

Abstract
Background and Aims
The processes and mechanisms underlying lineage diversification are major topics in evolutionary biology. Eurasian goldenrod species of the Solidago virgaurea complex show remarkable morphological and ecological diversity in the Japanese Archipelago, with ecotypic taxa well adapted to specific environments (climate, edaphic conditions and disturbance regimes). The species complex is a suitable model to investigate the evolutionary processes of actively speciating plant groups, due to its ability to evolve in relation to environmental adaptation and its historical population dynamics.
Methods
Two chloroplast markers, 18 nuclear microsatellite markers and ddRAD-sequencing were used to infer population genetic demography of S. virgaurea complex with its related species/genera.
Key Results
Our analysis showed that populations in Japan form an evolutionary unit, which was genetically diverged from adjacent continental populations. The phylogenetic structure within the archipelago strongly corresponds to the geography, but interestingly there is no concordance between genetic structure and ecotypic boundaries; neighbouring populations of distinct ecotypes share a genetic background.
Conclusions
We propose that the traits specific to the ecotypic entities are maintained by natural selection or are very recently generated and have little effect on the genomes, making genome-wide genetic markers unsuitable for detecting ecotypic differentiation. Furthermore, some sporadically distributed taxa (found as rheophytes and alpine plants) were repeatedly generated from a more widespread taxon in geographically distant areas by means of selection. Overall, this study showed that the goldenrod complex has a high ability to evolve, enabling rapid ecological diversification over a recent timeframe.
Keywords: Ecotype, environmental adaptation, parallel evolution, population genetics, Solidago
INTRODUCTION
The processes and mechanisms underlying lineage diversification are major topics in evolutionary biology. Phylogeographic analysis is an essential tool to understand the speed and spatial scale of an organism’s diversification (Crisp et al., 2011; Donoghue and Edwards, 2014). An array of studies has demonstrated a general trend of allopatric diversification in plants, where the population divergence was triggered by geographic barriers and historical range shifts (Magri et al., 2006; Sakaguchi et al., 2012; Sork et al., 2016). Ecological factors, such as divergent habitats and pollinator fauna, also play significant roles in adaptive divergence, which can limit gene exchanges between populations in alternative environments and then drive the evolution of prezygotic barriers (Rundle and Nosil, 2005; Rieseberg and Willis, 2007). Hence, evaluating the relative roles of geographic and ecological factors in plant diversification will not only provide insights into how the observed plant diversity has been shaped, but also highlight the significance of natural selection and the evolvability of organisms. This is particularly important for the plant groups that speciated into diverse ecological niche space (Givnish, 2010).
The goldenrod species of the Solidago virgaurea L. complex (Asteraceae) (2n = 18) (Watanabe, 2017) are perennial herbs with a broad distribution range across Eurasia, extending from Europe to East Asia (Kawano, 1988; Semple, 2016; Iwatsuki et al., 1995). While the centre of species diversity of the genus is located in North America, where 153 lower taxa are recognized (Semple and Cook, 2006), the Eurasian species S. virgaurea is also polymorphic and includes intraspecific ecotypic entities in its ranges (Slavík, 2004; Kiełtyk and Mirek, 2014; Jang et al., 2012) and has long been the subject of ecological studies (Abe and Takasu, 1983; Turesson, 1925; Nishizawa et al., 2001; Bergstern, 2009; Takahashi and Matsuki, 2016). It is ecologically and morphologically diverse in the Japanese Archipelago (Fig. 1), an island chain of extensive temperature gradients both vertically (from sea level to over 3000 m above sea level) and horizontally (~24–45°N) and disturbance regimes such as river floods, wind velocity and volcanic activity. The territory harbours nine taxa: S. v. subsp. asiatica var. asiatica (syn. S. decurrens Lour.); S. horieana; S. v. subsp. asiatica var. insularis; S. v. subsp. leiocarpa var. leiocarpa; S. minutissima; S. nipponica (syn. S. v. subsp. gigantea (Nakai) Kitam.); S. v. subsp. leiocarpa var. ovata; S. v. subsp. leiocarpa var. praeflorens; and S. yokusaiana (Honda, 1937; Iwatsuki et al., 1995; Kadota, 2008; Semple, 2013) (shown as asiatica, horieana, insularis, leiocarpa, minutissima, nipponica, ovata, praeflorens and yokusaiana, respectively, in Fig. 1). In terms of plant size, the northern populations (S. nipponica and var. asiatica) sometimes reach 1 m in height, while S. minutissima, which includes the wetland plants, grows to at most 3 cm. The complex is considered to have expanded into harsher environments, such as the alpine area (var. leiocarpa) and the serpentine (S. horieana) and volcanic barrens (var. praeflorens), leading to the generation of distinct ecotypic taxa (Iwatsuki et al., 1995; Ohba, 1999) (Fig. 1). Their ecotypic differentiation is closely associated with local adaptation; alpine populations (var. leiocarpa) have a stunted stature with a decreased number of flower heads and thinner leaves, which are all adaptions to cooler temperatures and strong wind conditions at higher elevations (Takahashi and Matsuki, 2016). Narrow leaves are independently selected in rheophytic taxa (S. yokusaiana and S. horieana), with the former having many linear-lanceolate cauline leaves (2–5 mm wide), enabling the plants to resist the strong flow of water in the frequently flooded river systems (Takasu, 1978).
Fig. 1.

(A) Distribution of the Solidago virgaurea complex in the Japanese Archipelago. The distribution of wide-ranging taxa is indicated by differently coloured areas, while those of narrow endemics (horieana, ovata and minutissima) are shown as squares. (B) Geography of the Japanese Archipelago. (c) Plant morphology of each taxon of the S. virgaurea complex in the archipelago.
