

Abstract
Parasitic “wing lice” (Phthiraptera: Columbicola) and their dove and pigeon hosts are a well-recognized model system for coevolutionary studies at the intersection of micro- and macroevolution. Selection on lice in microevolutionary time occurs as pigeons and doves defend themselves against lice by preening. In turn, behavioral and morphological adaptations of the lice improve their ability to evade host defense. Over macroevolutionary time wing lice tend to cospeciate with their hosts; yet, some species of Columbicola have switched to new host species. Understanding the ecological and evolutionary factors that influence coadaptation and codiversification in this system will substantially improve our understanding of coevolution in general. However, further work is hampered by the lack of a robust phylogenetic framework for Columbicola spp. and their hosts. Previous attempts to resolve the phylogeny of Columbicola based on sequences from a few genes provided limited support. Here, we apply a new approach, target restricted assembly, to assemble 977 orthologous gene sequences from whole-genome sequence data generated from very small, ethanol-preserved specimens, representing up to 61 species of wing lice. Both concatenation and coalescent methods were used to estimate the species tree. These two approaches yielded consistent and well-supported trees with 90% of all relationships receiving 100% support, which is a substantial improvement over previous studies. We used this new phylogeny to show that biogeographic ranges are generally conserved within clades of Columbicola wing lice. Limited inconsistencies are probably attributable to intercontinental dispersal of hosts, and host switching by some of the lice. [aTRAM; coalescent; coevolution; concatenation; species tree.]
Parasites comprise about half of the diversity of life on earth (Windsor 1998; Poulin and Morand 2000, Poulin and Morand 2004; Dobson et al. 2008; Mora et al. 2011). Documenting this diversity, and understanding the evolutionary processes responsible for it, requires accurate information on the phylogenetic history of parasites. A robust phylogenetic tree can be used to reconstruct events triggering parasite diversification. Examples include cospeciation of parasites with hosts, or the switching of parasites between hosts, followed by parasite speciation. One host–parasite system in which studies of coevolutionary processes have been linked to macroevolutionary patterns consists of pigeons and doves (Aves: Columbidae; hereafter “doves”) and their ectoparasitic lice (Clayton and Johnson 2003; Johnson et al. 2009; reviewed by Clayton et al. 2016).
Doves are parasitized by 90 known species of wing lice belonging to the genus Columbicola (Phthiraptera: Ischnocera; Bush et al. 2009; Gustafsson and Bush 2015). Columbicola species are “permanent” parasites that complete all stages of their life cycle on the body of the host, where they feed primarily on the downy feathers (Marshall 1981). Damage to feathers by Columbicola causes energetic stress, which reduces host survival and mating success (Clayton et al. 2016). In response to such damage, doves have coevolved defenses against Columbicola, such as preening behavior that is very effective at controlling populations of lice (Clayton et al. 2005). Host preening reciprocally selects for the long, thin body shape of Columbicola. This shape enables lice to escape preening by hiding between the barbs of the large flight feathers of the wings and tail (Clayton 1991; Clayton et al. 2003). Comparative and experimental studies show that lice on “wrong” sized feathers are more vulnerable to preening; hence, preening reinforces the host specificity of Columbicola (Clayton et al. 1999; Johnson et al. 2005; Bush and Clayton 2006; Bush et al. 2006; Malenke et al. 2009). Some Columbicola spp. are also known to match the color of their host’s feathers, making them more difficult for the bird to see the lice when preening (Bush et al. 2010). Thus, host preening has had a selective effect on the size, shape, and color of Columbicola species.
The selective effect of host preening on Columbicola reinforces host specificity, which, in turn, reinforces congruence between host and parasite phylogenies over macroevolutionary time. However, a previous study (Johnson et al. 2003) found that host and parasite phylogenies in this system are not always congruent, suggesting that some lineages of Columbicola have switched hosts. Some species of Columbicola also parasitize more than one host species (Johnson et al. 2002; Malenke et al. 2009; Bush et al. 2009). Host switching, along with host-imposed selection, may facilitate reproductive isolation and specialization by Columbicola [see Althoff et al. (2014) for a review of factors affecting coevolutionary diversification]. Together, these findings make the dove-louse system ideal for investigating the link between microevolutionary processes and macroevolutionary patterns of diversification (Clayton and Johnson 2003; Johnson et al. 2009; Clayton et al. 2016).
A crucial step in understanding the coevolutionary diversification of Columbicola is to have a well-supported phylogeny for the lice. Johnson et al. (2007) attempted to reconstruct the phylogenetic history of Columbicola using three genes (two mitochondrial and a single nuclear gene). While this work supported many of the morphologically recognized species groups within Columbicola, relationships among the species groups were largely unresolved (Johnson et al. 2007). The reasons for the lack of resolution are unclear. However, it is possible that lice radiated rapidly, leaving few phylogenetically informative DNA substitutions (synapomorphies). Alternatively, rapid radiation may have led to the incomplete sorting of alleles, which resulted in conflicting phylogenetic signals.
Recent phylogenomic approaches using hundreds or thousands of genes have shown great potential to resolve difficult phylogenetic problems (Jarvis et al. 2014; Misof et al. 2014; Prum et al. 2015). However, Columbicola and other lice pose substantial technical challenges for collecting large-scale genomic data. For example, some high-throughput sequencing methods require large quantities of high-quality genomic DNA (e.g., target capture methods; Faircloth et al. 2012, Faircloth et al. 2015; Lemmon et al. 2012; McCormack et al. 2012) or RNA (transcriptome sequencing; e.g., Misof et al. 2014). Lice are very small (1–2 mm long), dorsoventrally compressed, and encased in a hardened exoskeleton. In most cases, only a relatively small amount of DNA (< 50 nanograms, ng) can be extracted from a single louse for sequencing. Combining samples is not a viable option because an individual dove can harbor more than one (potentially cryptic) species of louse (Malenke et al. 2009). In addition, fresh specimens of most species of lice are difficult to obtain. DNA from museum samples of lice (e.g., ethanol-preserved) is often highly degraded and RNA is often completely degraded. Consequently, the long fragments of DNA required for de novo genome sequencing and assembly are typically unavailable. However, the quantity and quality of DNA available from archival specimens is often sufficient for constructing a single short-insert next-generation library using Illumina technology. Although sequences from such libraries are insufficient for complete genome assembly, they contain sufficient coverage of the genome to mine informative data for phylogenetic reconstruction.
The recently developed software, automated Target Restricted Assembly Method (aTRAM), provides a new tool for mining genomic libraries for phylogenomic data (Johnson et al. 2013; Allen et al. 2015). This software focuses on localized assemblies of targeted genomic regions that contain highly conserved protein-coding genes. BLAST (Altschul et al. 1990) searches are used to identify reads and assemble contigs potentially belonging to single-copy, one-to-one orthologous genes. These reads are assembled to produce small contigs containing the gene. The gene sequences are then available for phylogenetic study. This approach is optimal for phylogenetic reconstruction because it reduces assembly complexity and takes advantage of highly informative gene sequences that are conserved across taxa (Allen et al. 2015). Therefore, useful phylogenetic sequence data can be recovered even when the DNA sample is degraded and genome-sequencing coverage is uneven.
In the current study, we used aTRAM to assemble 977 orthologous genes from up to 61 different species of Columbicola wing lice. We used several approaches to determine the species relationships within the genus and used the resulting phylogeny to test for conservation of biogeographic regions within the genus. Our results provide robust insights into how Columbicola diversified across the global landscape.
Materials and Methods
Library Preparation and Genome Sequencing
We extracted total genomic DNA from 61 specimens of Columbicola preserved in ethanol, yielding sufficient gDNA for whole-genome sequencing (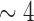 –50 ng). These 61 specimens represented 45 named species, 7 undescribed species, and 9 additional specimens that represent potentially cryptic species within named species (designated with a number after the species name; Malenke et al. 2009; Supplementary Table S1, available on Dryad at http://dx.doi.org/10.5061/dryad.4812p). This sampling represents about 50% of named Columbicola species, as well as additional suspected species. Total genomic DNA was extracted from each louse using the Qiagen DNA micro-extraction kit (see Supplementary Materials for complete extraction methods, available on Dryad) and was provided to the WM Keck Center, University of Illinois Urbana-Champaign in 37
–50 ng). These 61 specimens represented 45 named species, 7 undescribed species, and 9 additional specimens that represent potentially cryptic species within named species (designated with a number after the species name; Malenke et al. 2009; Supplementary Table S1, available on Dryad at http://dx.doi.org/10.5061/dryad.4812p). This sampling represents about 50% of named Columbicola species, as well as additional suspected species. Total genomic DNA was extracted from each louse using the Qiagen DNA micro-extraction kit (see Supplementary Materials for complete extraction methods, available on Dryad) and was provided to the WM Keck Center, University of Illinois Urbana-Champaign in 37 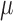 L of buffer EB for library preparation and DNA sequencing (submission occurred between April and August 2015). The DNA was sheared on a Covaris M220 instrument to target a mean fragment size of 400 bp or 650 bp (actual fragment sizes ranged from 80 to 1200 bp depending on library). From the sheared DNA, sequencing libraries were prepared using the Kapa Library Preparation kit (Kapa Biosystems, Wilmington, MA). Each DNA extract received a 6-bp barcode during library preparation so that four samples could be pooled and sequenced on a single sequencing lane (see Supplementary Table S1, available on Dryad, for barcode and pooling information). The libraries were pooled in equimolar concentrations (as verified by qPCR) and paired-end sequenced for 2 x 160 cycles on an Illumina HiSeq2500 instrument using the TruSeq SBS Rapid sequencing kit v.2 or the HiSeq SBS Sequence Kit v.4. Fastq files were generated using Casava v.1.8.2–1.8.4 using Illumina 1.9 quality score encoding.
