

Abstract
Parkinson’s disease (PD) is the result of complex interactions between genetic and environmental factors. Two chemically distinct environmental stressors relevant to PD are the metal manganese and the pesticide rotenone. Both are thought to exert neurotoxicity at least in part via oxidative stress resulting from impaired mitochondrial activity. Identifying shared mechanism of action may reveal clues towards an understanding of the mechanisms underlying PD pathogenesis. Here we compare the effects of manganese and rotenone in human-induced pluripotent stem cells-derived postmitotic mesencephalic dopamine neurons by assessing several different oxidative stress endpoints. Manganese, but not rotenone caused a concentration and time-dependent increase in intracellular reactive oxygen/nitrogen species measured by quantifying the fluorescence of oxidized chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate (DCF) assay. In contrast, rotenone but not manganese caused an increase in cellular isoprostane levels, an indicator of lipid peroxidation. Manganese and rotenone both caused an initial decrease in cellular reduced glutathione; however, glutathione levels remained low in neurons treated with rotenone for 24 h but recovered in manganese-exposed cells. Neurite length, a sensitive indicator of overall neuronal health was adversely affected by rotenone, but not manganese. Thus, our observations suggest that the cellular oxidative stress evoked by these 2 agents is distinct yielding unique oxidative stress signatures across outcome measures. The protective effect of rasagiline, a compound used in the clinic for PD, had negligible impact on any of oxidative stress outcome measures except a subtle significant decrease in manganese-dependent production of reactive oxygen/nitrogen species detected by the DCF assay.
Keywords: dopamine neuron, human-induced pluripotent stem cells, manganese, oxidative stress, rasagiline, rotenone
Parkinson’s disease (PD) is a chronic, progressive, noncurable disease and like other neurodegenerative diseases, is the result of complex interactions between genetic and environmental factors (Kalia and Lang, 2015). The genetic make-up of a patient not only determines the risk level of developing the disease, but also plays a role in age of onset, disease progression, responsiveness to therapeutics and importantly sensitivity and response to environmental factors. Human-induced pluripotent stem cells (iPSC) provide a model system to study patient-specific effects of disease-related environmental stressors on human mature neurons in the context of genetic factors underlying stressor sensitivities (Kumar etal., 2012).
Oxidative stress, which can be caused by a variety of genetic and environmental factors that often act in concert, is believed to be 1 of the major factors involved in the pathogenesis of PD. Different sources of PD-associated cellular oxidative stress have been suggested, but dysfunctional mitochondria are thought to be the predominant source of reactive oxygen/nitrogen species (RONS) (Jiang etal., 2016). Two chemically distinct environmental risk factors relevant to PD are the transition metal manganese and the pesticide rotenone (Nandipati and Litvan, 2016; Ratner etal., 2014). Both these molecules affect mitochondrial function and owe their toxicity at least partially to the oxidative stress resulting from impaired mitochondrial activity (Fernandes etal., 2017; Gavin etal., 1992; Gunter etal., 2009; Xiong etal., 2012). Indeed, both these compounds have been reported to inhibit mitochondrial Complex I (Degli Esposti, 1998; Galvani etal., 1995; Xiong etal., 2012; Zhang etal., 2004). The Ki values for complex I-inhibition of isolated rat brain mitochondrial by rotenone have been reported in the low nano-molar range and as low as 1 nM (Degli Esposti, 1998). Manganese concentrations of 50–70 µM, a concentration range that lies at the transition from normal to toxic manganese levels in the human brain (Bowman and Aschner, 2014; Peres etal., 2016), have been reported to cause a 50% inhibition of complex I activity in isolated rat brain mitochondria (Heron etal., 2001; Zhang etal., 2004). The commonalities of the mechanism of action of manganese and rotenone may provide clues towards an understanding of the mechanisms underlying the demise of dopamine neurons characteristic for PD pathogenesis.
Manganese, is an essential trace element and plays critical roles as a co-factor for enzymes, in the modulation of the immune system, cell adhesion, protein, and carbohydrate metabolism and is required for brain development yet excessive exposure is associated with neurotoxicity (Benedetto etal., 2009; Chen etal., 2014). Thus, excessive occupational (miners and welders), medical (total parenteral nutrition and contrast agents) or environmental (manganese containing pesticides and drinking water) exposure to manganese can lead to accumulation of this metal in the brain, especially the basal ganglia and can result in a disease termed manganism (Bouabid etal., 2016). Although manganism shares many common features with PD such as bradykinesia and rigidity, there is little resting tremor and long-term dopaminergic pharmacotherapy is mostly ineffective (Chen etal., 2015; Neal and Guilarte, 2013). Manganese exposure in animals does not produce an overt PD phenotype (nor do most genetic mouse models of familial PD), but does affect the functionality of the mesencephalic dopaminergic system (Neal and Guilarte, 2013). The observations that the prevalence of PD is increased in areas where the population is exposed to naturally or industrially elevated levels of manganese (Lucchini etal., 2007; Racette etal., 2012) and, that several genes associated with increased risk for PD (ATP13A2, parkin, DJ-1, α-synuclein, SLC39A14, SLC30A10) also regulate manganese homeostasis and/or toxicity suggest a potential relevance for manganese-gene interactions for PD pathology (Chen etal., 2014; Tuschl etal., 2016).
Manganese toxicity has been attributed to alterations in a variety of cellular functions including impairment of iron homeostasis, excitotoxicity, neurotransmitter concentrations, protein aggregation, formation of RONS, and foremost mitochondrial dysfunction (Hernandez etal., 2011; Peres etal., 2016). Mitochondria are the main intracellular storage site for manganese (Maynard and Cotzias, 1955), into which it is taken up via the mitochondrial Ca2+ uniporter (Gavin etal., 1999). In comparison to other cellular organelles, neuronal, and astrocytic mitochondria accumulate the highest relative levels of manganese after chronic exposures (Morello etal., 2008). Increased mitochondrial manganese has been reported to result in the inhibition of oxidative phosphorylation (Gavin etal., 1992), increased mitochondrial matrix Ca2+ concentration (Gavin etal., 1990) and inhibition of respiratory complexes I–IV in brain mitochondria (Galvani etal., 1995; Zhang etal., 2003) all of which will ultimately result in increased RONS formation and thus oxidative stress.
Rotenone, a commonly used pesticide, has been associated with increased risk for PD (Chin-Chan etal., 2015). It is a classical, high affinity inhibitor of mitochondrial complex 1 (Degli Esposti, 1998) and believed to be a strong RONS producer (McLennan and Degli Esposti, 2000). Systemic chronic exposure of rats to rotenone causes a selective degeneration of the substantia nigra (DA) neurons, in spite of its relatively uniform CNS distribution (Betarbet etal., 2000; Greenamyre etal., 1999; Sherer etal., 2003). This degeneration occurs at rotenone levels which inhibit mitochondrial respiratory complex I (Betarbet etal., 2000).
Based on the shared inhibition of mitochondrial complex I and sensitivity of the dopaminergic system to these compounds, we hypothesized, that after adjusting for dose of exposure, cellular outcome measures of oxidative stress would show similarities in DA neurons following rotenone or manganese poisoning. Here we compared the effects of manganese and rotenone on mesencephalic DA neurons derived from hiPSC and, contrary to our hypothesis, observed that these 2 toxicants, in spite of their shared action upon mitochondria, resulted in distinct neuronal oxidative stress signatures across 3 independent well established measures of oxidative stress (ie, DCF assay, generation of isoprostanes, and alteration in cellular glutathione [GSH] levels). In addition, we tested the hypothesis that rasagiline, a therapeutic used for PD (McCormack, 2014) protects iPSC-derived human mesencephalic DA neurons from manganese and rotenone-induced insults. Rasagiline is thought to afford its neuroprotective activity partially through its inhibition of monoamine oxygenase-B (MAO-B) but also by protecting mitochondrial integrity and the induction of antiapoptotic and neurotrophic factors (Naoi etal., 2013; Weinreb etal., 2011). We did not observe detectable neuroprotective effects of rasagiline in any of our assays except of a low-magnitude, but significant reduction in manganese evoked RONS detected by DCF assay.
MATERIALS AND METHODS
Derivation, validation and differentiation of hiPSCs
hiPSCs used for this study include 5 lines obtained from 4 healthy human subjects (CA11, CC3, CD10 and CD12 and CE6; 2 letter symbols are used to identify the subject from whom the cells were derived and the number identifies the derived hiPSC line). In brief, human dermal fibroblasts were obtained by skin biopsy after appropriate patient consent/assent under the guidelines of an approved IRB protocol (Vanderbilt No. 080369). About 6×105 cells were reprogrammed by electroporation with the CXLE plasmid vectors using the Neon Transfection System (Life Technologies, Carlsbad, California) following published methods (Okita etal., 2011). After 7 days in culture the cells were replated at a density of 2.0×103 cells/cm2 onto a SNL feeder cell layer. The next day, the medium was changed to hES medium (DMEM/F12 [Thermofisher, Waltham, Massachusetts], 20% Knockout serum replacement [Thermofisher], 2 mM Glutamax [Thermofisher], nonessential amino acids [Sigma-Aldrich, St. Louis, Missouri], penicillin/streptomycin [Mediatech Inc., Manassas, Virginia], 55 μM β-mercaptoethanol [Sigma-Aldrich], and recombinant human FGF-2 [Promega, Madison, Wisconsin]). After about 30 days the hiPSC colonies were big enough to be isolated and propagated.
