] or [Cl−] remained less than ∼4 mmol/L.
FIGURE 4.

Blood gas analysis and electrolytes concentration in Slc26a4+/+, Slc26a4Δ/Δ and Tg[E];Tg[R];Slc26a4Δ/Δ mice during doxycycline administration or after doxycycline withdrawal. Mice that do not expressed pendrin have significantly higher blood [] and lower blood [Na+] and [Cl−]. However, the changes were of limited amplitude (<4 mmol/L). Statistical significance was tested by ANOVA followed by Sidak’s post hoc test for multiple comparisons (n=6, 9 and 9, for Slc26a4+/+, Slc26a4Δ/Δ and Tg[E];Tg[R];Slc26a4Δ/Δ mice, respectively).
Effects of the presence of pendrin on blood pressure in basal conditions
We tested whether the chronic presence or absence of pendrin alters blood pressure in mice. Mice were fed a normal-concentration sodium (0.3% Na+) diet. Systolic and diastolic blood pressure and heart rate were monitored continuously by radiotelemetry.
Figure 5 shows that Tg[E];Tg[R];Slc26a4Δ/Δ mice fed a normal salt diet and given doxycycline for at least 2–3 weeks exhibited no significant differences in systolic and diastolic blood pressure or heart rate compared with their controls (Slc26a4+/+, Tg[E];Slc26a4Δ/Δ or Tg[R];Slc26a4Δ/Δ). This is consistent with previous studies performed with mice with constitutive genetic deletion of pendrin that observed normal blood pressure under basal conditions [8, 9].
FIGURE 5.

(A and B) Blood pressure and (C) heart rate in basal conditions in Slc26a4+/+, Tg[E];Tg[R];Slc26a4Δ/Δ Tg[R];Slc26a4Δ/Δ, Tg[E];Slc26a4Δ/Δ and Slc26a4Δ/Δ mice. Systolic and diastolic blood pressures were continuously recorded by radiotelemetry. During all the experiments, mice were fed a normal salt (0.3% Na+) diet and were administrated doxycycline during at least 2–3 weeks. Data are presented as the mean ± standard error of the values obtained during either the active (night) or inactive (day) period; n = 4–6 per group. The differences in heart rate, systolic and diastolic blood pressures were tested by one-way ANOVA.
Acute depletion of pendrin decreases blood pressure
In this study, our primary goal is to determine whether acute depletion of pendrin is an effective strategy to lower blood pressure. However, no specific inhibitors of pendrin, or simply, of renal chloride transport that can be administered in vivo, have been identified. Therefore, our aim was to use the model developed by Choi et al. [16] to test the effect of the acute depletion of pendrin as an alternative strategy to pharmacological inhibition. Tg[E];Tg[R];Slc26a4Δ/Δ mice were treated for 2–3 weeks with doxycycline to induce robust expression of pendrin in the kidney. The duration of doxycycline administration was designed to allow the diminution of compensatory mechanisms that limit the effects of renal pendrin depletion and that are likely to account for the normal blood pressure observed in pendrin-deficient mice in the preceding experiments. Figure 6 shows that upon withdrawal of doxycycline, systolic and diastolic blood pressure dropped significantly in Tg[E]; Tg[R];Slc26a4Δ/Δ mice, while it had no effects on blood pressure in either Slc26a4+/+ or Tg[E];Slc26a4Δ/Δ mice (that are on a pendrin-knockout background). Taken together, the results shown in Figures 5 and 6 demonstrate that whereas congenital pendrin deletion has limited effects on blood pressure, likely due to the recruitment of compensatory mechanisms, acute depletion of pendrin is an effective intervention to modulate blood pressure.
FIGURE 6.

Effects of doxycycline withdrawal on the time course of (A and C) systolic (B and D) and diastolic blood pressures of Slc26a4+/+, Tg[E];Tg[R];Slc26a4Δ/Δ, Tg[E];Slc26a4Δ/Δ mice. Systolic and diastolic blood pressures were continuously recorded by radiotelemetry. During all the experiments, mice were fed a normal salt (0.3% Na+) diet. Mice were administrated doxycycline for at least 2–3 weeks and then the time course of blood pressure was monitored after doxycycline withdrawal. Data are presented as the mean ± standard error of the values obtained during either the active (night) or inactive (day) period; n = 4–6 per group. The differences were tested using a two-way ANOVA for repeated measures followed by Student–Newman–Keuls post hoc test. Both systolic and diastolic blood pressures significantly (P < 0.05) dropped from Day 1 in Tg[E];Tg[R];Slc26a4Δ/Δ mice after doxycycline withdrawal versus the control period (Day 0), while they were not affected in Slc26a4+/+ or Tg[E];Slc26a4Δ/Δ control mice. * indicates statistical significance (P < 0.05 or lower) versus Slc26a4+/+ mice; # indicates statistical significance (P < 0.05 or lower) versus Tg[E];Slc26a4Δ/Δ mice.