L of buffer EB for library preparation and DNA sequencing (submission occurred between April and August 2015). The DNA was sheared on a Covaris M220 instrument to target a mean fragment size of 400 bp or 650 bp (actual fragment sizes ranged from 80 to 1200 bp depending on library). From the sheared DNA, sequencing libraries were prepared using the Kapa Library Preparation kit (Kapa Biosystems, Wilmington, MA). Each DNA extract received a 6-bp barcode during library preparation so that four samples could be pooled and sequenced on a single sequencing lane (see Supplementary Table S1, available on Dryad, for barcode and pooling information). The libraries were pooled in equimolar concentrations (as verified by qPCR) and paired-end sequenced for 2 x 160 cycles on an Illumina HiSeq2500 instrument using the TruSeq SBS Rapid sequencing kit v.2 or the HiSeq SBS Sequence Kit v.4. Fastq files were generated using Casava v.1.8.2–1.8.4 using Illumina 1.9 quality score encoding.
Sequence Data Quality Control
Illumina sequence data were first analyzed using FastQC v.0.10.1 (Babrahams Bioinformatics) to check for irregularities or sequencing errors. To further control for sequencing error, we removed duplicated-read pairs using the fastqSplitDups.py script available from the Mcscript Github package (https://github.com/McIntyre-Lab/mcscript and dependencies https://github.com/McIntyre-Lab/mclib; obtained 31 March 2015). The “distinct” read files were retained from the fastqSplitDups results. This file contained all unique read pairs as well as a single copy of each duplicated read pair. Next, 5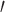 and 3
and 3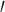 Illumina sequencing adapters were removed using Fastx_clipper v.0.0.14 (FASTX-Toolkit; http://hannonlab.cshl.edu/fastx_toolkit/). Following clipping, reads were “hard” clipped to remove the first 5 nt from the 5
Illumina sequencing adapters were removed using Fastx_clipper v.0.0.14 (FASTX-Toolkit; http://hannonlab.cshl.edu/fastx_toolkit/). Following clipping, reads were “hard” clipped to remove the first 5 nt from the 5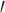 -end using Fastx_trimmer v.0.0.14 (FASTX-Toolkit). Reads were then “soft” trimmed from the 3
-end using Fastx_trimmer v.0.0.14 (FASTX-Toolkit). Reads were then “soft” trimmed from the 3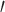 -end to remove bases with a phred score less than 28 using Fastq_quality_trimmer v.0.0.14 (trimming window
-end to remove bases with a phred score less than 28 using Fastq_quality_trimmer v.0.0.14 (trimming window  1 nt; FASTX-Toolkit). Those reads less than 75 nt after quality trimming were removed from the fastq files. The resulting fastq files were then re-analyzed using FastQC to check for additional sources of error not removed by our quality control process.
1 nt; FASTX-Toolkit). Those reads less than 75 nt after quality trimming were removed from the fastq files. The resulting fastq files were then re-analyzed using FastQC to check for additional sources of error not removed by our quality control process.
Assembly of Orthologous Sequences
We used aTRAM v.1.0.4 (Allen et al. 2015) to assemble protein coding gene sequences from the quality controlled Illumina sequence data. First, each library of Illumina data was prepared using the format_sra.pl script available in the aTRAM package. Next, we obtained a reference set of 1107 translated protein coding gene sequences from Pediculus humanus humanus strain USDA (PhumU2 assembly; Kirkness et al. 2010). These genes were identified as single copy orthologs in Culex pipiens, Aedes aegypti, Anopheles gambiae, Drosophila melanogaster, Tribolium castaneum, Nasonia vitripennis, Apis mellifera, P. h. humanus, and Acyrthosiphon pisum (Johnson et al. 2013). These reference sequences served as the initial starting sequence for aTRAM to assemble potentially orthologous genes including intron and untranslated regions from our Illumina sequence data. aTRAM was run for three iterations, using the ABySS (Simpson et al. 2009) option for de novo assembly, and using the “protein” option to define our starting reference as a translated amino acid sequence (aTRAM options are as follows: -max_processes 16, -iterations 3, -assembler ABySS, -protein). The resulting contigs from aTRAM (found in the “best” file output) were processed using a post-aTRAM exon stitching processing pipeline (https://github.com/juliema/phylogenomic_pipeline). This process uses Exonerate (v.2.2.0; Slater and Birney 2005) and custom perl scripts to identify and stitch together the exons of each gene. Finally, we used reciprocal best BLAST hits (using blastx v.2.2.28) between the assembled genes and all known P. h. humanus translated proteins to verify that the genes assembled were orthologs of our original reference sequences. The aTRAM runs were either completed on local 4 AMD Opteron with 16 2.4-GHz processors providing 64 CPU servers or the Texas Advanced Computing Center Stampede system (https://portal.xsede.org/tacc-stampede; Towns et al. 2014). Analyses were conducted on Stampede using Launcher for serial implementation of aTRAM on different nodes (Wilson and Fonner 2014).
Sequence Alignment and Data Matrix Construction
Gene sequences from aTRAM that passed the reciprocal-best BLAST filter were then grouped into files of orthologous genes (1107 different genes). In some instances, we recovered a gene sequence from only a subset of the taxa sampled. To avoid large blocks of missing data, we excluded genes for which we obtained an assembly for 50% or less of the lice, leaving 1066 genes for analysis. We further filtered the data by removing any genes that contained 50% or more ambiguous characters, leaving 977 genes. A %GC bias in third codon positions could violate the model assumptions of the GTR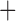 gamma substitution model for sequence evolution. To examine whether there was evidence for base composition biases, we examined the %GC content by codon position in each gene for each louse sampled. There was limited variation in GC content at any position in our data (see Supplementary Fig. S1, available on Dryad; the standard deviation of mean %GC content across lice was 0.029), and thus all codon positions were retained for phylogenetic analysis. Each gene was aligned separately using UPP v.2.0 (Nguyen et al. 2015) according to its translated amino acid sequence. The aligned data were then back-translated into its DNA sequence. Finally, the GTR
gamma substitution model for sequence evolution. To examine whether there was evidence for base composition biases, we examined the %GC content by codon position in each gene for each louse sampled. There was limited variation in GC content at any position in our data (see Supplementary Fig. S1, available on Dryad; the standard deviation of mean %GC content across lice was 0.029), and thus all codon positions were retained for phylogenetic analysis. Each gene was aligned separately using UPP v.2.0 (Nguyen et al. 2015) according to its translated amino acid sequence. The aligned data were then back-translated into its DNA sequence. Finally, the GTR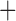 gamma parameters were calculated for each codon position in the gene alignment using RAxML (v.8.1.3; Stamatakis 2014). There were 76 first and second codon positions found to have abnormally high rate parameters (
gamma parameters were calculated for each codon position in the gene alignment using RAxML (v.8.1.3; Stamatakis 2014). There were 76 first and second codon positions found to have abnormally high rate parameters (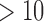 standard deviations from the mean rate parameters). These individual codon positions were removed from the concatenated alignment.
standard deviations from the mean rate parameters). These individual codon positions were removed from the concatenated alignment.