For all 5 cell lines the lack of plasmid integration into the genomic DNA was demonstrated by qPCR. Karyotype analyses were performed using standard protocols with at least 20 metaphase spreads per cell line (Genetics Associates, Nashville, Tennessee). The pluripotency of the hiPSC lines was validated by Pluritest (Muller etal., 2011), and their capacity to differentiate into neural lineages (Aboud etal., 2014; Kumar etal., 2014; Neely etal., 2012; Tidball etal., 2015).
Differentiation of hiPSC to a mesencephalic DA lineage was performed exactly as described (Kriks etal., 2011) except that LDN193189 (Stemgent, Lexington, Massachusetts) was used at 0.4 µM and the cells were maintained on matrigel (BD Biosciences, San Jose, California) throughout the differentiation and toxicant exposures.
Toxicant exposures
On days 21–24 of differentiation the neurons were dissociated with accutase and then replated onto matrigel (BD Biosciences) coated 6-well or 96-well plates at 0.5×106 cells/ml and maintained in floor plate neuralization medium (Kriks etal., 2011) (Neurobasal medium [Thermofisher] supplemented with B27 [Thermofisher], glutamax [Thermofisher], BDNF [brain-derived neurotrophic factor; R&D, Minneapolis, MN, 20 ng/ml], GDNF [glial cell line-derived neurotrophic factor; R&D, 20 ng/ml], TGFβ3 [transforming growth factor β3 R&D, 1 ng/ml], ascorbic acid [Sigma, 200 µM], DAPT [Tocris, Bristol, UK, 10 µM], dibutyryl cAMP [Sigma, 0.5 mM], and ROCK-inhibitor [Tocris, 10 µM]). The next day the neurons were transferred into the same medium, but lacking ROCK-inhibitor. Between days 25–27, the neurons were exposed to rotenone (Sigma, No. R8875, purity HPLC ≥ 95%) prepared fresh for every experiment as 1000-fold concentrated stock solutions in DMSO ensuring that the final DMSO concentration was 0.1% in all exposure conditions, or manganese (Fisher Scientific, No. M87-500, MnCl2, purity: assay percentage range 98%–101%; Fe: 5 ppm max; Zinc: 0.005% max; heavy metals [as Pb]: 5 ppm max) and/or rasagiline (TEVA pharmaceuticals, Netanya, Israel, heavy metals ≤0.002% [USP <231> METHOD II and EP <2.4.8> METHOD C, these methods are limit tests and do not show real content of metals <0.002% level]) which were both prepared as 1000-fold stock solutions in H2O. The rasagiline stock solution was prepared fresh for every experiment. Exposure to 1 mM H2O2 (Sigma, No. H1009) (diluted from a 30% [wt/wt] stock into H2O just before use) was used as a positive control in the DCF assay (see below). Exposure to the toxicants/rasagiline was either in HBSS containing 25 mM glucose (for the DCF assay, see below) or floor plate neuralization medium (see above) lacking ascorbic acid.
Immunofluorescence and high content imaging
For all immunofluorescence analysis DA neurons were plated into 96-well plates (Greiner Bio-One, Monroe, North Carolina, µclear) and immunofluorescence performed as described (Neely etal., 2012). Briefly, the cells were fixed in PBS containing 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PZ) for 30 min at room temperature, permeabilized with 0.2% Triton X-100 for 20 min at room temperature and then incubated in PBS containing 5% donkey serum (Jackson ImmunoResearch, West Grove, Pennsylvania) and 0.05% Triton X-100 for 2 h at room temperature or overnight at 4 °C. The following primary antibodies were used: mouse anti-Foxa2 antibody (BD Pharmingen, San Jose, California, 561580, 1:100), rabbit anti-Lmx1A (Millipore, Billerica, Massachusetts, AB10533, 1:1000), mouse anti-b3-tubulin antibody (Thermo Scientific, Rockfoed, Illinois, MA1-19187, 1:500), rabbit anti-tyrosine hydroxylase (TH) (Pel-Freez, Rogers, Arkansas; P40101, 1:500) and sheep anti-TH (Pel-Freez; P60101, 1:250). Secondary antibodies conjugated to DyLight 488 (1:800), DyLight 549 (1:800) and Dylight 649 (1:800) were purchased from Jackson ImmunoResearch. Images were obtained with a Zeiss ObserverZ1 microscope and AxioVs40 software (version 4.7.2). For high content imaging images were acquired using a Molecular Device’s ImageXpress Micro XL system and MetaXpress software available at the Vanderbilt High-throughput Screening Core Facility, an institutionally supported core with assistance provided by Joshua Bauer. In each experiment, approximately 32 000 neurons per subject (approximately 1000 cells per image for 16 images per well in duplicate cultures) were analyzed. The absolute neurite lengths measured were subject to considerable variability, which is likely a result of the different time-line of neural differentiation and rate of neurite extension between different hiPSC lines. Thus, we observed a 2- to 3-fold difference in neurite length between different cell lines assessed within the same experiment and an up to a 3.5-fold difference in neurite length for the same cell line assessed in different experiments. The experiment to experiment variability was partially due to the fact that the interval between the replating of the neurons and neurite length assessment varied by about 24 h between experiments. Therefore, in order to make meaningful determinations of the response of the neurites to toxicants in the different experiments and different cell lines we normalized the neurite length in each experiment to the length measured in vehicle-treated control neurons.
Quantitative reverse transcription PCR (RT-qPCR)
For validation of hiPSC-lines and assessment of neural differentiation, total RNA was prepared using the RNeasy kit (Qiagen, Valencia, California) according to the manufacturer’s instructions. Isolated RNA was reverse transcribed into cDNA on a Mycycler Thermal Cycler (Bio-Rad, Hercules, California) using SuperScriptIII First-Strand Synthesis System with oligo(dT)20 (ThermoFisher) according to the protocol provided by manufacturer. Primer sequences used for RT-qPCR are provided in Supplementary Table 1. The expression of housekeeping genes GAPDH, PGK1, UBC, and ACTIN were assessed and ACTIN and UBC expression found to be the most consistent; all mRNA signals were normalized to the expression of ACTIN mRNA. RT-qPCR was performed using a Power SYBR Green Master Mix (Applied Biosystems, Carlsbad, California) on an ABI 7900HT fast real-time PCR detection system (Applied Biosystems) in the Vanderbilt VANTAGE Core which is supported in part by CTSA Grant (5UL1 RR024975-03), the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956).
Cell viability assay
To assess cell viability, we used the Cell Titer Blue assay (Promega, kit G8081) following the protocol provided by the manufacturer. This assay uses the indicator dye resazurin that is reduced in viable cells to the highly fluorescent product resorufin. Fluorescence was measured using a POLARstar Omega plate reader (BMG LABTECH, Cary, North Carolina) or a Beckman coulter DTX 880 multimode plate reader (Beckman Coulter, Brea, California). This assay is based on readings of fluorescence intensity in a plate reader and thus there are day to day variabilities resulting from type of plate reader used, and cell line dependent variabilities resulting from slight differences in cell densities and dye turnover. In order to present the data in a cohesive way and to accommodate the biological and technical variabilities we encountered all data were normalized to the vehicle treated control values.
Chloromethyl 2′, 7′-dichlorodihydrofluorescein diacetate assay
Neurons that had been replated into black 96-well plates (Corning, Corning, New York No. 3603) were washed twice with Hanks Balanced Salt Solution containing Ca2+ and Mg2+ (HBSS), before they were loaded with 2 µM CM-H2DCFDA (ThermoFisher No. C6827) in HBSS containing 25 mM glucose for 25 min at room temperature in the dark. The cells were then washed again 2 times with HBSS before exposure. All chemicals used were diluted in HBSS containing 25 mM glucose, H2O2 (Sigma) at 1 mM was used as a positive control. Fluorescence intensities were measured using a Beckman coulter DTX 880 multimode (Beckman Coulter) or a POLARstar Omega plate reader (BMG LABTECH) plate reader starting just before dye-loading (background) and then immediately after adding the toxicants (time 0 min) and then every 20 min for 4 h. This assay also is based on readings of fluorescence intensity in a plate reader and thus there are cell line-specific variabilities and day to day variabilities resulting from type of plate reader used, slight differences in cell densities, dye loading efficiencies. Thus, for the DCF assays the data are expressed (after subtracting the background fluorescence) as a fold increase relative to vehicle treated cells. Cell viability was assessed at the 4-h time point using the Cell Titer Blue assay described earlier.