DISCUSSION
Hypertension is one of the most common diseases affecting the human population. It is a major risk factor for many cardiovascular diseases including stroke, ischemic cardiopathy, chronic renal failure and ischemic arteriopathy. For example, 50% of myocardial infarctions and 62% of strokes are caused by high blood pressure. As a consequence, preventing the raise in blood pressure, or lowering high blood pressure, is expected to have enormous impact on health costs, and hence, is considered as health priority worldwide.
However, despite the availability of different and complementary classes of antihypertensive therapies, only half of hypertensive patients exhibit an appropriate control of their blood pressure. Most patients (>75%) require a combination of at least two antihypertensive drugs. Others require up to five different compounds. Resistant hypertension, which is defined by a blood pressure that remains >140/90 mmHg in patients receiving three independent classes of antihypertensive drugs, affects up to 30% of the patients. Moreover, the actual effectiveness of some antihypertensive treatments to decrease blood pressure, and more importantly, to increase the lifespan of the patients is still a matter of discussion. Thus, the development of new drugs targeting blood pressure is clearly required to establish effective therapies for hypertension.
Depending on the molecule, different diuretics lead directly or indirectly to a reduced NaCl absorption along the nephron. Acetazolamide inhibits carbonic anhydrase, which leads to a reduced sodium absorption via the apical Na+/H+ exchanger of the proximal tubule. Loop diuretics inhibit the apical Na+/K+/2Cl− cotransporter NKCC2 of the thick ascending limb (TAL). Thiazides are blocker of the NaCl cotransporter NCC of the distal convoluted tubule. Finally, K-sparing diuretics are blockers of the sodium channel ENaC, which is present in principal cells of the connecting tubule and of the collecting duct. The beneficial effects of one particular diuretic is generally limited, however, either by a decrease in the glomerular filtration rate (particularly, if sodium absorption by the proximal tubule is inhibited) or more generally by an adaptative increase of the activity of the various sodium transporters that are not targeted by the molecule. Indeed, many physiological studies in rodents have confirmed the crucial role of kidney in salt-sensitive hypertension when transport activity is increased [26] and its ability to compensate when it is decreased. For example, deletion of NCC is compensated by a marked upregulation of pendrin, Na-driven chloride bicarbonate exchanger (NDCBE) and ENaC [4, 6, 27, 28]. In the same way, disruption of ROMK1 [29, 30], which affects NaCl absorption by TAL cells, is compensated by upregulation of NCC, and possibly ENaC. This has important consequences in clinical practice as the same phenomenon can occur and lead to ‘diuretic resistance’ in hypertensive patients. Indeed, a recent study in humans strongly suggests that the simultaneous blockade of several renal NaCl transport systems is an effective strategy to optimize volume depletion and hence the control of volume-dependent hypertension [31]. Several studies have recently shown that constitutive disruption or overexpression of the chloride transporter pendrin can significantly affect blood pressure [32]. Indeed, mice with pendrin disruption are resistant to mineralocorticoid-induced hypertension [9]. They were also unable to adapt to a low-NaCl diet or selective Cl− restriction, and developed hypotension in these settings [7, 8]. Conversely, overexpression of pendrin in renal-intercalated cells leads to chloride-sensitive hypertension [20]. It was also reported that pendrin disruption markedly potentiated the effects of NCC disruption and that mice with dual knockout of both NCC and pendrin had very severe volume depletion [27]. No measurements of blood pressure were reported in this model [27].
A recent study using a high throughput screen of small molecules describes the identification of two novel pendrin inhibitors [33]. One of these compounds increases the natriuretic effects of furosemide, a loop diuretic, indicating that it might be useful to limit resistance to diuretics. However, this pendrin blocker had no effects when administrated alone, suggesting that either pendrin is active only in volume-depleted states or that the dose used in this study was not optimal. The effects of this drug on blood pressure were not investigated [33].