Inferring Species Relationships
Both concatenation and coalescent-based species tree estimation approaches were used to determine the phylogenetic relationships within Columbicola. First, phylogenetic relationships were inferred from all sequence data simultaneously using a data concatenation method. To control for rate heterogeneity across genes, we grouped the codon positions of the genes into separate partitions. The partitions were computed by first running a principal coordinates analysis (PCoA) on the GTR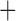 gamma parameters of each codon position for each gene (Supplementary Fig. S1, available on Dryad; Wickett et al. 2014). Next, codon positions were grouped into partitions based upon a k-means clustering of the PCoA. We found that 13 data partitions explained most of the variation between the clusters. Finally, we built a concatenated supermatrix alignment by combining these 13 data partitions into a single alignment. We generated an initial maximum-likelihood (ML) tree from the supermatrix alignment using FastTree-2 under GTR
gamma parameters of each codon position for each gene (Supplementary Fig. S1, available on Dryad; Wickett et al. 2014). Next, codon positions were grouped into partitions based upon a k-means clustering of the PCoA. We found that 13 data partitions explained most of the variation between the clusters. Finally, we built a concatenated supermatrix alignment by combining these 13 data partitions into a single alignment. We generated an initial maximum-likelihood (ML) tree from the supermatrix alignment using FastTree-2 under GTR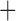 CAT (v.2.1.7; Price et al. 2010), and then performed a partitioned ML analysis using RAxML under GTR
CAT (v.2.1.7; Price et al. 2010), and then performed a partitioned ML analysis using RAxML under GTR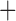 gamma (v.8.1.3; Stamatakis 2014), using the FastTree-2 ML tree as an initial starting tree. Support for the branches in the ML tree was computed by estimating 100 bootstrap replicate trees. Second, to determine whether incomplete lineage sorting (ILS) or other biological phenomena may have affected phylogenetic reconstruction, we reconstructed the phylogeny using coalescent-based species tree estimation methods (Mirarab et al. 2014; Vachaspati and Warnow 2015). These methods are designed to reconcile gene tree–species tree conflict caused by ILS. ML gene trees were estimated from each gene alignment using RAxML under GTR
gamma (v.8.1.3; Stamatakis 2014), using the FastTree-2 ML tree as an initial starting tree. Support for the branches in the ML tree was computed by estimating 100 bootstrap replicate trees. Second, to determine whether incomplete lineage sorting (ILS) or other biological phenomena may have affected phylogenetic reconstruction, we reconstructed the phylogeny using coalescent-based species tree estimation methods (Mirarab et al. 2014; Vachaspati and Warnow 2015). These methods are designed to reconcile gene tree–species tree conflict caused by ILS. ML gene trees were estimated from each gene alignment using RAxML under GTR gamma. The species tree was then estimated using ASTRAL (v.4.9.7; Mirarab et al. 2014; Mirarab and Warnow 2015) and ASTRID (v.1.4; Vachaspati and Warnow 2015) from the set of gene trees. Support for the branches in the ASTRAL and ASTRID trees were computed using local posterior probabilities based on the gene tree quartet frequencies (Sayyari and Mirarab 2016). To summarize our phylogenetic results, we calculated a strict consensus tree from concatenation, ASTRAL, and ASTRID phylogenies. Prior to calculating the consensus tree, any node with less than 95% bootstrap or local support was collapsed into a polytomy. Any conflict between any of the trees (including unresolved nodes) resulted in a polytomy within the consensus tree.
gamma. The species tree was then estimated using ASTRAL (v.4.9.7; Mirarab et al. 2014; Mirarab and Warnow 2015) and ASTRID (v.1.4; Vachaspati and Warnow 2015) from the set of gene trees. Support for the branches in the ASTRAL and ASTRID trees were computed using local posterior probabilities based on the gene tree quartet frequencies (Sayyari and Mirarab 2016). To summarize our phylogenetic results, we calculated a strict consensus tree from concatenation, ASTRAL, and ASTRID phylogenies. Prior to calculating the consensus tree, any node with less than 95% bootstrap or local support was collapsed into a polytomy. Any conflict between any of the trees (including unresolved nodes) resulted in a polytomy within the consensus tree.
Biogeography
To determine whether the Columbicola phylogeny is significantly structured according to biogeographic region, we conducted a Maddison–Slatkin test (Maddison and Slatkin 1991). This test used the tree reconstructed from the concatenation analysis (the consensus tree was not used to avoid a polytomy that could complicate analysis) and was implemented using an R script that randomly assigns character states to phylogeny tips; thus, calculating the number of character transitions for each randomization (as in Bush et al. 2016, https://github.com/juliema/publications/tree/master/BrueeliaMS; obtained April 2016). The distribution of each taxon was classified based on its occurrence in one of the following four biogeographic regions–-Eurasia, Australasia (including islands in SE Asia, and the Pacific), Africa, or the New World. Tip character states (biogeographic regions) were then randomized 999 times. Next, we tested six different biogeographic pattern models and inferred ancestral areas using BioGeoBEARS v.0.2.1 (Matzke 2013) implemented in R v.3.2.4 (R Core Team; available from http://r-project.org/) based on the concatenation tree (Fig. 1). Since this analysis requires ultrametric trees, we estimated new ultrametric branch lengths in BEAST v.2.3.2 under a lognormal clock (files prepared in BEAUTi; Drummond et al. 2012) while holding the concatenated tree topology constant. The width of the concatenated data matrix used to construct the original tree exceeded that allowed by the software. Therefore, we used trimAl v.1.2 (Capella-Gutierrez et al. 2009) to reduce the matrix to only those columns that contained 10% or fewer gap characters. This left us with 388,048 alignment columns with which to infer branch lengths. Using the reduced alignment, we ran BEAST on the CIPRES Science Gateway (Miller et al. 2010) for 12 million MCMC generations, sampling every 1000 generations and discarding the first 50% as a burn-in. Outgroup taxa were then removed and each Columbicola taxon was coded as belonging to one of the four biogeographic regions used in the Maddison–Slatkin test. We inferred the ancestral range at each node using six different unconstrained models in BioGeoBEARS (DEC, DEC J, DIVALIKE, DIVALIKE
J, DIVALIKE, DIVALIKE J, BAYAREALIKE, and BAYAREALIKE
J, BAYAREALIKE, and BAYAREALIKE J) with maximum biogeographic regions set to four. To identify the optimal biogeographic model for our data, we compared the reconstructions of ancestral biogeography under each model using the Akaike Information Criterion (AIC).
J) with maximum biogeographic regions set to four. To identify the optimal biogeographic model for our data, we compared the reconstructions of ancestral biogeography under each model using the Akaike Information Criterion (AIC).
Figure 1.

Maximum-likelihood tree showing the relationships of Columbicola species based on the concatenation of 977 single copy orthologous gene sequence alignments. The tree was calculated using RAxML and support was calculated from bootstrap replicates. Numbers at nodes indicate support as percent of 100 bootstrap replicates that also contain that node. Names at tips of branches identify the species of Columbicola, followed by the dove or pigeon species from which that sample was collected. Images of Columbicola (arrows) are also shown. Columbicola sp. designates undescribed species of Columbicola. Branches to out-group taxa have been shortened (denoted by *) for presentation. Letters “a–d” in circles designate major clades discussed in the text.
Validating Species Identification
Lice used in this study were identified by host associations (Supplementary Table S1, available on Dryad), as well as morphology. Many of the taxa in the study were included in previous studies that sequenced the mitochondrial Cytochrome c oxidase I (COI) gene. To validate that our identifications were consistent with previous studies, we assembled the COI gene from each Columbicola sample using aTRAM. A partial, translated COI sequence from C. columbae (gi15419110, gbAAK96907.1) served as the reference sequence. The resulting contigs assembled by aTRAM were compared with the COI sequences available on NCBI using the blastn web interface (http://blast.ncbi.nlm.nih.gov/Blast.cgi; accessed February 2016). The best hits for each contig are reported in Supplementary Table S1, available on Dryad.
Results
DNA Sequencing and Assembly
Whole-genome sequencing produced a mean of 73,611,688 (range 25,082,170–133,845,542) raw 160-nt reads for each sample with a mean duplication rate of 6.02% (range 0.43–24.74%). Of the 1107 targeted genes, we assembled a mean of 1101 candidate orthologs (range of 1069–1107), either in part or whole, from each louse. After filtering to remove potentially non-orthologous contigs and gene sets with more than 50% missing data, 977 ortholog genes remained for phylogenetic analysis. Percent GC content was slightly elevated in third codon positions, but this bias was consistent across all taxa; therefore, third positions were retained. These assembly and filtering steps yielded 1,596,995 sites for phylogenetic analyses.
Dove Wing Louse Species Relationships
The relationships among species of wing lice were determined by both simultaneous analysis of all data (concatenation method) and by gene tree summary methods (coalescent method using ASTRAL and ASTRID). The concatenation method produced a very well supported tree at most nodes (mean bootstrap of 97.8%, range of 51–100%; Fig. 1). Of the 61 ingroup nodes, 54 (89%) were supported in 100% of bootstrap replicates, and 59 (97%) were supported in 80% or more of bootstrap replicates. Two species, Columbicola fortis and C. triangularis, formed the sister clade to all other Columbicola. The remaining Columbicola fell into four major, well-supported clades (labeled a–d in Fig. 1). However, the relationship of these four major clades to one another was not resolved. In contrast, the relationships within these clades were well supported, with nearly all branches receiving 100% bootstrap support. Only clade d had five branches with less than 100% bootstrap support, all in the range 88–99%; all other clades had 100% support on all branches.