GSH assay
Neurons were plated into black 96-well plates and cellular GSH levels assessed using the GSH-Glo Glutathione Assay kit (Promega, kit V6912) according to the manufacturer’s protocol with the following modifications. The cells were lysed with 120 µl of 1× passive lysis buffer (Promega, No. E1941). After a brief mixing 40 µl of the cell lysates were transferred into empty wells and 40 µl of 2× GSH-Glo reagent added and the plates mixed and then incubated for 30 min in the dark at room temperature. Thereafter, 80 µl of Luciferin Detection reagent was added, the plates were incubated for 15 min at room temperature in the dark and then the luminescent signal was quantified using a POLARstar Omega plate reader. For each assay, we performed a standard curve with the GSH standards provided with the kit by the manufacturer; this standard curve was used to ensure that the measurements from the cell lysates were performed within the linear range of the assay and as a quality control of the kit components from day to day.
Isoprostane assay
Cells that had been replated into 6-well plates at 1×106 cells per well were exposed to manganese or rotenone, washed once with HBSS, then scraped off the plates, centrifugated at 200×g for 3 min, cell pellets flash frozen in liquid nitrogen and stored at −80 °C. Total F2-Isoprostanes (F2-IsoPs) from cell pellets containing 1×106 cells were isolated and quantified using gas chromatography/mass spectrometry with selective ion monitoring (GC/MS) by the Vanderbilt University Eicosanoid Core Laboratory as described in detail previously (Milatovic etal., 2011; Milne etal., 2013). In brief, cell pellets were resuspended in 3 ml of Folch solution, sonicated, lipids extracted, and dried. The dried lipids were resuspended in 0.5 ml of methanol containing 0.005% butylated hydroxytoluene, vortexed, and then subjected to chemical saponification using 15% KOH to hydrolyze bound F2-IsoPs. The cell lysates were adjusted to pH 3, and 1 ng of 4H2-labeled 15-F2a-IsoP d4-15-F2t-IsoP (8-iso-PGF2α) internal standard (Cayman Chemical; Cat. No. 316351) added as internal standard. F2-IsoPs were then purified by sequential C18- and silica Sep-Pak extraction, derivatized to the corresponding pentafluorobenzyl esters, and the derivatives separated by thin layer chromatography. The purified pentafluorobenzyl ester derivatives were further derivatized to trimethylsilyl ether derivatives, and then quantified via GC/MS. Quantification of F2-IsoPsis achieved with a precision of ±6% and an accuracy of 96%. The lower limit of quantitation is 0.002 ng/ml. The content of all our samples were well within the detection limit.
Statistical analysis
DCF assay data were analyzed with 2-way ANOVA for time and treatment. Comparisons of relative changes from normalized data (cell viability, GSH level, isoprostane quantification, and neurite length) were performed by testing for nonoverlap of the 95% confidence interval with the control value (normalized = 1). To assess the effects of rasagiline, comparisons of samples treated ± rasagiline were performed by a 1-way ANOVA followed by SIDAK multiple comparison tests using GraphPad Prism software (version 5.0b, La Jolla, California).
RESULTS
Differentiation of hiPSC into Mesencephalic DA Neurons
hiPSCs were differentiated into the mesencephalic DA lineage for 21–27 days. The differentiation was performed in 2 stages, a dual SMAD inhibition combined with ventral midbrain patterning resulting in floor plate cells (mesencephalic neural precursors) followed by neuronal differentiation and maturation resulting in β3-tubulin, TH, and Lmx1A expressing dopaminergic neurons (Chambers etal., 2009; Kriks etal., 2011). The efficiency of differentiation was assessed by quantification of the expression of lineage-specific markers by immunofluoresence and qPCR (Figure 1). At the final time point of differentiation β3-tubulin was expressed in 88 ± 2.6%, Lmx1A in 89.1 ± 2.1% and TH in 57.8 ± 2.9% of the cells as assessed by high content imaging (Figs. 1A and B). As observed by others, Foxa2 expression was seen early peaking at day 11 and was maintained at a slightly lower level until day 25 (Figure 1C;Kumar etal., 2014). Levels for β3-tubulin, a marker for postmitotic neurons and Lmx1A, Pitx3, AADC, and TH, all markers for midbrain DA neurons were highest on day 25 (Figure 1C) (Kriks etal., 2011; Kumar etal., 2014; Ryan etal., 2013).
Figure 1.

Differentiation of hiPSCs into mesencephalic DA neurons. A, The expression of β3-tubulin (red), tyrosine hydroxylase (TH, green) and Lmx1A (purple) in cultures of dopamine neurons differentiated for 21–27 days was assessed by immunocytochemistry, all cultures were counterstained with a nuclear stain, Hoechst (light blue) (shown here is cell line CA11, Scale bar = 50 µm). B, β3-tubulin-, Lmx1A- and TH-positive cells were quantified by high content imaging. Quantification for β3-tubulin and TH expression was from 18, and Lmx1A from 4 individual experiments including cell lines derived from 4 different control subjects. (shown are means ± SEM). C, Expression of DA neuron markers were also quantified by qRT-PCR, cell lines from 3 control subjects were used (shown are means ± STD, n= 8).
Manganese and Rotenone-Induced Oxidative Stress
We measured manganese and rotenone-induced oxidative stress by 3 independent measures within the same exposure time frame. None of the tested rotenone, manganese, and/or rasagiline exposure paradigms described in the following experiments resulted in loss of neuronal viability as measured by the Cell Titer Blue assay (Figs. 2A and B). In order to present the data in a cohesive way and to accommodate the biological and technical variabilities we encountered in this assay all data were normalized to the vehicle treated control values. Examples of the variability encountered between cell lines (within 1 experiment) and day to day variability are provided in Supplementary Figures 1A and B, respectively.
Figure 2.

Manganese or rotenone exposures used in this study do not result in loss of neuronal viability. A, Except for H2O2 (1 mM), none of the compounds applied to the neurons in the DCF assay caused loss of neuronal viability under the conditions and in time frame used in the assay. Mean and 95% confidence intervals are shown, n= 6–24. B, The 24-h exposures to manganese or rotenone at the concentrations used in any of the experiments described below do not result in loss of neuronal viability. Means and 95% confidence intervals are shown, n = 3.
In a first set of experiments DA neurons were preloaded with DCFH-DA and then exposed to increasing concentrations of manganese or rotenone. The oxidation of DCFH-DA to the fluorescent product DCF was measured every 20 min within a 4-h time frame. Manganese caused a concentration- and time-dependent increase in cellular RONS as measured by the DCF assay in DA neurons differentiated from 4 different hiPSC lines from 4 different control subjects. Finteraction (72, 728) = 3.278, P < .0001; Ftime (12, 728) = 63.71, P < .0001; FMn conc. (6, 728) = 123.2; P < .0001; mean and SEM are shown, n = 9 (Figure 3A). Rasagiline on its own did not affect DCF fluorescence, but caused a small but significant inhibition of the manganese-induced RONS formation Fras conc. (5, 858) = 494.3 (P < .0001) in DA neurons (Figure 3B). Turkey’s multiple comparisons test indicates that the degree of inhibition was not significantly different for the 3 concentrations of rasagiline tested (Figure 3B). In contrast to manganese, rotenone did not induce a significant RONS production as measured by the DCF assay (Figure 3C). Bonferroni multiple comparisons test indicated no significant difference in RONS formation between vehicle and rotenone treated cultures, whereas H2O2 used as a positive control did increase RONS formation significantly (P < .0001). The manganese-induced RONS production was specific to neurons, because no increase in a DCF signal was observed in hiPSCs exposed to manganese (Figure 3D), thus Bonferroni multiple comparisons test indicated no significant difference in RONS formation in control and manganese treated hiPSC cells, whereas H2O2, the positive control increased the DCF signal significantly (P < .0001). As detailed in the Methods section, all data obtained are expressed as a fold increase over vehicle-treated control cultures in order to accommodate the variabilities we observed. Examples of the variabilities we encountered with the DCF assay between cell lines within 1 experiment and from day to day (between different experiments) are provided in Supplementary Figures 2A and B.
Figure 3.