The ancestral habitat of the Japanese S. virgaurea complex is proposed to have been the ecotone between deciduous woodlands and open grasslands since this area is currently occupied by the most widespread taxon, S. v. subsp. asiatica var. asiatica (Kawano, 1988) (Fig. 1A), and to have expanded later into more specialized habitats. However, no phylogeographic studies have been undertaken to clarify how the species complex diversified; either the complex suddenly diversified over a short period of time (Kawano, 1988) or diversified hierarchically to generate ecotypic taxa. The former hypothesis predicts that ecotypic taxa were established over a recent timeframe in the archipelago, whereby the demographic signals would be detected as shallow branches in a phylogenetic tree, followed by sudden population growth. On the other hand, as the alpine ecotypic populations also occur in Europe (Turesson, 1925; Bergstern, 2009), some ecotypic taxa may have already diverged from the lowland populations before expanding into the Japanese Archipelago, which would result in hierarchical lineage divergence concordant with the taxonomic boundary (Kikuchi et al., 2010; Qi et al., 2012; Higashi et al., 2013). Similarly, the rheophytic taxon (S. yokusaiana) is distributed across a wide range (Fig. 1), but the local populations occur sporadically and are sometimes isolated from each other by hundreds of kilometres (Fig. 1A). Because historical configurations of the river systems show no possible connections between these populations (Kimura, 1996) and the strong water flow could potentially select for narrow-leaved populations in distant areas, the potential role of parallel evolution in generating these ecotypic taxa should be considered.
In this study, we examined the biogeographic processes of the ecologically diverse S. virgaurea complex in the Japanese Archipelago as follows: (1) phylogenetic analysis to test whether the Japanese goldenrods constitute a monophyletic group, which addresses whether the ecotypic taxa were differentiated within the archipelago after arrival of common ancestor(s); (2) population genetic analysis to reconstruct the demographic history of the Japanese goldenrods; the analyses would identify whether the species complex diversified over a short period of time or whether the ecotypic taxa diverged in a hierarchical manner; and (3) testing alternative scenarios of vicariance, long-distance dispersal and parallel evolution to infer the origin of the disjunct distribution of the ecotypic taxa.
MATERIALS AND METHODS
Population sampling
To comprehensively evaluate the genetic and taxonomic variation in the S. virgaurea complex in the Japanese Archipelago and adjacent regions, 223 individuals were collected from 129 populations for population genetic analyses (Table 1, Fig. 1, Supplementary Data Tables S1 and S2). The sample set included all the Japanese intraspecific taxa, and plant materials from the East Asian S. virgaurea complex (from Taiwan, mainland China, the Korean Peninsula and the Russian Far East) and European S. virgaurea s.s. were also included. The East Asian taxa included S. spiraeifolia in Kamchatka, S. cf. dahurica from the Russian Far East, a disjunct S. nipponica-like population in Ulleungdo Island, S. v. subsp. asiatica var. asiatica from China and the Korean Peninsula, and S. v. subsp. leiocarpa var. leiocarpa from Taiwan. There are some unsampled taxa outside Japan (e.g. S. pacifica Juz. and S. kurilensis Juz. in Kuril Islands and Far East), which should be closely related to Japanese populations based on their morphologies (Takasu, 1982), although the taxonomic correspondence between Japanese and Russian treatments is not confirmed (J. Semple, pers. comm.). Euthamia graminifolia and two diploid plants of North American Solidago species (S. flexicaulis and S. ulmifolia) were included as outgroup taxa in this study.
Table 1.
Intraspecific taxa and samples of Solidago virgaurea complex analysed in this study
| Taxon | Distribution | Habitat | Plant height | Number of samples analysed | ||||
|---|---|---|---|---|---|---|---|---|
| ddRAD phylogeny | nSSR population genetics in East Asia | cpDNA population genetics | nSSR population genetics in Japan | ddRAD population genetics | ||||
| S. horieana Kadota | Hokkaido, Japan | Serpentine riverside | ≤ 60 cm | 0 | 3 | 8 | 8 | 3 |
| S. minutissima (Makino) Kitam. | Yakushima, Japan | Alpine grassland | ≤ 10 cm | 1 | 3 | 7 | 7 | 4 |
| S. nipponica Semple | North Japan | Coastal grassland | ≤ 80 cm | 1 | 13 | 15 | 15 | 2 |
| S. spiraeifolia Fisch. ex Herder | Russian Far East and Kamchatka | Woodland and grassland | ≤ 70 cm | 0 | 15 | 0 | 0 | 0 |
| S. virgaurea subsp. asiatica var. asiatica Nakai ex H.Hara | East Asia | Woodland and grassland | ≤ 80 cm | 7 | 149 | 121 | 121 | 113 |
| S. virgaurea subsp. asiatica var. insularis (Kitam.) H.Hara | Ryukyu Islands, Japan | Coastal grassland | ≤ 50 cm | 0 | 6 | 10 | 10 | 3 |
| S. virgaurea subsp. dahurica (Kitag.) Kitag. | Central and East Asia | Woodland and grassland | ≤ 100 cm | 1 | 2 | 17 | 17 | 0 |
| S. virgaurea subsp. leiocarpa var. leiocarpa (Benth.) A.Gray | East Asia | Alpine grassland | ≤ 70 cm | 2 | 9 | 21 | 21 | 9 |
| S. virgaurea subsp. leiocarpa var. ovata (Honda) Ohba | Central Japan | Coastal grassland | ≤ 30 cm | 0 | 2 | 5 | 5 | 2 |
| S. virgaurea subsp. leiocarpa var. praeflorens Nakai | Central Japan | Volcanic barren | ≤ 25 cm | 0 | 2 | 8 | 8 | 6 |
| S. virgaurea subsp. virgaurea | Europe | Woodland and grassland | ≤ 100 cm | 2 | 0 | 0 | 0 | 0 |
| S. yokusaiana Makino | Japan | Riverside rock surface | ≤ 60 cm | 2 | 19 | 60 | 60 | 31 |
| S. flexicaulis L. | North America | Woodland | ≤ 90 cm | 1 | 0 | 0 | 0 | 0 |
| S. ulmifolia Muhl. ex Willd. | North America | Woodland | ≤ 120 cm | 1 | 0 | 0 | 0 | 0 |
| Euthamia graminifolia (L.) Nutt. | North America | Grassland | ≤ 150 cm | 1 | 0 | 0 | 0 | 0 |
| Total | 19 | 223 | 272 | 272 | 173 | |||
Molecular experiments
The collected leaf samples were immediately dried using silica gel and kept in the dark at room temperature before DNA extraction. Plant DNA was extracted using a slightly modified cetyltrimethylammonium bromide (CTAB) method (Murray and Thompson, 1980).