The lack of resolution among the four major clades in the concatenation tree described above could be due to ILS, and it is important to account for this possibility. Therefore, we built two coalescent trees (using ASTRAL and ASTRID). These trees were similar to each other as well as to the tree from concatenated analysis. Only branches that were weakly supported by some of the analyses differed among trees from the three methods. In particular, the coalescent tree built using ASTRAL yielded slightly different relationships from the concatenated tree among the four main clades (i.e., differences in the short branches connecting these clades; Fig. 2). The ASTRAL coalescent tree had higher support for these short branches than the concatenation tree. However, support for the coalescent tree was assessed as local support and it is difficult to compare these values to bootstrap values. In most other respects the coalescent tree was identical to the concatenation analysis. Like the concatenation tree, most relationships within the four major clades in the coalescent tree were also highly supported (100% local support). Only one branch within a major clade was supported at less than 100% posterior probability, and there was only one topological difference between the trees within a major clade (an alternative arrangement of C. arnoldi, C. beccarii, and C. exilicornis was supported). The second coalescent tree, built using ASTRID (Fig. 3), was again similar to both the concatenation tree and the ASTRAL tree. Again, we found an alternate relationship among the four well-supported clades connected by short branches. Within these four clades, the relationships are identical among ASTRAL and ASTRID trees. Finally, a strict consensus of concatenation, ASTRAL, and ASTRID trees shows that all three approaches yielded nearly identical results (Fig. 4) with relationships within major Columbicola clades being resolved, but the relationships among major clades remaining unresolved.
Figure 2.

Coalescent tree showing relationships of Columbicola species based on 977 single-copy ortholog gene trees. Tree was calculated using ASTRAL from gene trees calculated using RAxML with support based local posterior probabilities. Letters “a–d” in circles designate major clades described in Figure 1.
Figure 3.

Coalescent tree showing relationships of Columbicola species based on 977 single-copy ortholog gene trees. The tree was calculated using ASTRID from gene trees calculated using RAxML and support based local posterior probabilities. Letters “a–d” in circles designate major clades described in Figure 1.
Figure 4.

Strict consensus of concatenation, ASTRAL, and ASTRID trees with all resolved nodes receiving at least 95% support in all three trees. Tree is presented as a cladogram and branch lengths are not informative. Letters “a–d” in circles designate major clades described in Figure 1.
Compared with the most comprehensive phylogenetic study of Columbicola to date (Johnson et al. 2007), the consensus tree (Fig. 4) presented here shows dramatic improvement in both resolution and support. The previous study was based on three genes (two mitochondrial and one nuclear; Johnson et al. 2007), while our study sampled 977 genes from across the nuclear genome. A comparison of taxa common to both studies (Fig. 5) shows that 83% of nodes are now resolved with greater than 95% support in all trees. By comparison, only 48% of nodes were resolved with at least 95% Bayesian posterior probability in the previous study.
Figure 5.

Comparison of (a) Bayesian tree based on three genes from Johnson et al. (2007) and (b) consensus tree reconstructed from genome-wide data in the current study. Only those taxa analyzed in both studies are shown (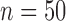 ). Nodes with less than 0.95 posterior probability in tree A and 95% bootstrap support in tree B were collapsed. “Columbicola sp.” indicates an undescribed species of Columbicola from Streptopelia semitorquata. Letters “a–d” in circles designate major clades described in Figure 1. Dotted lines join species with a consistent phylogenetic position in each tree.
). Nodes with less than 0.95 posterior probability in tree A and 95% bootstrap support in tree B were collapsed. “Columbicola sp.” indicates an undescribed species of Columbicola from Streptopelia semitorquata. Letters “a–d” in circles designate major clades described in Figure 1. Dotted lines join species with a consistent phylogenetic position in each tree.
The arrangement of many nodes differs between our consensus tree and the Bayesian tree of Johnson et al. (2007); however, only two of these conflicting nodes were strongly supported (Johnson et al. 2007). In this particular conflict, the three-gene tree supported a clade of the Australasian phabine dove lice (C. mjobergi and C. rodmani) as sister to lice from New World ground doves and pigeons (C. extinctus, C. adamsi, and C. macrourae from Patagioenas and Zenaida columbid species). This resulted in a paraphyletic assemblage of the lice from Australasian phabine genera sampled in the prior study (Phaps, Geophaps, Ocyphaps, Petrophassa, and Geopelia). In contrast, the consensus tree from our genome-wide data recovers the lice of these Australasian phabine doves as a monophlyetic group within clade d. In another case, the two species in clade d that parasitize different species of Phaps (C. harbisoni and C. tasmaniensis) are sister taxa in our new tree, but were paraphyletic in the previous Bayesian tree. Thus, in both cases, relationships in the current phylogenomic trees are more concordant with host relationships.
Biogeographic Patterns
BioGeoBEARS supported the DEC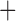 J biogeographic model as optimal for our data (Table 1). This model is similar to the Dispersal–Extinction–Cladogenesis of Lagrange, but with an extra parameter that models founder events (Matzke 2013). In addition, all models that included the founder event parameter scored better than similar models lacking this component (Table 1). Collectively, this supports the hypothesis that wing lice dispersed into different regions and then speciated. The results of the Maddison–Slatkin test were highly significant, with none of the character state randomizations having an equal or lower number of state transitions (biogeographic regions) than the observed (
J biogeographic model as optimal for our data (Table 1). This model is similar to the Dispersal–Extinction–Cladogenesis of Lagrange, but with an extra parameter that models founder events (Matzke 2013). In addition, all models that included the founder event parameter scored better than similar models lacking this component (Table 1). Collectively, this supports the hypothesis that wing lice dispersed into different regions and then speciated. The results of the Maddison–Slatkin test were highly significant, with none of the character state randomizations having an equal or lower number of state transitions (biogeographic regions) than the observed (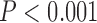 ). Thus, both ancestral area reconstruction and the Maddison–Slatkin test indicate that the biogeographic distribution of the lice is phylogenetically conserved and that there is limited exchange between regions (Fig. 6).
). Thus, both ancestral area reconstruction and the Maddison–Slatkin test indicate that the biogeographic distribution of the lice is phylogenetically conserved and that there is limited exchange between regions (Fig. 6).
Table 1.
Comparison of different biogeographic models tested in BioGeoBEARS
| Model | Ln 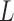
|
Params |
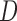
|
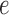
|
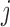
|
AIC | AIC_wt |
|---|---|---|---|---|---|---|---|
| DEC | –59.97 | 2 | 0.5 | 1.9E 8 8 |
0 | 123.9 | 9.5E 8 8 |
DEC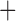 J J |
–43.39 | 3 | 1E 12 12 |
1E 12 12 |
0.036 | 92.78 | 0.55 |
| DIVALIKE | –62.89 | 2 | 0.9 | 5.2E 9 9 |
0 | 129.8 | 5.1E 9 9 |
DIVALIKE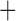 J J |
–44.06 | 3 | 1E 12 12 |
1E 12 12 |
0.038 | 94.12 | 0.28 |
| BAYAREALIKE | –87 | 2 | 0.56 | 5 | 0 | 178 | 1.7E 19 19 |
BAYAREALIKE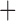 J J |
–44.61 | 3 | 1E 4 4 |
1E 4 4 |
0.037 | 95.22 | 0.16 |
Note: Ln 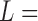 natural log, Params
natural log, Params  parameters, and AIC
parameters, and AIC  Akaike information criterion.
Akaike information criterion.
Figure 6.

Ancestral area reconstruction modeled in BioGeoBEARS using a DEC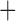 J model. Colors at tips represent current biogeographic area and pie charts represent predicted ancestral areas at cladogenesis. Red
J model. Colors at tips represent current biogeographic area and pie charts represent predicted ancestral areas at cladogenesis. Red  New World, Yellow
New World, Yellow  Australasia, Green
Australasia, Green  Africa, Blue
Africa, Blue  Eurasia, Orange
Eurasia, Orange  hypothetical ancestral area of New World
hypothetical ancestral area of New World Australasia. Letters “a–d” in circles designate major clades described in Figure 1.
Australasia. Letters “a–d” in circles designate major clades described in Figure 1.
We found that New World Columbicola are restricted to two major clades (c and d; Fig. 6), with the exception of C. triangularis. Similarly, Australasian Columbicola are restricted to two major clades (b and d), with the exception of a three species cluster in clade a. The New Guinean species C. fortis plus New World C. triangularis are sister to most other Columbicola (Fig. 1). Three other taxa from Australasia (C. waiteae, C. guimaraesi 1, and C. guimaraesi 3) are embedded within a clade dominated by African and Eurasian species (clade a). This suggests there may have been ancient movement of Columbicola from Africa or Eurasia into Australasia, with subsequent speciation. Members of clade a appear to have undergone extensive movement between Eurasia and Africa. Together, our analysis favors a New World or the New World plus Australasia as the ancestral region for dove lice. However, the latter scenario seems unlikely, because doves diversified in the Cenozoic (Steadman 2008; Prum et al. 2015), making vicariance an unlikely explanation. However, a lack of well-supported nodes near the base of the phylogenetic tree (probable ancient distribution shifts within monophyletic groups) together with mixed geographic distributions within modern clades makes it difficult to resolve with confidence the ancestral geographic origin for this group of lice.