Manganese, but not rotenone leads to immediate RONS production as measured by the DCF assay, inhibition by rasagiline. A, Manganese causes a concentration and time dependent increase in cellular RONS as measured by the DCF assay in DA neurons differentiated from 4 different hiPSC lines from 4 different control subjects. Finteraction (72, 728) = 3.278; Ftime (12, 728) = 63.71; FMn conc. (6, 728) = 123.2); P < .0001; mean and SEM are shown, n = 9. B, Rasagiline significantly inhibits manganese-induced RONS formation F (5, 858) = 494.3 (P < .001) in DA neurons differentiated from 5 different hiPSC lines from 4 different control subjects. The degree of inhibition was not significantly different for the 3 concentrations of rasagiline tested. Mean and SEM are shown, n = 12. C, Rotenone does not cause a significant formation of cellular RONS over time F (12, 455) = 1.579 (P = .0943) in DA neurons differentiated from 3 different hiPSC lines from 3 different control subjects. Bonferroni multiple comparisons test indicated no significant difference in RONS formation among any of the treatment conditions (except for H2O2). Mean and SEM are shown, n = 6. D, Manganese does not increase cellular RONS in hiPSCs. A 2-way ANOVA analysis of this data indicated a time-dependent F (12, 1365) = 5.959 and treatment-dependent F (6, 1365) = 300.7 significant increase in RONS formation by H2O2. Bonferroni multiple comparisons test indicated no significant difference in RONS formation in control cells and cells treated with different manganese concentrations. Mean and SEM are shown, n = 16.
GSH is an important antioxidant protecting cells from cellular damage caused by free radicals, peroxides, and metals, and changes in intracellular levels of this tripeptide is frequently used as an indicator of the cellular redox state (Pompella etal., 2003). We thus assessed manganese and rotenone-induced oxidative stress by determining cellular GSH (reduced) content over a time course of 2.5–24 h (Figure 4). A 2.5-h exposure to either toxicant did not result in any significant changes in cellular GSH content (Figure 4A). The 5-h exposures to manganese or rotenone resulted in similar and significant decreases of cellular GSH levels (Figure 4B). Exposures of 24 h lead to a further loss of cellular GSH in rotenone exposed neurons, whereas, interestingly, in cells exposed to manganese for 24 h cellular GHS levels returned to control values (Figure 4C). Rasagiline on its own caused a slight, but significant decrease in GSH at 1 µM after a 2.5-h exposure and at 10 µM caused a small but significant increase at 5 and 24 h. An ANOVA followed by a Sidak multiple comparisons revealed no significant differences in cellular GSH levels between cultures treated with toxicants in the presence or absence of rasagiline (ANOVA followed by Sidak Multiple comparisons: FMn, 2.5h (2, 20) = 0.11, FRot 1µM, 2.5h (2, 20) = 0.796; FRot 10µM, 2.5h (2, 20) = 1.733; FMn, 5h (2, 21) = 0.247, FRot 1µM, 5h (2, 20) = 2.039; FRot 10µM, 5h (2, 19) = 1.731; FMn, 24h (2, 13) = 1.721; FRot 1µM, 24h (2, 11) = 5.833; FRot 10µM, 24h (2, 13) = 0.950) (Figs. 4B and C). In addition, the effects of rotenone at 1 and 10 µM were not statistically different from each other. The variances for the GSH values at the 5-h time point were higher than at either 2.5 or the 24-h time point; we interpret this as being possibly the result of the 5-h time point representing a critical turning point in the cellular response, because in some experiments we observed significantly decreased GSH levels, whereas in other experiments GSH levels were not significantly different at the 5-h time point.
Figure 4.

Differential effects of Rotenone and Manganese on cellular GSH content. A, A 2.5-h exposure to either rotenone or manganese does not induce a significant change in cellular GSH content (n = 3–8). B, A 5-h exposure to manganese or rotenone caused a small but significant decrease in cellular GSH (P < .05, n = 3–11). C, A 24-h exposure results in a significant loss of cellular GSH in rotenone treated cells (P < .05), whereas cultures exposed to manganese show GSH levels return to control value (n = 4–6). Shown are mean ± 95% confidence intervals. Rasagiline on its own caused a slight, but significant decrease in GSH at 1 µM after a 2.5-h exposure and at 10 µM a small but significant increase at 5 and 24 h; however, rasagiline did not affect the GSH levels in neurons treated with manganese or rotenone under any of the exposure paradigms (ANOVA followed by Sidak multiple comparisons test).
Isoprostanes are a unique series of prostaglandin-like molecules formed in a nonenzymatic free radical-initiated peroxidation of arachidonic acid and are thus a measure of lipid peroxidation. Their quantification has become a “gold standard” measure of oxidative stress (Milne etal., 2011). To determine yet another measure of oxidative stress we quantified cellular isoprostane levels in dopamine neurons exposed to manganese or rotenone. None of the manganese treatments caused any changes in cellular isoprostane levels, rotenone exposure for 5 or 24 h led to a concentration dependent increase in cellular isoprostane levels (Figure 5). Rasagiline did not protect the cells from this rotenone-induced increase in isoprostane levels nor did it affect isoprostane levels on its own (Figure 5).
Figure 5.
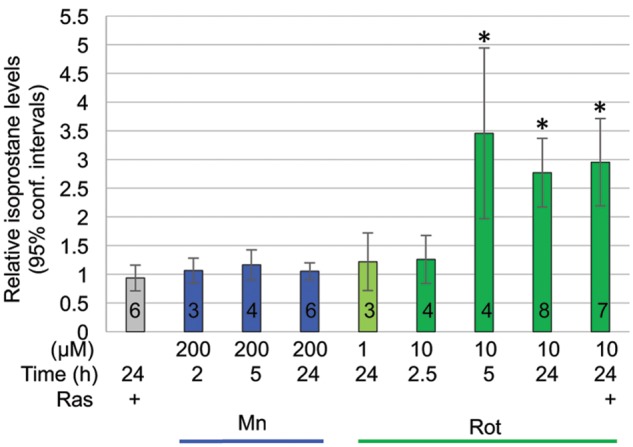
Isoprostane levels in days 26–29 DA neurons are elevated by rotenone, but not manganese. Rotenone, but not manganese caused a concentration and time dependent increase in cellular isoprostane levels. Rasagiline did not inhibit rotenone-induced isoprostane formation. Three cell lines from 3 different control subjects were assessed. Mean and 95% confidence intervals are shown; number of experiments is indicated by the black numbers in the according bars.
Effect of Manganese and Rotenone on Neurite Morphology
Changes in neurite morphology are a very sensitive measure of neurotoxicity (Krug etal., 2013). Thus, we assessed manganese and rotenone-induced changes on β3-tubulin and TH-positive neurites. A 2-h exposure to rotenone, but not manganese causes a significant decrease in β3-tubulin and TH-positive neurite length (Figs. 6A and B). Although Rasagiline (10 µM) appears to provide a minimal protection from this loss of β3-tubulin- and TH-positive neurites the observed trend was not statistically significant (ANOVA with Sidak’s multiple comparisons test, F (7, 25) = 4.658, P < .002 and TH F (7, 25) = 6.208, P < .0003).
Figure 6.

Rotenone, but not manganese causes loss of neurites. A and B, A 2-h exposure of days 26–27 DA neurons to rotenone (1 or 10 µM), but not manganese (100 or 200 µM) causes a significant loss of β3-tubulin and TH containing neurites. Rasagiline (+ = 10 µM) does not protect from rotenone-induced loss of β3-tubulin or TH-positive neurites (1-way ANOVA with Sidak’s multiple comparisons test). Shown are means and 95% confidence intervals, n = 3–6. C and D, A 5-h exposure of days 26–27 DA neurons to rotenone (1 or 10 µM), but not manganese (200 µM) causes a significant loss of β3-tubulin and TH containing neurites. Rasagiline (+ = 10 µM) does not protect from rotenone-induced loss of β3-tubulin or TH-positive neurites (1-way ANOVA with Sidak’s multiple comparisons test). Shown are means and 95% confidence intervals, n = 3. E and F, A 24-h exposure of days 26–27 DA neurons to rotenone (0.1–10 µM) causes a significant concentration-dependent loss of β3-tubulin-containing neurites. Rasagiline (+ = 1 µM, ++ = 10 µM) does not protect from rotenone-induced loss of β3-tubulin or TH-positive neurites (1-way ANOVA with Sidak’s multiple comparisons test). Manganese exposure for 24 h leads to a minimal loss (5%) of β3-tubulin-positive neurites at 100 µM, but not 50 or 200 µM, and a loss of TH-neurites at 100 and 200 µM. Shown are means and 95% confidence intervals, nMn = 3 and nRot, Ras = 5.