Chloroplast sequencing.
All the population samples were sequenced at two regions (trnH-psbA and rpl16) of the chloroplast genome using universal primers (Shaw et al., 2005). Polymerase chain reactions (PCRs) were performed in a 10-µL volume containing 20 ng of template DNA, 1 µL of ×10 PCR buffer, 0.8 µL of 2.5 mm dNTP, 0.05 µL of 50 µm each primer and 0.25 units of TaKaRa Ex Taq (TaKaRa, Otsu, Japan). The PCR cycle was as follows: template denaturation at 94 °C for 3 min then 30 cycles of denaturation at 94 °C for 1 min, annealing at 52 °C for 1 min and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 7 min. The PCR products were sequenced using an ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction kit v. 3.1 (Applied Biosystems, Foster City, CA, USA) and electrophoresed on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems).
Nuclear microsatellite genotyping.
All the population samples were genotyped using 15 expressed sequence tag simple sequence repeat (EST-SSR) markers developed for this complex (Sakaguchi and Ito, 2014) and three genomic SSR markers (Salt1, Salt3 and Salt17) for S. altissima (Sakata et al., 2013). For all loci, the forward primer was synthesised with one of three different M13 sequences (5′-CACGACGTTGTAAAACGAC-3′, 5′-TGTGGAATTGTGAGCGG-3′ and 5′-CTATAGGGCACG CGTGGT-3′) and the reverse primer was tagged with a pigtail sequence (5′-GTTTCTT-3′) to promote full adenylation (Brownstein et al., 1996). The PCR reaction was performed following Sakaguchi and Ito (2014). The PCR products were loaded onto an auto-sequencer (GenomeLab GeXP, Beckman Coulter, Brea, CA, USA) to assess fragment lengths using Fragment Analysis Software ver. 8.0 (Beckman Coulter).
Double-digest restriction-associated DNA sequencing.
A double-digest restriction-associated DNA (ddRAD) library (Peterson et al., 2012) was prepared for the phylogeny and population genetic samples (Supplementary Data Tables S1 and S2) using Peterson’s protocol with slight modifications. Briefly, 10 ng of genomic DNA was digested with EcoRI and BglII (New England Biolabs, Ipswich, MA, USA) and adapters were ligated at 37 °C overnight in a 10-µL volume containing 1 µL of ×10 New England Biolabs (NEB) buffer 2, 0.1 µL of ×100 bovine serum albumin (BSA) (NEB), 0.4 µL of 5 µmEcoRI adapter and BglII adapter, 0.1 µL of 100 mm ATP and 0.5 µL of T4 DNA Ligase (Enzymatics, Beverly, MA, USA). The reaction solution was then purified with AMPure XP (Beckman Coulter). Next, 3 µL of purified DNA was used in the PCR amplifications in a 10-µL volume containing 1 µL of each 10 µM index and TruSeq universal primer, 0.3 µL of KOD-Plus-Neo enzyme and 1 µL of ×10 PCR buffer (Toyobo, Osaka, Japan), 0.6 µL of 25 mm MgSO4 and 1 µL of 10 mm dNTP. For several DNA samples used in the phylogenetic analysis, the PCR amplification included 1 µL of each 10 µm index and TruSeq universal primer and 5 µL of ×2 KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA). Thermal cycling was initiated with a 94 °C step for 2 min followed by 20 cycles of 98 °C for 10 s, 65 °C for 30 s and 68 °C for 30 s. The PCR products were pooled and purified again with AMPure XP. The purified DNA was then loaded onto a 2.0 % agarose gel and fragments of around 320 base pairs (bp) were retrieved using E-Gel SizeSelect (Life Technologies, Carlsbad, CA, USA). After quality assessment using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), the library was sequenced with 51-bp single-end reads in one lane of an Illumina HiSeq2000 (Illumina, San Diego, CA, USA). Primer sequences used in this study are available in Sakaguchi et al. (2015).
Data analysis of chloroplast sequences and nuclear microsatellite genotypes
The chloroplast sequence data were edited and aligned using BioEdit v. 7.0.8.0 software (Hall, 1999). The alignment file was imported into GENALEX 6.3 software (Peakall and Smouse, 2006) to recover the chloroplast haplotypes. The relationship between haplotypes were inferred as a median-joining network using Network v. 5 software (Bandelt et al., 1999).