Discussion
Parasitic feather lice (Columbicola) and their dove hosts (Columbiformes) have emerged as a powerful system for studying the interface between microevolutionary processes and macroevolutionary patterns (Clayton et al. 2016). Nevertheless, relationships among the many species of Columbicola have been challenging to resolve (Johnson et al. 2003, Johnson et al. 2007). The phylogenomic methods used here provide a well-supported and largely resolved phylogeny for this important group. Resolving the phylogeny of Columbicola was facilitated both by extensive sampling of species (about 50% of all known species in the genus), and by analyzing a large number of genes (977 nuclear single copy orthologs). Trees produced by concatenation and coalescent summary methods were largely compatible and well supported, with the exception of a few weakly supported nodes (Fig. 4). That is, nodes with high support were consistent across methods and weakly supported nodes were inconsistent across analyses. The agreement of nodal support and methods suggests that (1) the phylogeny presented is stable and (2) support measures are a good gauge of confidence in this phylogeny. Additionally, a consensus tree of all analyses resolves 17 nodes within Columbicola that were unresolved in an earlier work, including resolving all but four major branches close to the root of the tree (Johnson et al. 2007; Fig. 5). In short, our phylogenomic tree provides a solid basis for future comparative and cophylogenetic studies of Columbicola species and their hosts.
Based on relative branch lengths, it appears that there was a long period of stasis (or perhaps extinction) between a pair of highly diverged lineages of Columbicola (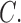 fortis and
fortis and 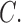 triangularis) versus all other Columbicola in our analysis (Fig. 1). The placement of C. fortis and C. triangularis as sister taxa to all other Columbicola was also recovered in a parsimony tree, but not in a Bayesian tree, by Johnson et al. (2007). The morphology of these lice is unusual in some respects. For example, C. triangularis and another closely related species, C. baculoides (not included in our analysis), are morphologically distinct from other Columbicola species (Johnson et al. 2007). Males of these two species have antennae resembling those of females, whereas other Columbicola species have sexually dimorphic antennae (Clayton and Price 1999). Columbicola fortis is also unusual, but in different ways: C. fortis has sexually dimorphic antennae like other species of Columbicola, but C. fortis has a distinctly large head (Adams et al. 2005). Neither of these features are synapomorphies shared with C. triangularis or C. baculoides. These three species also parasitize dove species in different regions of the world. Columbicola baculoides and C. triangularis parasitize New World doves, while C. fortis parasitizes the pheasant pigeon (Otidiphaps nobilis), which is endemic to New Guinea. Thus, the disjunct biogeographic distributions, morphological dissimilarity, and molecular divergence, all indicate an ancient divergence among the lice in this clade. Based on the short branches connecting the major clades a–d, we infer that this initial divergence was followed by an independent and relatively more rapid radiation, generating the rest of the diversity within Columbicola.
triangularis) versus all other Columbicola in our analysis (Fig. 1). The placement of C. fortis and C. triangularis as sister taxa to all other Columbicola was also recovered in a parsimony tree, but not in a Bayesian tree, by Johnson et al. (2007). The morphology of these lice is unusual in some respects. For example, C. triangularis and another closely related species, C. baculoides (not included in our analysis), are morphologically distinct from other Columbicola species (Johnson et al. 2007). Males of these two species have antennae resembling those of females, whereas other Columbicola species have sexually dimorphic antennae (Clayton and Price 1999). Columbicola fortis is also unusual, but in different ways: C. fortis has sexually dimorphic antennae like other species of Columbicola, but C. fortis has a distinctly large head (Adams et al. 2005). Neither of these features are synapomorphies shared with C. triangularis or C. baculoides. These three species also parasitize dove species in different regions of the world. Columbicola baculoides and C. triangularis parasitize New World doves, while C. fortis parasitizes the pheasant pigeon (Otidiphaps nobilis), which is endemic to New Guinea. Thus, the disjunct biogeographic distributions, morphological dissimilarity, and molecular divergence, all indicate an ancient divergence among the lice in this clade. Based on the short branches connecting the major clades a–d, we infer that this initial divergence was followed by an independent and relatively more rapid radiation, generating the rest of the diversity within Columbicola.
Biogeography
Biogeographic analysis of Columbicola species is important for understanding the history and diversification of the genus. Species of Columbicola sometimes parasitize more than one host species, and there are also cases where Columbicola have switched between distantly related dove species (Johnson et al. 2002, Johnson et al. 2003; Malenke et al. 2009; Bush et al. 2009). Therefore, while the dispersal and speciation of parasites often mirrors host diversification, host switches frequently lead to more complicated patterns of codiversifiation. With these factors in mind, several patterns emerge from our biogeographic analysis (Fig. 6). First, we find that biogeographic distributions are generally concordant with the major clades of Columbicola. In general, New World taxa are limited to two clades (c and part of d) and Old World taxa are limited to three clades (a, b, and part of d). Paleontological and molecular evidence indicates that the hosts of Columbicola radiated during the middle Cenozoic (Steadman 2008; Prum et al. 2015). This geologically recent timing rules out phylogenetically deep vicariance events in Columbicola that would be attributable to continental drift. A more likely scenario is movement between the Old and New World on dispersing hosts, followed by subsequent speciation in the new host range. The preferred biogeographic model in our analyses, which includes founder event speciation, supports this notion. However, bootstrap support near the base of our phylogeny is low, so we are unable to confidently determine the direction of these exchanges. Among Old World taxa, two clades are found exclusively in Australasia (clade b and part of clade d), while another occurs in Africa, Australasia, and Eurasia (clade a). Within this latter clade, we find evidence of movement from Eurasia into Australasia and between Eurasia and Africa, with at least one dispersal event occurring from Eurasia to Africa (the direction of other dispersal events in this clade is ambiguous). This movement might be explained by the high biogeographic connectivity between Eurasia and Africa.
Geography and Ecology of Host Switching
A comprehensive phylogenomic tree for the hosts of Columbicola is not yet available, thus making a thorough cophylogenetic analysis premature. However, based on previous phylogenetic studies of Columbiformes (Johnson and Clayton 2000; Pereira et al. 2007), some general co-evolutionary patterns emerge with respect to biogeography and host phylogeny. One predominant pattern is that monophyletic groups of Columbicola occur on monophyletic groups of doves, suggesting a general pattern of phylogenetic congruence. For example, the lice of small New World ground doves form a monophyletic group (C. passerinae, C. drowni, 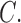 gymnopeliae, and
gymnopeliae, and 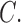 altamimiae; clade c), as do their hosts (Columbina spp. and Metriopelia spp.; Fig. 1; Johnson and Clayton 2000; Pereira et al. 2007; Sweet and Johnson 2015). The lice of most Australian phabine doves (Phaps, Geophaps, Ocyphaps, Petrophassa, and Geopelia) also form a clade (clade d). The only exception is Columbicola palmai, which is found on the Wonga Pigeon (Leucosarcia melanoleuca), a phabine dove whose louse falls within clade b. Most phabines are open country or dry forest species, but the Wonga Pigeon inhabits sub-tropical wet forests, along with distantly related fruit pigeons and doves (Gibbs et al. 2001). Lice that are closely related to C. palmai also occur in wet tropical/subtropical forests in Australasia and their hosts are only distantly related to the Wonga Pigeon (clade b; Johnson and Clayton 2000; Pereira et al. 2007). Since host–parasite phylogenies are not congruent in this case, this pattern suggests that C. palmai (or its ancestor) switched hosts.
altamimiae; clade c), as do their hosts (Columbina spp. and Metriopelia spp.; Fig. 1; Johnson and Clayton 2000; Pereira et al. 2007; Sweet and Johnson 2015). The lice of most Australian phabine doves (Phaps, Geophaps, Ocyphaps, Petrophassa, and Geopelia) also form a clade (clade d). The only exception is Columbicola palmai, which is found on the Wonga Pigeon (Leucosarcia melanoleuca), a phabine dove whose louse falls within clade b. Most phabines are open country or dry forest species, but the Wonga Pigeon inhabits sub-tropical wet forests, along with distantly related fruit pigeons and doves (Gibbs et al. 2001). Lice that are closely related to C. palmai also occur in wet tropical/subtropical forests in Australasia and their hosts are only distantly related to the Wonga Pigeon (clade b; Johnson and Clayton 2000; Pereira et al. 2007). Since host–parasite phylogenies are not congruent in this case, this pattern suggests that C. palmai (or its ancestor) switched hosts.
Another example of this pattern occurs in Africa. The African louse species C. smithae and C. fradei are closely related (within clade a), whereas their tropical forest dove hosts Turtur brehmeri and Aplopelia larvata are not. Moreover, the lice from other species of Turtur, which live in savannah habitats rather than tropical forests (Gibbs et al. 2001), are related to lice that parasitize distantly related birds in the same habitat. Collectively, our results suggest that ancient and persistent host–parasite associations have yielded congruent phylogenies between doves and lice, with incongruence resulting from periodic switching between unrelated species of doves that share the same habitat. Thus, both biogeographic and ecological overlap provide opportunities for host switching events, as shown in some other groups of birds and lice (Clayton et al. 2016).