A 5-h exposure to manganese did not result in a loss of neurite length; however, a 5-h exposure to rotenone led to a loss of β3-tubulin- (at 1 and 10 µM) and of TH-neurites (at 10 µM) (Figs. 6C and D). Rasagiline did not provide protection from this rotenone-induced neurite loss (ANOVA followed by Sidak multiple comparison test, Fβ3-tubulin (6, 14) = 10.37, P < .0002; FΤΗ (6, 14) = 3.571, P < .03)
The rotenone-induced β3-tubulin and TH-neurite loss was increased in cultures exposed for 24 h. In cultures treated with manganese a minimal (5%) β3-tubulin loss was observed at 100 µM, but not 200 µM, whereas an equally small (5%) TH-neurite loss was observed at both 100 and 200 µM (Figs. 6E and F). Rasagiline did not protect the neurons from rotenone-induced loss of neurite length (ANOVA followed by Sidak multiple comparison test Fβ3-tubulin (9, 34) = 22.46, P < .0001 and FΤΗ (9, 34) = 41.90, P < .0001).
DISCUSSION
Both manganese and rotenone induce oxidative stress mainly by interfering with mitochondrial function (Degli Esposti, 1998; Fernandes etal., 2017; Gavin etal., 1992; Gunter etal., 2009; Hernandez etal., 2011; Xiong etal., 2012; Zhang etal., 2004,Xiong etal., 2012; Fernandes etal., 2017). We thus hypothesized that exposure of DA neurons to these 2 toxicants would result in a similar pattern of cellular oxidative stress phenotypes.
The human DA neurons tested here showed no loss of viability after exposures of up to 200 µM manganese or 10 µM rotenone for 24 h; thus they are relatively more resistant to manganese than other neurons or neuronal cell lines (Stanwood etal., 2009; Stredrick etal., 2004; Yoon etal., 2011), a characteristic that we have previously observed in hiPSC-derived neural precursor cells (Aboud etal., 2012, 2014). We are not aware of any publications assessing manganese toxicity on postmitotic mesencephalic DA neurons differentiated from hiPSC. The reported sensitivity of hiPSC-derived DA neurons to rotenone varies. Although 1 group reported mitochondrial swelling but no cell death after a 24-h exposure to 100 µM rotenone (Shaltouki etal., 2015), several groups reported loss of hiPSC-derived DA neurons at rotenone concentrations in the range of 50–200 nM (Peng etal., 2013; Reinhardt etal., 2013; Schondorf etal., 2014; Zagoura etal., 2017). Strikingly, in all 4 of these studies the rotenone exposure was performed in the absence, or a very low level of GDNF (1 ng/ml). The protective effect of GDNF with respect to rotenone exposure in hiPSC-derived DA neurons was directly examined by Peng etal. (2013) who reported a 2- and 4-fold decrease in rotenone-induced dopamine neuronal death in the presence of 20 or 40 ng/ml of GDNF, respectively. In all our studies (except the DCF assay) GDNF was present at 20 ng/ml. In addition to the medium components, the substrate onto which the neurons are plated could also affect the response of neurons to toxicants. hiPSC-derived neurons are typically plated onto dishes coated with solutions containing laminin (1–10 µg/ml) and/or fibronectin (2–20 µg/ml) or matrigel (100 µg/ml). The different protein components and protein concentrations (and thus thickness) of these substrates could differentially bind and/or adsorb a toxicant in question and thus differentially affect the concentration of the “free” toxicant in the medium. Another variable is exposure time. We observed significant neurite loss after acute 1 µM rotenone exposures of 24 h. Fang etal., observed decreased neurite length after a 10-nM rotenone exposure but lasting 23 days, a developmental chronic exposure paradigm, during which the lipophilic rotenone could accumulate in the neurons and is thus very different from the acute 2- to 24-h exposures we used here (Fang etal., 2016).
In addition to culture and exposure conditions the characteristics of the DA neurons likely also determine the degree of sensitivity to an oxidant stressor. Thus, Schoendorf etal. (2014) observed rotenone-induced death of hiPSC-derived DA neurons only in cell lines carrying PD-relevant mutations, but not the control cell lines, and Ryan etal., found rotenone-induced mitochondrial oxidative stress (mitoSOX signal) only in hiPSC-derived DA neurons carrying PD-relevant mutations, but not in their isogenic control counter parts (Ryan etal., 2015). In addition to the effect of genetic mutations, the maturity and thus the intracellular levels of the highly oxidizable neurotransmitter DA may play a role (Chen etal., 2016; Farina etal., 2013). Primary mesencephalic rodent neurons and DA neurons derived from mouse ES cells (Xiong etal., 2012; Yamashita etal., 2006) have been reported to succumb to rotenone concentrations in the range of 0.5–30 nM and appear thus generally more sensitive than their human counter parts. However, as discussed earlier, culture conditions such as GDNF (which is not typically present in the medium used for rodent neuronal cultures) can greatly affect the Sensitivity of DA neurons to rotenone making it difficult to establish species-dependent differences in toxicant sensitivities from invitro neuronal cultures.
Here we compare manganese- and rotenone-induced oxidative stress by assessing 4 physiologically different endpoint measures. Manganese, but not rotenone caused an immediate RONS production as measured by the DCF assay, consistent with a recent report of increased H2O2 production in Mn exposed SHSY5Y human neuroblastoma cells (Fernandes etal., 2017). As in our DA neurons, in PC12 cells rotenone did not cause an increase in DCF-detectable cellular RONS within the first 4 h of exposure (Seyfried etal., 2000). Differential effects of manganese- and rotenone-induced DCFH oxidation was also observed in long-term exposures (24 h) of PC12 cells where rotenone-, but not manganese exposure resulted in elevated RONS (Hirata etal., 2006). In contrast, in hiPSC-derived neural precursor cells long-term (24 h) manganese exposure resulted in significant RONS production as determined by measuring DCF signals (Aboud etal., 2012). These observations suggest that in addition to toxicant exposure conditions (time), cellular DCFH oxidation upon toxicant exposure is likely also cell type specific (Bornhorst etal., 2013). Further supporting this conclusion are our observations that the amount of manganese accumulated by cells can depend on the developmental stage (Kumar etal., 2014) and that manganese exposure of undifferentiated hiPSCs (the same lines that we differentiated into neurons here) does not result in DCFH oxidation (Figure 3D).
Our observation of a lack of a DCF signal in hiPSC cells treated with manganese under the exact same conditions as the neurons were exposed and the lack of a signal in the rotenone treated neurons suggests that contaminating metals potentially present in our manganese and rotenone stock solutions do not contribute a detectable intracellular DCF signal. Importantly however, we cannot exclude the possibility that endogenous transition metals (such as Fe and/or Cu) affect the manganese-induced DFC signals measured in the neurons. Assessment of the effects of transition metals (iron) on manganese-induced ROS formation in mitochondrial-synaptosomal fractions assessed by the DCF assay demonstrate that different transition metals within the same sample can indeed affect each other’s redox state (HaMai and Bondy, 2004a,b; HaMai etal., 2001).
The lack of immediate RONS production during a 4-h rotenone exposure of DA neurons was somewhat surprising. However, similar observations have been reported for other cell types and isolated rat brain mitochondria (Esposti etal., 1999; Meinel etal., 2015; Seyfried etal., 2000). The difference between manganese- and rotenone-elicited RONS production could be due to the fact that rotenone rather specifically inhibits complex I, whereas manganese has been shown to also affect complexes II–IV and other mitochondrial enzymes (Degli Esposti, 1998; Gunter etal., 2010; Zhang etal., 2003, 2004). RONS production reported in cells exposed to rotenone for extended times (≥24 h) could be the result of secondary effects such as loss of stability of mitochondrial membrane potential, energy depletion, and cellular signaling (Pal etal., 2014; Radad etal., 2006).
Cellular GSH levels of hiPSC-derived DA neurons were also differentially affected by manganese and rotenone. Although short-term exposures (2.5 h) had no effect on GSH levels in cells treated with manganese or rotenone, 5-h exposures caused a similarly small and significant decrease in cellular GSH in manganese and rotenone exposed neurons. However, after a 24-h exposure GSH levels had recovered to control levels in manganese treated cells, but further decreased in rotenone exposed neurons. An initial decline, followed by a recovery of GSH levels has also been reported for manganese-exposed PC12 cells (Desole etal., 1997) and normal GSH levels were observed in primary cultures of rodent neurons after long-term (5 days) manganese exposures (Hernandez etal., 2011). The rotenone-induced persistent loss of GSH we observed in our study is similar to observations made in PC12 cells and human neuroblastoma cells (Seyfried etal., 2000; Sherer etal., 2002). These observations suggest that human neurons are able to, over time (here within 24 h), adapt to elevated manganese at least with regard to restoration of intracellular GSH levels, whereas in neurons treated with rotenone, this appears not to be the case, further suggesting that the oxidative stress pathways and their downstream targets are likely different for these 2 compounds.
F2-isoprostane levels, a measure of lipid peroxidation, were also differentially affected by manganese and rotenone. None of the manganese exposure paradigms tested resulted in a change of F2-isoprostanes in our hiPSC-derived DA neurons, whereas incubation with rotenone caused a concentration- and time-dependent increase in these indicators of lipid peroxidation. Elevated levels of F2-isoprostanes have been observed in brains of patients with neurodegenerative disease (Montine etal., 2004), but to our knowledge our study is the first to assess F2-isoprostane levels in hiPSC-derived neurons and to report distinctly different levels of these indicators of lipid peroxidation upon exposure to these 2 different PD-relevant oxidative stressors.