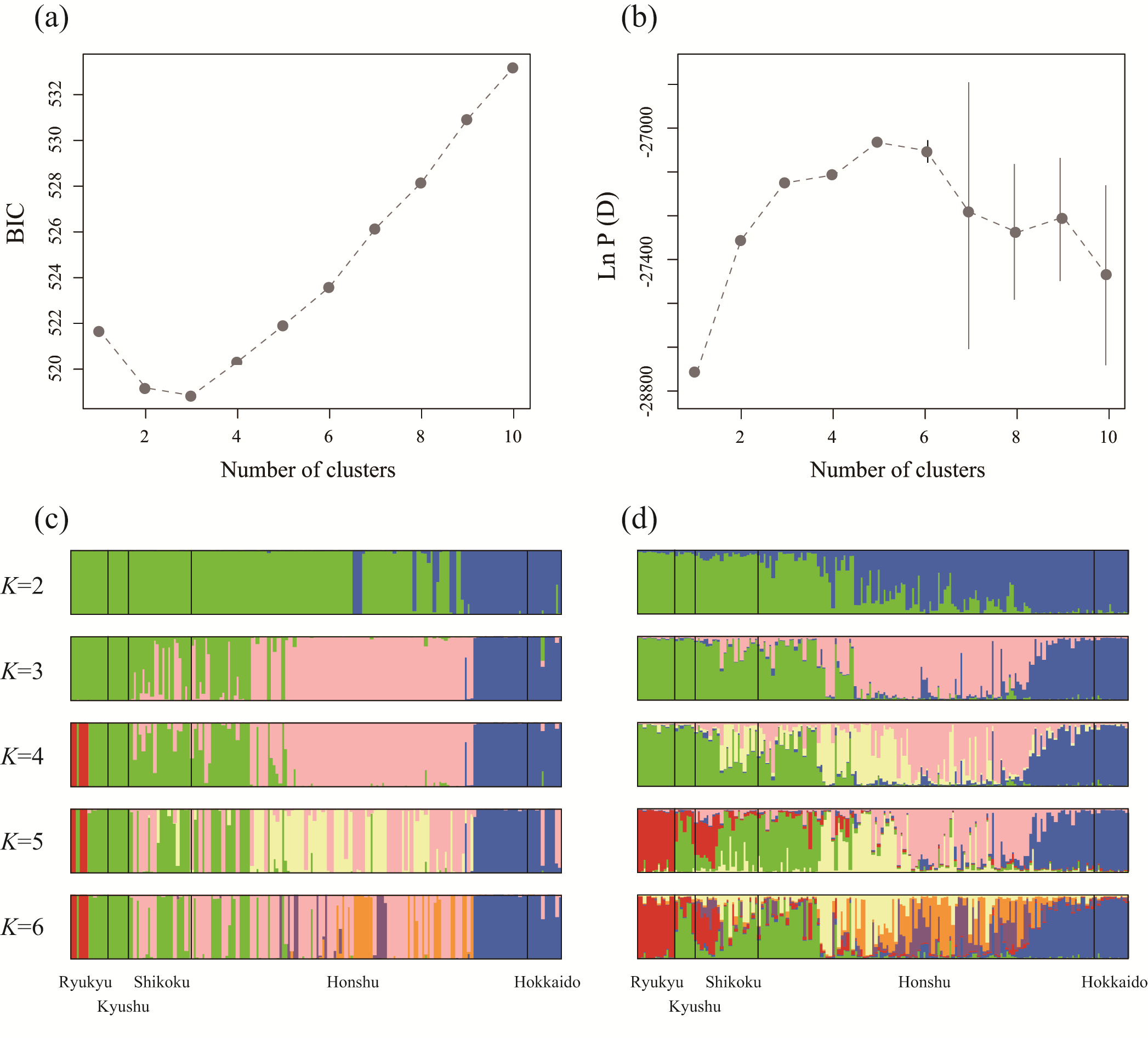
The microsatellite genotype data consisted of 18 unlinked markers used to infer the population genetic structure of the Japanese S. virgaurea complex populations. Allelic richness (El Mousadik and Petit, 1996) and expected heterozygosity (Nei, 1987) were calculated for the regional population groups divided by the geographic boundaries in Japan (Supplementary Data Table S2), using FSTAT 2.9.3 software (Goudet, 1995). To explore population genetic structure, a population genetic model using STRUCTURE software (Pritchard et al., 2000) was applied, which assumes Hardy–Weinberg disequilibrium within genetic clusters, because the species complex has self-incompatibility (Kühn et al., 2004) and population inbreeding is not usual (Sakaguchi and Ito, 2014). It was also assumed that each individual had admixed ancestral origins, that different gene pools that had retained their allele frequency correlated (Falush et al., 2003), and that the sampling locations were designated as prior information to obtain improved parameter estimates (Hubisz et al., 2009). Ten independent STRUCTURE simulations were run for each K (K = 1–10), with 50 000 burn-in steps followed by 100 000 Markov chain Monte Carlo steps. The STRUCTURE analysis was performed for the two data sets for East Asian-scale samples and the Japanese Archipelago-scale samples, separately (Table 1). Principal components analysis (PCA) was also applied to examine the phylogeographic structure of East Asian samples.
Data analysis of ddRAD-seq-derived short reads
Phylogenetic analysis using a ddRAD-seq read matrix.
To infer the phylogenetic relationship of the S. virgaurea complex, ddRAD-seq data of 19 taxa were used, including the ingroup of S. virgaurea samples and three outgroup species (Supplementary Data Table S2). A goldenrod species of Euthamia graminifolia was used as the most distant relative to the S. virgaurea complex to root the phylogenetic tree (Semple et al., 1984). Prior to the de novo assembly, raw reads were processed using Trimmomatic v. 0.32 software (Bolger et al., 2014) to remove the adapter and other Illumina-specific sequences and to cut out low-quality regions based on a quality score (<20), corresponding to a base call accuracy of 99 %. To obtain the ddRAD-seq single-nucleotide polymorphism (SNP) genotypes of the various plant genera, pyRAD v. 3.0 was used as this program allows reads containing indels (insertions and deletions) to be assembled and is therefore suitable for recovering larger numbers of loci when analysing phylogenetically distant groups (Cariou et al., 2013; Pante et al., 2015). The parameters that influence the assembly were set as follows: the minimum depth coverage for base calling at each locus was set at 6 and the similarity threshold for clustering reads within/across samples was set at 0.90. Potential paralogous loci were filtered out based on the number of samples with shared heterozygous sites (more than ten sites), and the polymorphic loci genotyped at more than seven samples were finally exported. The resulting super-matrix was then used to infer a maximum likelihood phylogenetic tree using RAxML v. 8.1.24 software (Stamatakis, 2014). In the RAxML analysis, the GTRGAMMA model was specified as the nucleotide evolution model and 1000 bootstrap replicates were generated to assess the node support values. The analysis was performed under the CIPRES Science Gateway portal (Miller et al., 2011). The program FigTree v. 1.3.1 (Rambaut, 2009) was used to visualize the phylogenetic tree.
Population genetic analysis.