To overcome potential drug-related pharmacokinetic and pharmacodynamics issues, we choose to use a genetic approach to study the acute effects of selective disabling of pendrin function [16]. Here, we show that the acute genetic disruption of pendrin is able to significantly lower blood pressure while the effects on acid-base status or on blood potassium concentration are negligible. The observation that pendrin blockade can affect blood pressure while the number of pendrin-expressing cells along the nephron is limited might appear to be quite surprising. However, it is worth noting that the blockade of this chloride transporter might actually affects simultaneously different sodium transport pathways; as previously shown, sodium and chloride transporters are strictly involved in the generation of salt-sensitive hypertension [34]. Indeed, we demonstrated that pendrin-dependent chloride absorption is coupled with NDCBE in the cortical collecting duct to mediate electroneutral thiazide-sensitive NaCl uptake [4, 35]. However, data from the groups of Mark Knepper and Susan Wall demonstrated that pendrin can also work in tandem with ENaC [36, 37]. The relative importance of either one pathway upon the other seems to depend upon the model used, and thus, is probably regulated by different factors like hormones such as the renin angiotensin system, bradykinin, tissue kallikrein or vasopressin [4, 9, 37–40]. Nevertheless, the fact that inhibition or disruption of pendrin can affect different sodium transporters might explain why the inactivation of the transporter has such an important impact on blood pressure.
In summary, our study adds evidence on the important role of pendrin in controlling blood pressure and candidates this strategy as potentially effective as an additional antihypertensive treatment in humans.
ACKNOWLEDGEMENTS
D.E. and J.T. are funded by grant ANR BLANC 14-CE12-0013-01/HYPERSCREEN from l’Agence Nationale de la Recherche (ANR); D.E. and J.T. are also funded by grant ECOS/CONICYT France-Chile 2014; D.E and B.V. are funded by grant CHLORBLOCK from the IDEX Sorbonne Paris Cité; R.C. is funded by grant ANR BLANC 2012-R13011KK; F.T. is funded by European Renal Association—European Dialysis and Transplantation Association (LTF141-2013); Y.K. is funded by grant H2020-MSCA-IF-2014-CLOPRESS. B.Y.C. and A.J.G. were supported by NIH intramural research fund Z01-DC-000060. P.W. is funded by grant NIH-R01-DC012151.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Scott DA, Wang R, Kreman TM. et al. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 1999; 21: 440–443 [DOI] [PubMed] [Google Scholar]
- 2. Reardon W, Trembath RC.. Pendred syndrome. J Med Genet 1996; 33: 1037–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pendred V. Deaf-mutism and goitre. Lancet 1896; 148: 532 [Google Scholar]
- 4. Leviel F, Hubner CA, Houillier P. et al . The Na+-dependent chloride-bicarbonate exchanger slc4a8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010; 120: 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quentin F, Chambrey R, Trinh-Trang-Tan MM. et al. The Cl-/ exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol 2004; 287: F1179–F1188 [DOI] [PubMed] [Google Scholar]
- 6. Vallet M, Picard N, Loffing-Cueni D. et al. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 2006; 17: 2153–2163 [DOI] [PubMed] [Google Scholar]
- 7. Verlander JW, Kim YH, Shin W. et al. Dietary Cl- restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol 2006; 291: F833–F839 [DOI] [PubMed] [Google Scholar]
- 8. Wall SM, Kim YH, Stanley L. et al. NaCl restriction upregulates renal slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension 2004; 44: 982–987 [DOI] [PubMed] [Google Scholar]
- 9. Verlander JW, Hassell KA, Royaux IE. et al. Deoxycorticosterone upregulates pds (slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003; 42: 356–362 [DOI] [PubMed] [Google Scholar]
- 10. Pech V, Wall SM, Nanami M. et al. Pendrin gene ablation alters ENaC subcellular distribution and open probability. Am J Physiol Renal Physiol 2015; 309: F154–F163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim YH, Pech V, Spencer KB. et al. Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am J Physiol Renal Physiol 2007; 293: F1314–F1324 [DOI] [PubMed] [Google Scholar]
- 12. Lazo-Fernandez Y, Aguilera G, Pham TD. et al. Pendrin localizes to the adrenal medulla and modulates catecholamine release. Am J Physiol Endocrinol Metab 2015; 309: E534–E545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amlal H, Soleimani M.. Pendrin as a novel target for diuretic therapy. Cell Physiol Biochem 2011; 28: 521–526 [DOI] [PubMed] [Google Scholar]
- 14. Eladari D, Chambrey R, Frische S. et al. Pendrin as a regulator of ecf and blood pressure. Curr Opin Nephrol Hypertens 2009; 18: 356–362 [DOI] [PubMed] [Google Scholar]
- 15. Wall SM, Pech V.. Pendrin and sodium channels: Relevance to hypertension. J Nephrol 2010; 23 (Suppl 16): S118–S123 [PubMed] [Google Scholar]