The Columbicola phylogeny contains other examples of probable host switching. For example, the lice (C. macrourae, C. adamsi, and 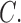 extinctus) of New World pigeons (Patagioenas) and New World mid-sized doves (Zenaida and Geotrygon) form a clade, but their hosts do not (within clade d; Johnson and Clayton 2000; Pereira et al. 2007; Johnson and Weckstein 2011). Better resolution of the directionality of host-switching events awaits a more comprehensive and well-supported phylogeny of pigeons and doves.
extinctus) of New World pigeons (Patagioenas) and New World mid-sized doves (Zenaida and Geotrygon) form a clade, but their hosts do not (within clade d; Johnson and Clayton 2000; Pereira et al. 2007; Johnson and Weckstein 2011). Better resolution of the directionality of host-switching events awaits a more comprehensive and well-supported phylogeny of pigeons and doves.
Utility of Targeted Assembly
For this study, we used a whole-genome sequencing approach, but assembled only gene-containing contigs for the phylogenetic analyses. We therefore recovered a portion of the genome that was directly comparable across Columbicola species, and we did so with very small quantities of DNA and at relatively low cost using conventional extraction and library construction methods. This approach allowed us to bypass the complex and time-consuming process of whole-genome assembly and annotation.
Other recent studies have implemented genome reduction methods to limit the portion of the genome that is sequenced in the first place (i.e., reduced representation sequencing; Ekblom and Wolf 2014). This strategy also alleviates some of the biological and technical challenges inherent in whole-genome assembly by capturing a limited number of conserved regions. These genome reduction methods, including selection for ultra-conserved elements (UCEs; Faircloth et al. 2012, Faircloth et al. 2015; McCormack et al. 2012), or anchored hybrid enrichment sequencing (Lemmon et al. 2012), only sequence regions of interest around conserved sites in the genome. These techniques are being widely applied currently to systematic problems for a diverse array of taxa. However, these methods require development of specialized DNA binding probes to capture conserved regions (Faircloth et al. 2012; Lemmon et al. 2012; McCormack et al. 2012). The performance of such probes can be difficult to assess a priori across an entire taxonomic group of interest. By using random shotgun sequencing of the entire genome and relying on target-restricted assembly (instead of targeted sequencing), we accomplished a similar goal. This was made possible by aTRAM, a recently developed software package that does not require specialized sequencing-library preparation or marker development. In addition, given that we sequenced the entire genome of each louse, we can return to the raw data to recover additional or different regions of the genome in the future. Thus, subsequent investigations using the same raw data are not limited to the particular set of genes we used.
Insect genomes vary widely in size, from 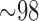 Mb to 16,528 Mb (Gregory et al. 2007). Parasitic lice, like those studied here, have small genomes compared with other insects; for example, the human body louse (Pediculus humanus) has a genome of only 110.78 Megabases (Mb; Kirkness et al. 2010). The genome of Columbicola (
Mb to 16,528 Mb (Gregory et al. 2007). Parasitic lice, like those studied here, have small genomes compared with other insects; for example, the human body louse (Pediculus humanus) has a genome of only 110.78 Megabases (Mb; Kirkness et al. 2010). The genome of Columbicola (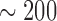 Mbp, unpublished data) appears to be relatively similar in size and composition to the closely related Pediculus. Because of this small genome size, we utilized multiplexing (simultaneous sequencing of samples) to reduce sequencing costs. We combined four samples per sequencing lane, but given our sequencing yield after error correction (Supplementary Table S1, available on Dryad), we could have increased multiplexing without decreasing sequencing depth below suitable levels. This would have further reduced the cost of generating the necessary phylogenomic data. Similar projects, focusing on organisms with larger genomes would likely be limited in the degree of multiplexing to achieve sufficient coverage (
Mbp, unpublished data) appears to be relatively similar in size and composition to the closely related Pediculus. Because of this small genome size, we utilized multiplexing (simultaneous sequencing of samples) to reduce sequencing costs. We combined four samples per sequencing lane, but given our sequencing yield after error correction (Supplementary Table S1, available on Dryad), we could have increased multiplexing without decreasing sequencing depth below suitable levels. This would have further reduced the cost of generating the necessary phylogenomic data. Similar projects, focusing on organisms with larger genomes would likely be limited in the degree of multiplexing to achieve sufficient coverage (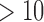 times coverage, Allen et al. in press). However, we observed that if sequencing coverage is low or inconsistent, aTRAM can still recover partial target assemblies. In this study we designed a test case in which aTRAM was used to rapidly collect phylogenomic data with preserved museum specimens yielding limited starting genomic DNA. The performance of aTRAM with larger genomes and different genome sequencing approaches warrants further investigation.
times coverage, Allen et al. in press). However, we observed that if sequencing coverage is low or inconsistent, aTRAM can still recover partial target assemblies. In this study we designed a test case in which aTRAM was used to rapidly collect phylogenomic data with preserved museum specimens yielding limited starting genomic DNA. The performance of aTRAM with larger genomes and different genome sequencing approaches warrants further investigation.
The aTRAM approach is particularly useful with limited or low quality DNA. Sample condition was inconsistent in our project and sequence data from each sample presented unique challenges for assembly. Despite the potential limitations of our study due to sample degradation, aTRAM provided results that were consistent across samples. Our results suggest that aTRAM is a viable phylogenomics pipeline for the analyses of closely related lineages, as well as more ancient patterns of divergence. It promises to be a useful tool for many other groups of organisms.
Supplementary Material
Acknowledgements
The authors would like to thank Jeff Haas and Bemi Ekwjunor and the rest of UIUC Life Science Biocomputing Support Team for their help with server maintenance and software installation; Alvaro Hernandez and Chris Wright of the UIUC WC Keck Center for overseeing Illumina sequencing; Philip Blood of the Pittsburgh Supercomputing Center for help implementing software on the XSEDE Blacklight system; Antonio Gomez of the Texas Advanced Computing Center for implementing aTRAM on the XSEDE Stampede system and for writing a Launcher script for parallel implementation of aTRAM; Justin Fear of NIH for his help implementing the Mcscript package; Kim Walden UIUC and Jonathan Trow NCBI for their help depositing our data to NCBI-SRA; Robert Moyle, Jason Weckstein, Terry Chesser, Ian Mason, John Wombey, Robert Palmer, Vincent Smith, Kevin McCracken, Robert Wilson, Brett Benz, David Steadman, David Willard, Mark Robbins, Andy Kratter, Selvino de Kort, Jack Dumbacher, Robert Faucett, and Ben Marks for assistance in obtaining lice used in this study; and the anonymous reviewers for their comments.
Supplementary Material
Raw sequence data collected from the Columbicola samples isolated in this study have been deposited in the NCBI short-read archive under the study SRP069898 and are tied to the BioProject identifier PRJNA296666. Individual BioSample and SRA run data can be found in Supplementary Table S1, available on Dryad. Data for Columbicola columbae (collected from Columba livia) and outgroup samples are being deposited as part of other studies. Nucleotide data matrix and phylogenetic trees have been deposited in TreeBASE http://purl.org/phylo/treebase/phylows/study/TB2:S19776.
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.4812p.
Funding
This work was supported by grants from the National Science Foundation [DEB1342600 to D.H.C., S.E.B., and M.D.S.; DEB1342604, DEB1239788, and TG-DEB150004 to K.P.J.; DEB1310824 to B.M.B.; TG-DEB160002 to K.P.J., B.M.B., and J.M.A.; III:AF:1513629 and ABI-1458652 to T.W.; and ASC160042 to N.D.N.] This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575.