Changes in cell morphology, and neurite extension in particular, are key indicators of neuronal health (Krug etal., 2013). While manganese had only a very slight effect after a 24-h exposure, rotenone induced a time, and concentration-dependent decrease in neurite length. Rotenone-induced neurite loss occurred after exposure times and at rotenone concentrations, for which we did not see indication of oxidative stress as measured by DCF, GSH or F2-isoprostane levels suggesting that rotenone-induced neurite retraction occurs independent of oxidative stress. In addition, to its well characterized inhibition of mitochondrial complex I activity, rotenone binds directly to tubulin and causes microtubule depolymerization (Jiang etal., 2006b; Ren etal., 2005). The earliest pathological feature of invivo rotenone exposed rodents is the loss of distal DA processes (Betarbet etal., 2000; Jiang etal., 2006a). Rotenone-induced neurite loss in rodent mesencephalic primary cultures has been shown to be a result of microtubule depolymerization, and both, loss of neurites and microtubules, were ameliorated by BDNF or GDNF (Jiang etal., 2006b; Ren etal., 2005). The DA neurons in our study were exposed to rotenone in the presence of BDNF and GDNF at concentrations similar to the ones reported to be protective in cultured rodent neurons, an observation that suggests that either these neurotrophic factors do not serve the same function, or higher concentrations are required for the protection of human neurons. Neurite loss caused by a direct interaction between rotenone and tubulin would also explain the similar sensitivity of β3-tubulin- and TH-positive neurites observed in our study.
Rasagiline, a therapeutic used in the clinic for PD provided a small, but significant protection against the initial manganese-induced RONS formation as measured by the DCF assay. Rasagiline did not provide protection against rotenone- or manganese-induced loss of cellular GSH, formation of isoprostanes or loss of neurites. Rasagiline, at concentrations similar to the ones we used have been reported to afford a moderate protection in hiPSC-derived DA neurons and PC12 cells from rotenone-induced loss of neuronal viability as measured by MTT assay and/or immunostaining (Milusheva etal., 2010; Peng etal., 2013).
We show here that manganese and rotenone, which both are believed to interfere with mitochondrial function, particularly inhibition of mitochondrial complex I, exert distinctive cellular oxidative stress phenotypes on human dopaminergic neurons as determined by 3 methodologically and mechanistically different measures. Although rotenone is viewed as a classical complex I inhibitor (but also interferes with microtubules see the Discussion section above), manganese has been reported to affect several other cellular functions. Manganese 2+/3+ have very different valence states and unusual ionic properties when compared with other transition metals such as iron 2+/3+ and Copper 1+/2+. Manganese is known to affect the redox balance of other transition metals such as iron and copper, and indirectly modulates redox reactions driven by these metals (HaMai and Bondy, 2004a,b; HaMai etal., 2001). Thus, variations in intracellular manganese concentrations could globally impact the redox environment of the cell, indeed manganese has been referred to as the “hub within the global redox network” (Smith etal., 2017). Such an effect on transition metals by rotenone is not likely. Excess manganese can also dysregulate cellular iron regulation thereby increasing the intracellular labile iron pool (Kwik-Uribe etal., 2003). In addition to its effects on other cellular metals, manganese can cause oxidative stress in dopaminergic neurons by catalyzing the autoxidation of dopamine, a reaction which produces cytotoxic quinones and free radicals (Chen etal., 2016; Farina etal., 2013). The accumulation of manganese has also been postulated to lead to an exhaustion of cellular antioxidant defenses, result in oxidative damage of proteins, polyunsaturated fatty acids and nuclear and mitochondrial DNA (HaMai and Bondy, 2004a; Jellinger, 2013). Lastly, RONS resulting from excess manganese could interfere with redox-sensitive signaling pathways (HaMai and Bondy, 2004a; Jellinger, 2013).
Thus, albeit manganese and rotenone share an inhibition of mitochondrial complex I, some/or all of the above-outlined mechanisms by which manganese can cause oxidative stress could contribute to the different oxidative stress signatures we observed for rotenone and manganese. Our studies thus support the hypothesis that oxidative stress manifests in different ways depending on the stressor of choice, exposure condition and cell types assessed. As such, the determination of the capacity and potency of a compound to induce oxidative stress in a cell type of choice is more accurately described using a combination of oxidative stress measures, to define its cellular oxidative stress signature. This point is exemplified by the failure of the “gold-standard” oxidative stress measure, isoprostane analysis (Milatovic etal., 2011; Milne etal., 2011; Yin, 2008), to detect manganese-induced oxidative stress which otherwise manifested itself in alterations in cellular GSH levels and RONS production quantified by the DCF assay.
Interestingly, our observations in early postmitotic human DA neurons invitro mirror the dissimilarities of DA neuron responses to manganese and rotenone invivo. Manganese has been shown to affect dopamine release in some invivo models but it does not appear to affect DA synaptic morphology or neurite degeneration, and loss of nigral DA neurons is not a common feature (Guilarte, 2010; Kwakye etal., 2015). Chronic low dose exposure of rodents to rotenone on the contrary has an early and severe effect on mesencephalic DA nerve terminals and axons followed by a later selective loss of TH-positive cell bodies in the substantia nigra (Betarbet etal., 2000; Greenamyre etal., 1999; Sherer etal., 2003). The most sensitive rotenone-induced neuronal response (neurite retraction) we reported here is thus similar to what has been reported in mouse models of PD and is observed in brains of PD patients, where the loss of dopamine is significantly greater in the striatum than in the substantia nigra and DA axons and their terminals appear to be the earliest and principal site of PD pathology (Cheng etal., 2010; Hornykiewicz, 1966).
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
Research Award by TEVA Pharmaceuticals (to A.B.B.) and NIH/NIEHS grant RO1 ES010563 (to A.B.B. and M.A.).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank TEVA Pharmaceuticals for kindly providing rasagiline and a research grant, which partially funded this work. The Vanderbilt University Eicosanoid Core Laboratory is supported by the Center in Molecular Toxicology (NIH P30 ES000267) and we thank Ginger Milne for her assistance in performing the F2-isoprostane analysis. We thank Joshua Bauer for his assistance in performing the high content imaging at the Vanderbilt High-throughput Screening Core Facility. A portion of the funds for this project were provided from a grant by TEVA pharmaceuticals to A.B. Bowman. Further, A.B. Bowman has previously served on a scientific advisory board meeting for TEVA Pharmaceuticals. TEVA also provided, without charge, rasagiline for this study. Finally, TEVA Pharmaceuticals, by prior agreement with the authors in the context of funding a portion of this work, has approved submission of this work for publication to ensure no compromise of their intellectual property.