After read trimming using Trimmomatic v. 0.32 software (Bolger et al., 2014), the short-read assembler Stacks 1.08 (Catchen et al., 2011) was used to process the ddRAD-seq reads from the Japanese samples of the S. virgaurea complex with the following parameter settings: the minimum number of identical reads required to create a stack (m = 3); the nucleotide mismatches between loci within a single individual (M = 2); and the mismatches between loci when building the catalogue (n = 1). The SNP genotype for each individual was exported with a minimum read depth of 6, using the ‘populations’ command (Catchen et al., 2011). The exported genotype data were then processed using PLINK v. 1.07 software (Purcell et al., 2007) and markers with a minor allele frequency <0.05, missing individual rate >0.8, missing locus rate >0.6 and significant deviation from Hardy–Weinberg equilibrium (P < 0.01) were filtered out. Allelic richness and expected heterozygosity were calculated using nuclear microsatellite markers (nSSRs) for the regional population groups divided by the Japanese geographical boundaries. To infer population genetic structure, a STRUCTURE analysis of unlinked SNPs with 50 000 burn-in steps was performed, followed by 100 000 Markov chain Monte Carlo steps (Pritchard et al., 2000; Falush et al., 2003; Hubisz et al., 2009). Ten independent runs were performed for each K value, ranging from 1 to 10. Complementary to the STRUCTURE analysis, a multivariate clustering method of discriminant analysis of principal components (DAPC) was also performed, which assumes no population genetic model (Jombart et al., 2010). A DAPC analysis was performed for each value of K (1–10).
Population demographic analysis.
The population demography of the Japanese S. virgaurea complex was analysed by applying the five alternative demographic models (Fig. 5A). In model 1, an ancestral population was assumed to be split into five regional populations (as inferred from the STRUCTURE analysis; defined in Supplementary Data Table S2) at a given time (model 1). The model was assumed based on the finding that Japanese populations did not show a hierarchical divergence pattern and was supported by high bootstrap values for the regional nodes (see Results section). Models 2 and 3 considered the demographic scenarios where regional populations diverged from the northern (model 2) and southern (model 3) parts of the archipelago, respectively. The alternatives assumed more complicated histories, where the Solidago populations first split between the northern and southern regions (model 4), and subsequently the two lineages were assumed to have contacted at the central part of Honshu Island at an admixture rate to generate a new population (model 5).
Fig. 5.

(A) Graphic representation of population demographic models 1–5 (M1–M5) with posterior probabilities (PP). (B) Posterior distribution of demographic parameters (left, population size; middle, time parameter; right, admixture ratio).
We employed the Approximate Bayesian Computation (ABC) algorithm implemented in DIYABC 2.01 software (Cornuet et al., 2014) to estimate the posterior parameter distributions for these alternative modes, based on the ddRAD-seq SNP data. After trial runs under different combinations of priors, the prior distributions were set for all the population size parameters as a uniform distribution ranging from 10 000 to 150 000, under which a PCA showed the observed data to be surrounded by the simulated data sets. The summary statistics used for this analysis included (1) mean gene diversity across polymorphic loci, (2) mean FST across loci between any population pairs, and (3) variance of FST across loci between any population pairs. In total, 1 000 000 data sets were simulated for each model, and model checking was carried out by generating 1000 sets of summary statistics (0.01 %) that were compared with the observed values. Model selection was performed based on the posterior probabilities of each model, as described in Sakaguchi et al. (2013). The posterior density of each demographic parameter was estimated based on 5000 simulated data sets (0.05 %) with summary statistics that were closest to the observed data and transformed by logit regression.
RESULTS
Phylogenetic position and structure of East Asian goldenrods
The ddRAD-seq analysis of the East Asian S. virgaurea complex and related taxa resulted in a super-matrix consisting of 4656 loci with 14 565 variable sites. In the reconstructed phylogeny of the S. virgaurea complex (clade 1, Fig. 2), two fully supported clades were recovered: European S. virgaurea s.s. (clade 2) and East Asian S. virgaurea samples (clade 3). The latter had diverged into two subclades: one included all the Japanese samples and a sample from neighbouring southern Korean Peninsula, while the other consisted of the samples from the Russian Far East and Taiwan. Nuclear microsatellite analysis of East Asian population samples (genotyping rate 96.0 %) showed that the Japanese populations were genetically distinct from the surrounding regional populations, with the exception of the shared ancestry in Korean populations at the southern tip of the peninsula (Fig. 3). Although there are vast geographic areas from which no samples were collected, the results of PCA (Fig. 3A) and STRUCTURE analysis (clustering pattern for K = 6 is shown in Fig. 3B) revealed that the phylogeographic structure was strongly dependent on geography. In terms of genetic diversity, relatively high estimates were obtained for the populations in southern Japan and the continental Korean Peninsula and Primorsky area (Fig. 3C). The northernmost species’ range in Kamchatka and one isolated population in Ulleungdo Island harboured unique genetic clusters, which are characterized by the highest levels of genetic drift (FST = 0.46 and 0.36, respectively).
Fig. 2.

Maximum likelihood phylogenetic tree of the Solidago virgaurea complex. The tree represents nucleotide sequences from 4656 restriction-associated DNA (RAD) loci, containing 14 565 variable sites. The numbers on the tree branches show the bootstrap values (only values >95 are shown).
Fig. 3.

Genetic structure of East Asian Solidago virgaurea complex populations. (A) Individual plots of PCA and (B) population structure inferred by STRUCTURE analysis at K = 6 with the relationship between genetic clusters shown as a neighbour-joining (NJ) tree (C). In the NJ tree, the sizes of the circles represent the estimated heterozygosity in genetic clusters and the values indicate the FST values from the assumed ancestral genetic clusters. The geographic map represents the current land area (black) and the area during the last glacial maximum (grey), assuming sea level depression by 130 m.