References
- Adams R.J.,, Price R.D.,, Clayton D.H. 2005. Taxonomic revision of Old World members of the feather louse genus Columbicola (Phthiraptera: Ischnocera), including descriptions of eight new species. J. Nat. Hist. 39:3545–6318. [Google Scholar]
- Altschul S.F.,, Gish W.,, Miller W.,, Myers E.W.,, Lipman D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Allen J.M.,, Huang D.I.,, Cronk Q.C.,, Johnson K.P. 2015. aTRAM—automated target restricted assembly method: a fast method for assembling loci across divergent taxa from next-generation sequencing data. BMC Bioinform. 16:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J.M.,, Boyd B.M.,, Nguyen N.,, Vachaspati P.,, Warnow T.,, Huang D.I.,, Gero P.,, Bell K.C.,, Cronk Q.C.B.,, Mugisha L.,, Pittendrigh B.R.,, Leonardi M.S.,, Reed D.L.,, Johnson K.P. 2017. Phylogenomics from whole genome sequences using aTRAM. Syst. Biol. 66:786–798. [DOI] [PubMed] [Google Scholar]
- Althoff D.M.,, Segraves K.A.,, Johnson M.T.J. 2014. Testing for coevolutionary diversification: linking pattern with process. Trends Ecol. Evol. 29:82–89. [DOI] [PubMed] [Google Scholar]
- Bush S.E.,, Clayton D.H. 2006. The role of body size in host specificity: reciprocal transfer experiments with feather lice. Evolution 60:2158–2167. [PubMed] [Google Scholar]
- Bush S.E.,, Sohn E.,, Clayton D.H. 2006. Ecomorphology of parasite attachment: experiments with feather lice. J. Parasitol. 92(1):25–31. [DOI] [PubMed] [Google Scholar]
- Bush S.E.,, Kim D.,, Reed M.,, Clayton D.H. 2010. Evolution of cryptic coloration in ectoparasites. Am. Nat. 176:529–535. [DOI] [PubMed] [Google Scholar]
- Bush S.E.,, Price R.D.,, Clayton D.H. 2009. Descriptions of eight new species of feather lice in the genus Columbicola (Phthiraptera: Philopteridae), with a comprehensive world checklist. J. Parasitol. 95:286–294. [DOI] [PubMed] [Google Scholar]
- Bush S.E.,, Weckstein J.D.,, Gustafsson D.R.,, Allen J.,, DiBlasi E.,, Shreve S.M.,, Boldt R.,, Skeen H.R.,, Johnson K.P. 2016. Unlocking the black box of feather louse diversity: a molecular phylogeny of the hyper-diverse genus Brueelia. Mol. Phylogenet. Evol. 94:737–751. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S.,, Silla-Martinez J.M.,, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D.H. 1991. Coevolution of avian grooming and ectoparasite avoidance. In: Loye J.E.,, Zuk M., editors. Bird–parasite interactions: ecology, evolution and behaviour.Oxford (UK): Oxford University Press; p. 258–289. [Google Scholar]
- Clayton D.H.,, Bush S.E.,, Goates B.M.,, Johnson K.P. 2003. Host defense reinforces host–parasite cospeciation. Proc. Natl. Acad. Sci. USA. 100:15694–15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D.H.,, Bush S.E.,, Johnson K.P. 2016. Coevolution of life on hosts: integrating ecology and history. Chicago: University Chicago Press; 294 p. [Google Scholar]
- Clayton D.H.,, Johnson K.P. 2003. Linking coevolutionary history to ecological process: doves and lice. Evolution 57:2335–2341. [DOI] [PubMed] [Google Scholar]
- Clayton D.H., Lee P.L.M., Tompkins D.M., Brodie E.D. III.. 1999. Reciprocal natural selection on host–parasite phenotypes. Am. Nat. 154:261–270. [DOI] [PubMed] [Google Scholar]
- Clayton D.H.,, Moyer B.R.,, Bush S.E.,, Jones T.G.,, Gardiner D.W.,, Rhodes B.B.,, Goller F. 2005. Adaptive significance of avian beak morphology for ectoparasite control. Proc. R. Soc. B 272:811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D.H.,, Price R.D. 1999. Taxonomy of New World Columbicola (Phthiraptera: Philopteridae) from the Columbifomres (Aves), with descriptions of five new species. Ann. Entomol. Soc. Am. 92:675–685. [Google Scholar]
- Dobson A.,, Lefferty K.D.,, Kuris A.M.,, Hechinger R.F.,, Jetz W. 2008. Colloquium paper: homage to Linnaeus: how many parasites? How many hosts? Proc. Natl. Acad. Sci. USA. 105:11482–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J.,, Suchard M.A.,, Xie D.,, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R.,, Wolf B.W. 2014. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 7:1026–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth B.C.,, McCormack J.E.,, Crawford N.G.,, Harvey M.G.,, Brumfield R.T.,, Glenn T.C. 2012. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 61:717–726. [DOI] [PubMed] [Google Scholar]
- Faircloth B.C.,, Branstetter M.G.,, Whites N.D.,, Brady S.G. 2015. Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera. Mol. Ecol. 15:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs D.,, Barnes E.,, Cox J. 2001. Pigeons and doves: a guide to the pigeons and doves of the world. London: Christopher Helm; 615 p. [Google Scholar]
- Gustafsson D.R.,, Bush S.E. 2015. The chewing lice (Insecta: Phthiraptera: Ischnocera, Amblycera) of Japanese pigeons and doves (Columbiformes), with descriptions of three new species. J. Parasitol. 101:304–313. [DOI] [PubMed] [Google Scholar]
- Gregory T.R.,, Nicol J.A.,, Tamm H.,, Kullman B.,, Leitch I.J.,, Murray B.G.,, Kapraun D.F.,, Greihuber J.,, Bennett M.D. 2007. Eukaryotic genome size databases. Nucleic Acids Res. 35:D332–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis E.D.,, Mirarab S.,, Aberer A.J.,, Li B.,, Houde P.,, Li C.,, Ho S.Y.,, Faircloth B.C.,, Nabholz B.,, Howard J.T.,, Suh A.,, Weber C.C.,, da Fonseca R.R.,, Li J.,, Zhang F.,, Li H.,, Zhou L.,, Narula N.,, Liu L.,, Ganapathy G.,, Boussau B.,, Bayzid M.S.,, Zavidovych V.,, Subramanian S.,, Gabaldón T.,, Capella-Gutiérrez S.,, Huerta-Cepas J.,, Rekepalli B.,, Munch K.,, Schierup M.,, Lindow B.,, Warren W.C.,, Ray D.,, Green R.E.,, Bruford M.W.,, Zhan X.,, Dixon A.,, Li S.,, Li N.,, Huang Y.,, Derryberry E.P.,, Bertelsen M.F.,, Sheldon F.H.,, Brumfield R.T.,, Mello C.V.,, Lovell P.V.,, Wirthlin M.,, Schneider M.P.,, Prosdocimi F.,, Samaniego J.A.,, Vargas Velazquez A.M.,, Alfaro-Núñez A.,, Campos P.F.,, Petersen B.,, Sicheritz-Ponten T.,, Pas A.,, Bailey T.,, Scofield P.,, Bunce M.,, Lambert D.M.,, Zhou Q.,, Perelman P.,, Driskell A.C.,, Shapiro B.,, Xiong Z.,, Zeng Y.,, Liu S.,, Li Z.,, Liu B.,, Wu K.,, Xiao J.,, Yinqi X.,, Zheng Q.,, Zhang Y.,, Yang H.,, Wang J.,, Smeds L.,, Rheindt F.E.,, Braun M.,, Fjeldsa J.,, Orlando L.,, Barker F.K.,, Jønsson K.A.,, Johnson W.,, Koepfli K.P.,, O’Brien S.,, Haussler D.,, Ryder O.A.,, Rahbek C.,, Willerslev E.,, Graves G.R.,, Glenn T.C.,, McCormack J.,, Burt D.,, Ellegren H.,, Alström P.,, Edwards S.V.,, Stamatakis A.,, Mindell D.P.,, Cracraft J.,, Braun E.L.,, Warnow T.,, Jun W.,, Gilbert M.T.,, Zhang G. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P.,, Williams B.L.,, Drown D.M.,, Adams R.J.,, Clayton D.H. 2002. The population genetics of host specificity: genetic differentiation in dove lice (Insecta: Phthiraptera). Mol. Ecol. 11:25–38. [DOI] [PubMed] [Google Scholar]
- Johnson K.P.,, Adams R.J.,, Page R.D.M.,, Clayton D.H. 2003. When do parasites fail to speciate in response to host speciation? Syst. Biol. 52:37–47. [DOI] [PubMed] [Google Scholar]
- Johnson K.P.,, Bush S.E.,, Clayton D.H. 2005. Correlated evolution of host and parasite body size: test of Harrison’s Rule using birds and lice. Evolution 59:1744–1753. [PubMed] [Google Scholar]
- Johnson K.P.,, Clayton D.H. 2000. Nuclear and mitochondrial genes contain similar phylogenetic signal for pigeons and doves (Aves: Columbiformes). Mol. Phylogenet. Evol. 14:141–151. [DOI] [PubMed] [Google Scholar]
- Johnson K.P.,, Malenke J.R.,, Clayton D.H. 2009. Competition promotes the evolution of host generalists in obligate parasites. Proc. R. Soc. B. 276:3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P.,, Reed D.L.,, Parker S.L.H.,, Kim D.,, Clayton D.H. 2007. Phylogenetic analysis of nuclear and mitochondrial genes supports species groups for Columbicola (Insecta: Phthiraptera). Mol. Phylogenet. Evol. 45:508–518. [DOI] [PubMed] [Google Scholar]