- 16. Choi BY, Kim HM, Ito T. et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of slc26a4 expression for hearing acquisition. J Clin Invest 2011; 121: 4516–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Everett LA, Belyantseva IA, Noben-Trauth K. et al . Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 2001; 10: 153–161 [DOI] [PubMed] [Google Scholar]
- 18. Ito T, Li X, Kurima K. et al . Slc26a4-insufficiency causes fluctuating hearing loss and stria vascularis dysfunction. Neurobiol Dis 2014; 66: 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X, Sanneman JD, Harbidge DG. et al . Slc26a4 targeted to the endolymphatic sac rescues hearing and balance in slc26a4 mutant mice. PLoS Genet 2013; 9: e1003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacques T, Picard N, Miller RL. et al . Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 2013; 24: 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stehberger PA, Shmukler BE, Stuart-Tilley AK. et al. Distal renal tubular acidosis in mice lacking the ae1 (band3) cl-/hco3- exchanger (slc4a1). J Am Soc Nephrol 2007; 18: 1408–1418 [DOI] [PubMed] [Google Scholar]
- 22. Breton S, Wiederhold T, Marshansky V. et al . The B1 subunit of the H+ATPase is a PDZ domain-binding protein. Colocalization with NHE-RF in renal B-intercalated cells. J Biol Chem 2000; 275: 18219–18224 [DOI] [PubMed] [Google Scholar]
- 23. Eladari D, Cheval L, Quentin F. et al . Expression of RhCG, a new putative NH(3)/NH(4)(+) transporter, along the rat nephron. J Am Soc Nephrol 2002; 13: 1999–2008 [DOI] [PubMed] [Google Scholar]
- 24. Kim YH, Pham TD, Zheng W. et al. Role of pendrin in iodide balance: going with the flow. Am J Physiol Renal Physiol 2009; 297: F1069–F1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Royaux IE, Wall SM, Karniski LP. et al. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 2001; 98: 4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trepiccione F, Zacchia M, Capasso G.. The role of the kidney in salt-sensitive hypertension. Clin Exp Nephrol 2012; 16: 68–72 [DOI] [PubMed] [Google Scholar]
- 27. Soleimani M, Barone S, Xu J. et al . Double knockout of pendrin and Na−Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 2012; 109: 13368–13373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinning A, Radionov N, Trepiccione F. et al. Double knockout of the Na+-driven Cl−/HCO3 exchanger and NaCl cotransporter induces hypokalemia and volume depletion. J Am Soc Nephrol 5 May 2016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cantone A, Yang X, Yan Q. et al . Mouse model of type ii Bartter's syndrome. I. Upregulation of thiazide-sensitive Na−Cl cotransport activity. Am J Physiol Renal Physiol 2008; 294: F1366–F1372 [DOI] [PubMed] [Google Scholar]
- 30. Wagner CA, Loffing-Cueni D, Yan Q. et al. Mouse model of type II Bartter's syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol 2008; 294: F1373–F1380 [DOI] [PubMed] [Google Scholar]
- 31. Bobrie G, Frank M, Azizi M. et al . Sequential nephron blockade versus sequential renin-angiotensin system blockade in resistant hypertension: a prospective, randomized, open blinded endpoint study. J Hypertens 2012; 30: 1656–1664 [DOI] [PubMed] [Google Scholar]
- 32. Chambrey R, Trepiccione F.. Relative roles of principal and intercalated cells in the regulation of sodium balance and blood pressure. Curr Hypertens Rep 2015. ; 17: 538. [DOI] [PubMed] [Google Scholar]
- 33. Cil O, Haggie PM, Phuan PW. et al. Small-molecule inhibitors of pendrin potentiate the diuretic action of furosemide. J Am Soc Nephrol 6 May 2016, [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Capasso G, Rizzo M, Garavaglia ML. et al . Upregulation of apical sodium-chloride cotransporter and basolateral chloride channels is responsible for the maintenance of salt-sensitive hypertension. Am J Physiol Renal Physiol 2008; 295: F556–F567 [DOI] [PubMed] [Google Scholar]
- 35. Chambrey R, Kurth I, Peti-Peterdi J. et al. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 2013; 110: 7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pech V, Thumova M, Kim YH. et al. ENaC inhibition stimulates Cl− secretion in the mouse cortical collecting duct through an nkcc1-dependent mechanism. Am J Physiol Renal Physiol 2012; 303: F45–F55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Terada Y, Knepper MA.. Thiazide-sensitive nacl absorption in rat cortical collecting duct. Am J Physiol 1990; 259: F519–F528 [DOI] [PubMed] [Google Scholar]
- 38. Tomita K, Pisano JJ, Burg MB, et al.Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct. Evidence for an electroneutral sodium chloride transport pathway. J Clin Invest 1986; 77: 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tomita K, Pisano JJ, Knepper MA.. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest 1985; 76: 132–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. El Moghrabi S, Houillier P, Picard N. et al . Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci USA 2010; 107: 13526–13531 [DOI] [PMC free article] [PubMed] [Google Scholar]