Population genetic structure inferred from three types of molecular marker
Chloroplast DNA sequences of 198 Japanese samples (Supplementary Data Table S3) included rich genetic polymorphisms, from which 64 distinct haplotypes were detected (GenBank accession nunbers LC198369–LC198433) (Supplementary Data Table S4). The haplotype frequency was uneven, as the four most frequent haplotypes (shown as orange, light blue, green and pale yellow in Fig. 4A) accounted for 50 % of the analysed sample. The haplotype network analysis showed these major haplotypes in the centre, surrounded by many low-frequency derivatives. The major haplotypes were widespread across the archipelago, while the minor haplotypes tended to be population- or region-specific. The chloroplast DNA dataset (Fig. 4) did not correspond to any taxonomy or geography.
Fig. 4.

Phylogeographic structure of the Solidago virgaurea complex in the Japanese Archipelago. Populations with no markings indicate the most widespread taxon of asiatica, and those with coloured triangles and italicized characters show taxa other than asiatica. (A) Geographic distribution of 60 chloroplast haplotypes (cpDNA). A haplotype network connecting the recovered haplotypes is also shown. The four haplotypes with higher frequencies are shown in different colours while those with lower frequencies are white. (B) Geographic distribution of the four genetic clusters detected by STRUCTURE analysis (Pritchard et al., 2000) of 18 nSSRs. (C) Geographic distribution of the five genetic clusters of STRUCTURE analysis estimated based on 304 unlinked ddRAD-seq SNPs. For the STRUCTURE analysis of nSSRs and ddRAD-seq SNPs, the neighbour-joining trees of the detected clusters are also shown, with the circle sizes representing the genetic diversity of each cluster and the values showing the FST values from the assumed ancestral genetic clusters.
Nuclear microsatellite analysis of the Japanese samples recovered 166 alleles over 18 loci (genotyping rate 99.46 %). We did not find individuals with microsatellite genotypes showing more than three alleles at each locus, indicating that they would be diploid plants (although this was not investigated by cytological methods). The multilocus genotype data were used in the model-based clustering STRUCTURE analysis. The log-likelihood of the data for each K increased at a high rate from K = 3 to K = 4 and plateaued at K = 4, indicating that a minimum of four genetic clusters was needed to show the genetic structure of the data (Pritchard et al., 2009). The distribution of the genetic clusters had a geographic basis; e.g. blue was dominant in the northern regions (Hokkaido and the northern half of Honshu Island) and green dominated in the most southerly regions of south-west Honshu, Shikoku and the Kyushu Islands (Fig. 4B). The red cluster was confined to the southernmost Ryukyu Islands and possessed the most derived allele frequencies from the assumed common ancestor, as indicated by the exceptionally high FST value of 0.37. There was no concordance detected between taxonomy and genetic structure.
After filtering out the individuals and SNPs based on the thresholds of missing rates, the ddRAD-seq analysis resulted in a genotype matrix of 304 unlinked SNPs from 173 samples (genotyping rate 65.96 %). Genetic diversity statistics of the regional populations were significantly correlated between nuclear microsatellites and ddRAD-seq SNPs (Pearson’s correlation coefficient r = 0.80, P = 0.01 for allelic richness; r = 0.79, P = 0.01 for expected heterozygosity; Supplementary Data Fig. S1), supporting the power of both nuclear markers to evaluate the genetic variation of the S. virgaurea complex. Population genetic structure was assessed by multivariate analyses of DAPC and STRUCTURE (Supplementary Data Fig. S2). The clustering patterns from K = 2 to K = 6 were mostly consistent between the two analytical methods, but STRUCTURE tended to estimate the individual assignment probability as more admixed and the distribution of the genetic clusters as more geography-based than DAPC. The assignment probability of the STRUCTURE analysis at K = 5, at which the log-likelihood of the data was largest (Supplementary Data Fig. S2b), is shown on a geographic map in Fig. 3C. The spatial genetic structure showed that the five clusters distributed from the north to the south (blue, pink, pale yellow and green to red). Compared with the clustering of the nuclear microsatellite data (Fig. 4B), the distribution of each cluster based on the ddRAD-seq data showed stronger geographic trends (Fig. 4C), owing to an increased number of markers and its bi-allelic nature, which reflect genetic drift stronger than multi-allelic microsatellites (Haasl and Payseur, 2011). The genetic clusters of red (in the southernmost Ryukyu Islands) and blue (in the northern ranges of Hokkaido and northern Honshu Island) were estimated to have higher FST values than the others (FST = 0.16 and 0.13, respectively). In accordance with the microsatellite data, these genetic clusters did not correspond with any taxonomic boundaries for the widespread taxon of lowland S. v. subsp. asiatica var. asiatica, alpine S. v. subsp. leiocarpa var. leiocarpa (shown as l in Fig. 4C) and rheophytic S. yokusaiana (y in Fig. 4C). Taking a disjunct taxon of S. yokusaiana (y) as an example, the regional populations were assigned to different regional genetic clusters (i.e. pale yellow and green clusters in south-western Honshu and Shikoku, and the red cluster in the Ryukyu Islands).
Population genetic demography
Model selection based on posterior probabilities of demographic models showed an overwhelming support for model 5 (posterior probability 0.950; Fig. 5A). Model checking was performed for this model using 1000 data sets simulated from the posteriors, and the generated summary statistics were compared with the observed values. Among the 26 statistics tested, 24 did not deviate significantly from the observed values (P > 0.01). The mean and variance of FST between the blue and pink genetic clusters (northern Japan and central Honshu) showed significant deviations. Based on the demographic model, the extant population sizes were estimated to vary greatly among regions; the population groups of the southern main islands had relatively large population sizes (5.71–6.06 e+4 represented as medians), whereas the northern Japan and southernmost Ryukyu populations were smaller (2.32 and 1.55 e+4, respectively) (Fig. 5B). The first lineage split and population admixture event was estimated to have occurred at a timing of t4 [7950 (95 % quantiles of 4740–8980) generations ago] and t1 [1390 (258–2950) generations ago], respectively (Fig. 5B).