REFERENCES
- Aboud A. A., Tidball A. M., Kumar K. K., Neely M. D., Ess K. C., Erikson K. M., Bowman A. B. (2012). Genetic risk for Parkinson’s disease correlates with alterations in neuronal manganese sensitivity between two human subjects. Neurotoxicology 33, 1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboud A. A., Tidball A. M., Kumar K. K., Neely M. D., Han B., Ess K. C., Hong C. C., Erikson K. M., Hedera P., Bowman A. B. (2014). PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol. Dis. 73C, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetto A., Au C., Aschner M. (2009). Manganese-induced dopaminergic neurodegeneration: Insights into mechanisms and genetics shared with Parkinson’s disease. Chem. Rev. 109, 4862–4884. [DOI] [PubMed] [Google Scholar]
- Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Bornhorst J., Meyer S., Weber T., Boker C., Marschall T., Mangerich A., Beneke S., Burkle A., Schwerdtle T. (2013). Molecular mechanisms of Mn induced neurotoxicity: RONS generation, genotoxicity, and DNA-damage response. Mol. Nutr. Food Res. 57, 1255–1269. [DOI] [PubMed] [Google Scholar]
- Bouabid S., Tinakoua A., Lakhdar-Ghazal N., Benazzouz A. (2016). Manganese neurotoxicity: Behavioral disorders associated with dysfunctions in the basal ganglia and neurochemical transmission. J. Neurochem. 136, 677–691. [DOI] [PubMed] [Google Scholar]
- Bowman A. B., Aschner M. (2014). Considerations on manganese (Mn) treatments for invitro studies. Neurotoxicology 41, 141–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S. M., Fasano C. A., Papapetrou E. P., Tomishima M., Sadelain M., Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Chakraborty S., Peres T. V., Bowman A. B., Aschner M. (2015). Manganese-induced neurotoxicity: From C. elegans to humans. Toxicol. Res. 4, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Culbreth M., Aschner M. (2016). Exposure, epidemiology, and mechanism of the environmental toxicant manganese. Environ. Sci. Pollut. Res. Int. 23, 13802–13810. [DOI] [PubMed] [Google Scholar]
- Chen P., Parmalee N., Aschner M. (2014). Genetic factors and manganese-induced neurotoxicity. Front. Genet. 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H. C., Ulane C. M., Burke R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin-Chan M., Navarro-Yepes J., Quintanilla-Vega B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9, 124.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M. (1998). Inhibitors of NADH-ubiquinone reductase: An overview. Biochim. Biophys. Acta 1364, 222–235. [DOI] [PubMed] [Google Scholar]
- Desole M. S., Esposito G., Migheli R., Sircana S., Delogu M. R., Fresu L., Miele M., de Natale G., Miele E. (1997). Glutathione deficiency potentiates manganese toxicity in rat striatum and brainstem and in PC12 cells. Pharmacol. Res. 36, 285–292. [DOI] [PubMed] [Google Scholar]
- Esposti M. D., Hatzinisiriou I., McLennan H., Ralph S. (1999). Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J. Biol. Chem. 274, 29831–29837. [DOI] [PubMed] [Google Scholar]
- Fang D., Qing Y., Yan S., Chen D., Yan S. S. (2016). Development and dynamic regulation of mitochondrial network in human midbrain dopaminergic neurons differentiated from iPSCs. Stem Cell Rep. 7, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M., Avila D. S., da Rocha J. B., Aschner M. (2013). Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 62, 575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J., Hao L., Bijli K. M., Chandler J. D., Orr M., Hu X., Jones D. P., Go Y. M. (2017). Manganese stimulates mitochondrial H2O2 production in SH-SY5Y human neuroblastoma cells over physiologic as well as toxicologic range. Toxicol. Sci. 155, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani P., Fumagalli P., Santagostino A. (1995). Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur. J. Pharmacol. 293, 377–383. [DOI] [PubMed] [Google Scholar]
- Gavin C. E., Gunter K. K., Gunter T. E. (1990). Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem. J. 266, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin C. E., Gunter K. K., Gunter T. E. (1992). Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol. Appl. Pharmacol. 115, 1–5. [DOI] [PubMed] [Google Scholar]
- Gavin C. E., Gunter K. K., Gunter T. E. (1999). Manganese and calcium transport in mitochondria: Implications for manganese toxicity. Neurotoxicology 20, 445–453. [PubMed] [Google Scholar]
- Greenamyre J. T., MacKenzie G., Peng T. I., Stephans S. E. (1999). Mitochondrial dysfunction in Parkinson’s disease. Biochem. Soc. Symp. 66, 85–97. [DOI] [PubMed] [Google Scholar]
- Guilarte T. R. (2010). Manganese and Parkinson’s disease: A critical review and new findings. Environ. Health Perspect. 118, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter T. E., Gavin C. E., Gunter K. K. (2009). The case for manganese interaction with mitochondria. Neurotoxicology 30, 727–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter T. E., Gerstner B., Lester T., Wojtovich A. P., Malecki J., Swarts S. G., Brookes P. S., Gavin C. E., Gunter K. K. (2010). An analysis of the effects of Mn2+ on oxidative phosphorylation in liver, brain, and heart mitochondria using state 3 oxidation rate assays. Toxicol. Appl. Pharmacol. 249, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HaMai D., Bondy S. C. (2004a). Oxidative basis of manganese neurotoxicity. Ann. N.Y. Acad. Sci. 1012, 129–141. [DOI] [PubMed] [Google Scholar]
- HaMai D., Bondy S. C. (2004b). Pro- or anti-oxidant manganese: A suggested mechanism for reconciliation. Neurochem. Int. 44, 223–229. [DOI] [PubMed] [Google Scholar]
- HaMai D., Campbell A., Bondy S. C. (2001). Modulation of oxidative events by multivalent manganese complexes in brain tissue. Free Radic. Biol. Med. 31, 763–768. [DOI] [PubMed] [Google Scholar]
- Hernandez R. B., Farina M., Esposito B. P., Souza-Pinto N. C., Barbosa F. Jr., Sunol C. (2011). Mechanisms of manganese-induced neurotoxicity in primary neuronal cultures: The role of manganese speciation and cell type. Toxicol. Sci. 124, 414–423. [DOI] [PubMed] [Google Scholar]
- Heron P., Cousins K., Boyd C., Daya S. (2001). Paradoxical effects of copper and manganese on brain mitochondrial function. Life Sci. 68, 1575–1583. [DOI] [PubMed] [Google Scholar]
- Hirata Y., Meguro T., Kiuchi K. (2006). Differential effect of nerve growth factor on dopaminergic neurotoxin-induced apoptosis. J. Neurochem. 99, 416–425. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. (1966). Dopamine (3-hydroxytyramine) and brain function. Pharmacol. Rev. 18, 925–964. [PubMed] [Google Scholar]
- Jellinger K. A. (2013). The relevance of metals in the pathophysiology of neurodegeneration, pathological considerations. Int. Rev. Neurobiol. 110, 1–47. [DOI] [PubMed] [Google Scholar]
- Jiang Q., Yan Z., Feng J. (2006a). Activation of group III metabotropic glutamate receptors attenuates rotenone toxicity on dopaminergic neurons through a microtubule-dependent mechanism. J. Neurosci. 26, 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q., Yan Z., Feng J. (2006b). Neurotrophic factors stabilize microtubules and protect against rotenone toxicity on dopaminergic neurons. J. Biol. Chem. 281, 29391–29400. [DOI] [PubMed] [Google Scholar]
- Jiang T., Sun Q., Chen S. (2016). Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 147, 1–19. [DOI] [PubMed] [Google Scholar]
- Kalia L. V., Lang A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. [DOI] [PubMed] [Google Scholar]
- Kriks S., Shim J. W., Piao J., Ganat Y. M., Wakeman D. R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A. K., Balmer N. V., Matt F., Schonenberger F., Merhof D., Leist M. (2013). Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol. 87, 2215–2231. [DOI] [PubMed] [Google Scholar]
- Kumar K. K., Aboud A. A., Bowman A. B. (2012). The potential of induced pluripotent stem cells as a translational model for neurotoxicological risk. Neurotoxicology 33, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K. K., Lowe E. W. Jr., Aboud A. A., Neely M. D., Redha R., Bauer J. A., Odak M., Weaver C. D., Meiler J., Aschner M., et al. (2014). Cellular manganese content is developmentally regulated in human dopaminergic neurons. Sci. Rep. 4, 6801.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakye G. F., Paoliello M. M., Mukhopadhyay S., Bowman A. B., Aschner M. (2015). Manganese-induced Parkinsonism and Parkinson’s disease: Shared and distinguishable features. Int. J. Environ. Res. Publ. Health 12, 7519–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwik-Uribe C. L., Reaney S., Zhu Z., Smith D. (2003). Alterations in cellular IRP-dependent iron regulation by invitro manganese exposure in undifferentiated PC12 cells. Brain Res. 973, 1–15. [DOI] [PubMed] [Google Scholar]
- Lucchini R. G., Albini E., Benedetti L., Borghesi S., Coccaglio R., Malara E. C., Parrinello G., Garattini S., Resola S., Alessio L. (2007). High prevalence of Parkinsonian disorders associated to manganese exposure in the vicinities of ferroalloy industries. Am. J. Ind. Med. 50, 788–800. [DOI] [PubMed] [Google Scholar]
- Maynard L. S., Cotzias G. C. (1955). The partition of manganese among organs and intracellular organelles of the rat. J. Biol. Chem. 214, 489–495. [PubMed] [Google Scholar]
- McCormack P. L. (2014). Rasagiline: A review of its use in the treatment of idiopathic Parkinson’s disease. CNS Drugs 28, 1083–1097. [DOI] [PubMed] [Google Scholar]