- Johnson K.P.,, Walden K.K.,, Robertson H.M. 2013. Next-generation phylogenomics using a target restricted assembly method. Mol. Phylogenet. Evol. 66:417–422. [DOI] [PubMed] [Google Scholar]
- Johnson K.P.,, Weckstein J.D. 2011. The Central American land bridge as an engine of diversification in New World doves. J. Biogeogr. 38:1069–1076. [Google Scholar]
- Kirkness E.F.,, Haas B.J.,, Sun W.,, Braig H.R.,, Perotti M.A.,, Clark J.M.,, Lee S.H.,, Robertson H.M.,, Kennedy R.C.,, Elhaik E.,, Gerlach D.,, Kriventseva E.V.,, Elsik C.G.,, Graur D.,, Hill C.A.,, Veenstra J.A.,, Walenz B.,, Tubio J.M.C.,, Ribeiro J.M.C.,, Rozas J.,, Johnston J.S.,, Reese J.T.,, Popadic A.,, Tojo M.,, Rault D.,, Reed D.L.,, Tomoyasu Y.,, Kraus E.,, Mittapalli O.,, Margam V.M.,, Li H.M.,, Meyer J.M.,, Johnson R.M.,, Romero-Severson J.,, VanZee J.P.,, Alvarex-Ponce D.,, Vieira F.G.,, Aguade M.,, Guirao-Rico S.,, Anzola J.M.,, Yoon K.S.,, Strycharz J.P.,, Unger M.F.,, Christley S.,, Lobo N.F.,, Seufferheld M.J.,, Wang N.,, Dasch G.A.,, Struchiner C.J.,, Madey G.,, Hannick L.I.,, Bidwell S.,, Joardar V.,, Caler E.,, Shao R.,, Barker S.C.,, Cameron S.,, Bruggner R.V.,, Regier A.,, Johnson J.,, Viswanathan L.,, Utterback T.R.,, Sutton G.G.,, Lawson D.,, Waterhouse R.M.,, Venter J.G.,, Strausberg R.L.,, Berenbaum M.R.,, Collins F.R.,, Zdobnov E.M.,, Pittendrigh B.R. 2010. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. USA. 107:12168–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon A.R.,, Emme S.A.,, Lemmon E.M. 2012. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 61:727–744. [DOI] [PubMed] [Google Scholar]
- Maddison W.P.,, Slatkin M. 1991. Null models for the number of evolutionary steps in a character on a phylogenetic tree. Evolution 45:1184–1197. [DOI] [PubMed] [Google Scholar]
- Malenke J.R.,, Johnson K.P.,, Clayton D.H. 2009. Host specialization differentiates cryptic species of feather-feeding lice. Evolution 63:1427–1438. [DOI] [PubMed] [Google Scholar]
- Marshall A.G. 1981. The ecology of ectoparasitic insects. London: Academic Press. [Google Scholar]
- Matzke N.J. 2013. Probabilisitic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front. Biogeogr. 5:242–248. [Google Scholar]
- McCormack J.E.,, Faircloth B.C.,, Crawford N.G.,, Gowaty P.A.,, Brumfield R.T.,, Glenn T.C. 2012. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 22:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.A.,, Pfeiffer W.,, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), 14 November 2010, New Orleans, LA. p. 1–8. [Google Scholar]
- Mirarab S.,, Raez R.,, Zimmerman T.,, Swenson M.S.,, Warnow T. 2014. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30:i541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S.,, Warnow T. 2015. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Proc. Int. Conf. Intell. Syst. Mol. Biol. 31:i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B.,, Liu S.,, Meusemann K.,, Peters R.S.,, Donath A.,, Mayer C.,, Frandsen P.B.,, Ware J.,, Flouri T.,, Beutel R.G.,, Niehuis O.,, Petersen M.,, Izquierdo-Carrasco F.,, Wappler T.,, Rust J.,, Aberer A.J.,, Aspöck U.,, Aspöck H.,, Bartel D.,, Blanke A.,, Berger S.,, Böhm A.,, Buckley T.R.,, Calcott B.,, Chen J.,, Friedrich F.,, Fukui M.,, Fujita M.,, Greve C.,, Grobe P.,, Gu S.,, Huang Y.,, Jermiin L.S.,, Kawahara A.Y.,, Krogmann L.,, Kubiak M.,, Lanfear R.,, Letsch H.,, Li Y.,, Li Z.,, Li J.,, Lu H.,, Machida R.,, Mashimo Y.,, Kapli P.,, McKenna D.D.,, Meng G.,, Nakagaki Y.,, Navarrete-Heredia J.L.,, Ott M.,, Ou Y.,, Pass G.,, Podsiadlowski L.,, Pohl H.,, von Reumont B.M.,, Schütte K.,, Sekiya K.,, Shimizu S.,, Slipinski A.,, Stamatakis A.,, Song W.,, Su X.,, Szucsich N.U.,, Tan M.,, Tan X.,, Tang M.,, Tang J.,, Timelthaler G.,, Tomizuka S.,, Trautwein M.,, Tong X.,, Uchifune T.,, Walzl M.G.,, Wiegmann B.M.,, Wilbrandt J.,, Wipfler B.,, Wong T.K.,, Wu Q.,, Wu G.,, Xie Y.,, Yang S.,, Yang Q.,, Yeates D.K.,, Yoshizawa K.,, Zhang Q.,, Zhang R.,, Zhang W.,, Zhang Y.,, Zhao J.,, Zhou C.,, Zhou L.,, Ziesmann T.,, Zou S.,, Li Y.,, Xu X.,, Zhang Y.,, Yang H.,, Wang J.,, Wang J.,, Kjer K.M.,, Zhou X. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767. [DOI] [PubMed] [Google Scholar]
- Mora C.,, Tittensor D.P.,, Adi S.,, Simpson A.G.B.,, Worm B. 2011. How many species on earth and in the ocean? PLoS Biol. 9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N.D.,, Mirarab S.,, Kumar K.,, Warnow T. 2015. Ultra-large alignments using phylogeny-aware profiles. Genome Biol. 16:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira S.L.,, Johnson K.P.,, Clayton D.H.,, Baker A.J. 2007. Mitochondrial and nuclear DNA sequences support a Cretaceous origin of Columbiforms and a dispersal-driven radiation in the Paleogene. Syst. Biol. 56:656–672. [DOI] [PubMed] [Google Scholar]
- Poulin R.,, Morand S. 2000. The diversity of parasites. Q. Rev. Biol. 75:277–293. [DOI] [PubMed] [Google Scholar]
- Poulin R.,, Morand S. 2004. Parasite biodiversity. Washington, DC: Smithsonian Institution Press. [Google Scholar]
- Price M.N.,, Dehal P.S.,, Arkin A.P. 2010. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prum R.O.,, Berv J.S.,, Dornberg A.,, Field D.J.,, Townsend J.P.,, Lemmon E.M.,, Lemmon A.R. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 526:569–573. [DOI] [PubMed] [Google Scholar]
- Sayyari E.,, Mirarab S. 2016. Fast-coalescent-based computation of local branch support from quartet frequencies. arXiv:1601.07019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T.,, Wong K.,, Jackman S.D.,, Schein J.E.,, Jones S.J.M.,, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S.,, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steadman D.W. 2008. Doves (Columbidae) and cuckoos (Cuclidae) from the early Miocene of Florida. Bull. Fla. Mus. Nat. Hist. 48:1–16. [Google Scholar]
- Sweet A.D.,, Johnson K.P. 2015. Patterns of diversification in small New World ground doves are consistent with major geologic events. Auk 132:300–312. [Google Scholar]
- Towns J.,, Cockerill T.,, Dahan M.,, Foster I.,, Gaither K.,, Grimshaw A.,, Hazelwood V.,, Lathrop S.,, Lifka D.,, Peterson G.D.,, Roskies R.,, Scott J.R.,, Wilkins-Diehr N. 2014. XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 16:62–74. [Google Scholar]
- Vachaspati P.,, Warnow T. 2015. : accurate species trees from internode distances. BMC Genomics 16:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett S.,, Mirarab S.,, Nguyen N.,, Warnow T.,, Carpenter E.,, Matasci N.,, Ayyampalayam S.,, Barker M.S.,, Burleigh J.G.,, Gitzendanner M.A.,, Ruhfel B.R.,, Wafula E.,, Der J.P.,, Graham S.W.,, Mathews S.,, Melkonian M.,, Soltis D.E.,, Soltis P.S.,, Miles N.W.,, Rothfels C.J.,, Pokorny L.,, Shaw A.J.,, DeGironimo L.,, Stevenson D.W.,, Surek B.,, Villarreal J.C.,, Roure B.,, Philippe H.,, DePamphilis C.W.,, Chen T.,, Deyholos M.K.,, Baucom R.S.,, Kutchan T.M.,, Augustin M.M.,, Wang J.,, Zhang Y.,, Tian Z.,, Yan Z.,, Wu X.,, Sun X.,, Wong G.K.S.,, Leebens-Macka J. 2014. Phylotranscriptomic analysis of the origins and early diversification of land plants. Proc. Natl. Acad. Sci. USA. 111:E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windsor D.A. 1998. Most of the species on earth are parasites. Int. J. Parasitol. 28:1939–1941. [DOI] [PubMed] [Google Scholar]
- Wilson L.A.,, Fonner J.M. 2014. Launcher: a shell-based framework for rapid development of parallel parametric studies. Proceedings of the 2014 Annual Conference on Extreme Science and Engineering Discovery Environment Article No. 40, Atlanta, GA, USA: 10.1145/2616498.2616534. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.