DISCUSSION
Phylogeographic history of East Asian goldenrods
The East Asian S. virgaurea subclade had short branches in the ddRAD-seq phylogenetic tree (Fig. 2), suggesting that after splitting from the European lineage the East Asian populations diverged in a recent time scale. However, our population sampling between East Asia and Europe is inadequate, and more work is needed before drawing conclusions about the phylogenetic history of S. virgaurea complex. The phylogenetic history of S. virgaurea in the Japanese Archipelago seems relatively recent because the ancestral polymorphisms in the chloroplast haplotypes have been retained across the region (Fig. 4A), although the absolute timeframe is unknown in the absence of any calibration points. A star-like chloroplast haplotype network is evidence of population divergence accompanied by population expansions. In line with this, demographic analysis based on ddRAD-seq data showed how the effective population size increased from the ancestral population when the extant population sizes are summed across the regions (Fig. 5).
Significant spatial genetic structure was evident in both nuclear microsatellite and ddRAD-seq analyses of the Japanese S. virgaurea complex (Fig. 4). Reflecting the geographic arrangement of the archipelago, intraspecific lineages of temperate species are often reported to be differentiated along a north–south axis (Okaura et al., 2007; Sakaguchi et al., 2011;Sugahara et al., 2011; Clark et al., 2015; Tono et al., 2015). In the case of S. virgaurea complex, the demographic model, which suggested that the northern and southern lineages split first and then admixed in the central part of the island chain, was selected to account for genetic variation in the Japanese Archipelago. Assuming the observed generation time of 3–4 years (Kawano, 1988), the timing of the north–south lineage split may correspond to the last glacial period, during which the archipelago was once connected to the Asian continent via two land bridges (northern route through Sakhalin and southern route through the Korean Peninsula in Fig. 3). The independent migrations from the continental territories through these land bridges could be considered to generate the supported demographic model, which were subsequently contacted in the middle part of Honshu Island. Palynological evidence suggests that the temperate vegetation became more dominant after the climate improved in Hokkaido Island (~8 kya) (Igarashi et al., 2005). The northernmost blue cluster in the STRUCTURE analysis of the ddRAD-seq data showed a strong drift away from the ancestral population (Fig. 4C), which may have been influenced by recurrent range fluctuations in this area. The populations in the Ryukyu Islands had the lowest genetic diversity (Supplementary Data Fig. S1) and highest FST values compared with the common ancestor (Fig. 4B, C). Sequential dispersals and persistence as small isolated populations in the islands would have caused genetic bottlenecks and drift, respectively.
Discordance between genetic structure and taxonomic boundaries
Considering the remarkable morphological and ecological differentiation in the S. virgaurea complex in the archipelago, it is interesting to note that our phylogenetic analyses failed to detect any genetic structure concordant with taxonomic boundaries. For the narrow endemic taxa, this pattern may be explained simply by the scenario in which they diverged very recently and have not accumulated new mutations and/or they have retained their ancestral polymorphisms that are shared with more widespread taxa (Sakaguchi et al., 2017).
Apart from the local endemic cases, the disjunctly occurring ecotypic taxa (e.g. alpine S. v. subsp. leiocarpa var. leiocarpa and rheophytic S. yokusaiana) do not seem to support the scenario of a recent single origin, because they occur in at least three different regional genetic clusters (Fig. 4C). The shared genetic background among neighbouring or almost sympatric ecotypic taxa within the same regions indicates that phenotypic plasticity could be responsible for the observed phenotypic traits of the plants. However, the role of plasticity in Solidago species is limited compared with the genetic effects on morphology (Schmid and Bazzaz, 1990). Common garden studies of both European and Japanese S. virgaurea populations also demonstrated that genetic adaptation to local environments had overwhelming effects on plant phenotypic traits, compared with the roles of plasticity (Björkman and Holmgren, 1963; Bergstern, 2009; Hirano et al., 2017). These lines of evidence suggest that ecotypic variation in S. virgaurea is based largely on genetic components that were selected by locally differentiated environments.
One plausible explanation for the phylogeographic discordance is the parallel evolution of ecotypic taxa in different genetic clusters. This scenario assumes an ancestral population with genetic evolvability, and novel ecotypic populations evolved via mutations and/or new combinations of standing allelic variations at a set of adaptive loci. Solidago virgaurea is an outcrossing species with effective pollen and seed dispersal abilities and a short generation time (Kawano, 1988). These traits allow populations to effectively respond to natural selection (Charlesworth, 2009). Sakata et al. (2014) reported that Solidago species have evolved defensive traits against an introduced herbivorous insect over only a few generations, highlighting an evolutionary capability to rapidly adapt in the face of novel environments. Speciation can be driven by ecologically based divergent selection and proceeds even in the face of gene flow, depending on the strength of selection (Rundle and Nosil, 2005; Räsänen and Hendry, 2008). This results in a contrasting genomic divergence pattern between the regions surrounding the gene(s) under selection and other neutral regions (Nosil et al., 2009). Thus, the molecular markers from the vast majority of the genome poorly predict the levels of genetic differentiation in regions responsible for phenotypes targeted by divergent selection (Spitze, 1993), a pattern that was observed in this study.
Evolutionary biologists have been interested in the parallel evolution of sets of phenotypic traits in ecologically similar environments in independent populations, as this provides strong evidence for natural selection as the cause of evolution (Schluter and Nagel, 1995). A number of putative cases of parallel speciation have been reported from natural systems, but some factors limit the demonstration of parallel speciation (Nosil, 2012). Incomplete lineage sorting may result in phylogenetic signals consistent with parallel speciation. In this study, however, incomplete sorting is less likely to have had significant effects on the population genetics of the S. virgaurea complex, as we detected geographically well-sorted genetic variation in the nSSR and ddRAD-seq data. Second, gene flow exchanges alleles between ecotypic populations and could obscure the historical relationship between them. Under extensive gene flow, geographically close ecotypic populations share a genetic background in most of their genomes, independently of their historical origins (Quesada et al., 2007). This scenario can also account for the phylogeographic pattern with taxonomic discordance as observed in this study, regardless of the historical origin of the disjunctions (i.e. vicariance, dispersal and parallel evolution). Further testing of the roles of gene flow based on population samples combined with statistical approaches (e.g. ABC algorithms) is needed based on neighbouring population pairs of different ecotypic entities in distant regions.