- McLennan H. R., Degli Esposti M. (2000). The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr. 32, 153–162. [DOI] [PubMed] [Google Scholar]
- Meinel J., Radad K., Rausch W. D., Reichmann H., Gille G. (2015). Cabergoline protects dopaminergic neurons against rotenone-induced cell death in primary mesencephalic cell culture. Folia Neuropathol. 53, 29–40. [DOI] [PubMed] [Google Scholar]
- Milatovic D., Montine T. J., Aschner M. (2011). Measurement of isoprostanes as markers of oxidative stress. Methods Mol. Biol. 758, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne G. L., Gao B., Terry E. S., Zackert W. E., Sanchez S. C. (2013). Measurement of F2- isoprostanes and isofurans using gas chromatography-mass spectrometry. Free Radic. Biol. Med. 59, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne G. L., Yin H., Hardy K. D., Davies S. S., Roberts L. J. 2nd. (2011). Isoprostane generation and function. Chem. Rev. 111, 5973–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milusheva E., Baranyi M., Kormos E., Hracsko Z., Sylvester Vizi E., Sperlagh B. (2010). The effect of antiparkinsonian drugs on oxidative stress induced pathological [3H]dopamine efflux after invitro rotenone exposure in rat striatal slices. Neuropharmacology 58, 816–825. [DOI] [PubMed] [Google Scholar]
- Montine K. S., Quinn J. F., Zhang J., Fessel J. P., Roberts L. J. 2nd, Morrow J. D., Montine T. J. (2004). Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem. Phys. Lipids 128, 117–124. [DOI] [PubMed] [Google Scholar]
- Morello M., Canini A., Mattioli P., Sorge R. P., Alimonti A., Bocca B., Forte G., Martorana A., Bernardi G., Sancesario G. (2008). Sub-cellular localization of manganese in the basal ganglia of normal and manganese-treated rats An electron spectroscopy imaging and electron energy-loss spectroscopy study. Neurotoxicology 29, 60–72. [DOI] [PubMed] [Google Scholar]
- Muller F. J., Schuldt B. M., Williams R., Mason D., Altun G., Papapetrou E. P., Danner S., Goldmann J. E., Herbst A., Schmidt N. O., et al. (2011). A bioinformatic assay for pluripotency in human cells. Nat. Methods 8, 315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandipati S., Litvan I. (2016). Environmental exposures and Parkinson’s disease. Int. J. Environ. Res. Publ. Health 139, 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi M., Maruyama W., Inaba-Hasegawa K. (2013). Revelation in the neuroprotective functions of rasagiline and selegiline: The induction of distinct genes by different mechanisms. Expert Rev. Neurother. 13, 671–684. [DOI] [PubMed] [Google Scholar]
- Neal A. P., Guilarte T. R. (2013). Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely M. D., Litt M. J., Tidball A. M., Li G. G., Aboud A. A., Hopkins C. R., Chamberlin R., Hong C. C., Ess K. C., Bowman A. B. (2012). DMH1, a highly selective small molecule BMP inhibitor promotes neurogenesis of hiPSCs: Comparison of PAX6 and SOX1 expression during neural induction. ACS Chem. Neurosci. 3, 482–491. (PMC888888). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K., et al. (2011). A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412. [DOI] [PubMed] [Google Scholar]
- Pal R., Monroe T. O., Palmieri M., Sardiello M., Rodney G. G. (2014). Rotenone induces neurotoxicity through Rac1-dependent activation of NADPH oxidase in SHSY-5Y cells. FEBS Lett. 588, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Liu Q., Rao M. S., Zeng X. (2013). Using human pluripotent stem cell-derived dopaminergic neurons to evaluate candidate Parkinson’s disease therapeutic agents in MPP+ and rotenone models. J. Biomol. Screen. 18, 522–533. [DOI] [PubMed] [Google Scholar]
- Peres T. V., Schettinger M. R., Chen P., Carvalho F., Avila D. S., Bowman A. B., Aschner M. (2016). Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 17, 57.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompella A., Visvikis A., Paolicchi A., De Tata V., Casini A. F. (2003). The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 66, 1499–1503. [DOI] [PubMed] [Google Scholar]
- Racette B. A., Criswell S. R., Lundin J. I., Hobson A., Seixas N., Kotzbauer P. T., Evanoff B. A., Perlmutter J. S., Zhang J., Sheppard L., et al. (2012). Increased risk of parkinsonism associated with welding exposure. Neurotoxicology 33, 1356–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radad K., Rausch W. D., Gille G. (2006). Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 49, 379–386. [DOI] [PubMed] [Google Scholar]
- Ratner M. H., Farb D. H., Ozer J., Feldman R. G., Durso R. (2014). Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip. Toxicol. 7, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P., Schmid B., Burbulla L. F., Schondorf D. C., Wagner L., Glatza M., Hoing S., Hargus G., Heck S. A., Dhingra A., et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. [DOI] [PubMed] [Google Scholar]
- Ren Y., Liu W., Jiang H., Jiang Q., Feng J. (2005). Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 280, 34105–34112. [DOI] [PubMed] [Google Scholar]
- Ryan B. J., Hoek S., Fon E. A., Wade-Martins R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. [DOI] [PubMed] [Google Scholar]
- Ryan S. D., Dolatabadi N., Chan S. F., Zhang X., Akhtar M. W., Parker J., Soldner F., Sunico C. R., Nagar S., Talantova M., et al. (2013). Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schondorf D. C., Aureli M., McAllister F. E., Hindley C. J., Mayer F., Schmid B., Sardi S. P., Valsecchi M., Hoffmann S., Schwarz L. K., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028.. [DOI] [PubMed] [Google Scholar]
- Seyfried J., Soldner F., Kunz W. S., Schulz J. B., Klockgether T., Kovar K. A., Wullner U. (2000). Effect of 1-methyl-4-phenylpyridinium on glutathione in rat pheochromocytoma PC 12 cells. Neurochem. Int. 36, 489–497. [DOI] [PubMed] [Google Scholar]
- Shaltouki A., Sivapatham R., Pei Y., Gerencser A. A., Momcilovic O., Rao M. S., Zeng X. (2015). Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 4, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer T. B., Betarbet R., Stout A. K., Lund S., Baptista M., Panov A. V., Cookson M. R., Greenamyre J. T. (2002). An invitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 22, 7006–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer T. B., Kim J. H., Betarbet R., Greenamyre J. T. (2003). Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 179, 9–16. [DOI] [PubMed] [Google Scholar]
- Smith M. R., Fernandes J., Go Y. M., Jones D. P. (2017). Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 482, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanwood G. D., Leitch D. B., Savchenko V., Wu J., Fitsanakis V. A., Anderson D. J., Stankowski J. N., Aschner M., McLaughlin B. (2009). Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J. Neurochem. 110, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stredrick D. L., Stokes A. H., Worst T. J., Freeman W. M., Johnson E. A., Lash L. H., Aschner M., Vrana K. E. (2004). Manganese-induced cytotoxicity in dopamine-producing cells. Neurotoxicology 25, 543–553. [DOI] [PubMed] [Google Scholar]
- Tidball A. M., Bryan M. R., Uhouse M. A., Kumar K. K., Aboud A. A., Feist J. E., Ess K. C., Neely M. D., Aschner M., Bowman A. B. (2015). A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington’s disease. Hum. Mol. Genet. 24, 1929–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl K., Meyer E., Valdivia L. E., Zhao N., Dadswell C., Abdul-Sada A., Hung C. Y., Simpson M. A., Chong W. K., Jacques T. S., et al. (2016). Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 7, 11601.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb O., Amit T., Riederer P., Youdim M. B., Mandel S. A. (2011). Neuroprotective profile of the multitarget drug rasagiline in Parkinson’s disease. Int. Rev. Neurobiol. 100, 127–149. [DOI] [PubMed] [Google Scholar]
- Xiong N., Long X., Xiong J., Jia M., Chen C., Huang J., Ghoorah D., Kong X., Lin Z., Wang T. (2012). Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 42, 613–632. [DOI] [PubMed] [Google Scholar]
- Yamashita H., Nakamura T., Takahashi T., Nagano Y., Hiji M., Hirabayashi T., Amano T., Yagi T., Sakai N., Kohriyama T., et al. (2006). Embryonic stem cell-derived neuron models of Parkinson’s disease exhibit delayed neuronal death. J. Neurochem. 98, 45–56. [DOI] [PubMed] [Google Scholar]
- Yin H. (2008). New techniques to detect oxidative stress markers: Mass spectrometry-based methods to detect isoprostanes as the gold standard for oxidative stress invivo. Biofactors 34, 109–124. [DOI] [PubMed] [Google Scholar]
- Yoon H., Lee G. H., Kim D. S., Kim K. W., Kim H. R., Chae H. J. (2011). The effects of 3, 4 or 5 amino salicylic acids on manganese-induced neuronal death: ER stress and mitochondrial complexes. Toxicol. In Vitro 25, 1259–1268. [DOI] [PubMed] [Google Scholar]
- Zagoura D., Canovas-Jorda D., Pistollato F., Bremer-Hoffmann S., Bal-Price A. (2017). Evaluation of the rotenone-induced activation of the Nrf2 pathway in a neuronal model derived from human induced pluripotent stem cells. Neurochem. Int. 106, 62–73. [DOI] [PubMed] [Google Scholar]
- Zhang S., Fu J., Zhou Z. (2004). In vitro effect of manganese chloride exposure on reactive oxygen species generation and respiratory chain complexes activities of mitochondria isolated from rat brain. Toxicol. In Vitro 18, 71–77. [DOI] [PubMed] [Google Scholar]
- Zhang S., Zhou Z., Fu J. (2003). Effect of manganese chloride exposure on liver and brain mitochondria function in rats. Environ. Res. 93, 149–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.