Recent advances in high-throughput sequencing techniques have opened up a means to directly access the evolutionary dynamics of genomic regions responsible for ecotypic variation. This approach has successfully screened candidate adaptive genes of Arabidopsis species, which are convergently selected in similar environments in geographically distant areas (Turner et al., 2010; Kubota et al., 2015). Turner et al. (2010) re-sequenced the serpentine ecotypic populations of Arabidopsis lyrata in Europe and North America, and found different polymorphisms at two loci showing considerable differentiation between the soil types in the different geographic areas; this provided genetic evidence of convergent evolution. We anticipate that genome-wide scanning of the candidate genes will be sufficiently powerful to further resolve the complex processes that have generated the remarkable ecological and morphological diversity in the S. virgaurea complex by enabling us to examine the evolutionary dynamics of the responsible genetic polymorphisms.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Table S1: information on the plant samples used for phylogenetic analysis based on RAD-seq data. Table S2: information on the plant samples used for population genetic analysis of East Asian Solidago virgaurea complex based on nuclear microsatellite data. Table S3: information on the plant samples used for population genetic analysis within the Japanese Archipelago based on chloroplast DNA sequencing, nuclear microsatellite analysis and RAD-seq. Table S4: GenBank accession numbers for chloroplast DNA sequences for each haplotype. Figure S1: correlation of the diversity indices of regional populations between ddRAD-seq SNPs and nuclear microsatellite data. Figure S2: (a) Bayesian information criterion changes against the number of genetic clusters in the DAPC analysis of the ddRAD-seq data; (b) log-likelihood of the data as a function of the number of clusters in STRUCTURE analysis. (c) Clustering pattern in DAPC analysis (K = 2 to K = 6). (d) Clustering pattern in STRUCTURE analysis (K = 2 to K = 6). The individuals are ordered from north to south (right to left).
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors are grateful to A. Abe, N. Ando, Y. Azuma, T. Denda, K. Dohi, A. Ebihara, T. Fukuda, K. Fuse, Y. Hatanaka, S. Hayashi, K. Horie, M. Inoue, Y. Isagi, K. Ishida, W. Ishiduka, Y. Isshiki, Y. Ito, K. Iwamura, T. Iwasaki, S. Kaneko, G. Kokubugata, S. Kurata, Y. Matsuura, L. Mizusawa, S. Mori, H. Nagamasu, S. Nakagawa, N. Nakahama, J. H. Pak, Y. X. Qiu, M. Saito, Y. Sakata, T. Shiga, T. Shimada, A. Soejima, I. Tamaki, Y. Watanabe, T. Yahara and M. Yokogawa for their kind help in collecting the samples. Some materials from the Ryukyu Islands were provided by the Okinawa Churashima Research Center, the Okinawa Churashima Foundation and Tsukuba Botanical Gardens. The RAD-sequencing experiment was conducted using the Joint Usage/Research Program of the Center for Ecological Research, Kyoto University. Funding was provided by the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (JSPS KAKENHI 16H04827 and 25291085) and the Environmental Research and Technology Development Fund of the Ministry of the Environment SICORP Program of the Japan Science and Technology Agency (grant 4-1403).
LITERATURE CITED
- Abe N, Takasu H. 1983. Further studies on the variations in gross morphology and chromosome number of the Solidago virgaurea complex in Toyama prefecture, Honshu, Japan. Journal of Phytogeography and Taxonomy 31: 103–110. [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48. [DOI] [PubMed] [Google Scholar]
- Bergstern A. 2009. Population differentiation in Solidago virgaurea along altitudinal gradients. Thesis, Uppsala University, Sweden. [Google Scholar]
- Björkman O, Holmgren P. 1963. Adaptability of the photosynthetic apparatus to light intensity in ecotypes from exposed and shaded habitats. Physiologia Plantarum 16: 889–914. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein MJ, Carpten JD, Smith JR. 1996. Modulation of non-templated nucleotide addition by taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20: 1004–1006. [DOI] [PubMed] [Google Scholar]
- Cariou M, Duret L, Charlat S. 2013. Is RAD‐seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecology and Evolution 3: 846–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. 2011. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes – Genomes – Genetics 1: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nature Reviews. Genetics 10: 195–205. [DOI] [PubMed] [Google Scholar]
- Clark LV, Stewart JR, Nishiwaki A et al. . 2015. Genetic structure of Miscanthus sinensis and Miscanthus sacchariflorus in Japan indicates a gradient of bidirectional but asymmetric introgression. Journal of Experimental Botany 66: 4213–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet J-M, Pudlo P, Veyssier J et al. . 2014. DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30: 1187–1189. [DOI] [PubMed] [Google Scholar]
- Crisp MD, Trewick SA, Cook LG. 2011. Hypothesis testing in biogeography. Trends in Ecology & Evolution 26: 66–72. [DOI] [PubMed] [Google Scholar]
- Donoghue MJ, Edwards EJ. 2014. Biome shifts and niche evolution in plants. Annual Review of Ecology, Evolution, and Systematics 45: 547–572. [Google Scholar]
- Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givnish TJ. 2010. Ecology of plant speciation. Taxon 59: 1326–1366. [Google Scholar]
- Goudet J. 1995. FSTAT (Version 1.2): a computer program to calculate F-statistics. Journal of Heredity 86: 485–486. [Google Scholar]
- Haasl RJ, Payseur BA. 2011. Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity 106: 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- Higashi H, Sakaguchi S, Ikeda H, Isagi Y, Setoguchi H. 2013. Multiple introgression events and range shifts in Schizocodon (Diapensiaceae) during the Pleistocene. Botanical Journal of the Linnean Society 173: 46–63. [Google Scholar]
- Hirano M, Sakaguchi S, Takahashi K. 2017. Phenotypic differentiation of the Solidago virgaurea complex along an elevational gradient: insights from a common garden experiment and population genetics. Ecology and Evolution 7: 6949–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M. 1937. Nuntia ad Floram Japoniae. XXXIII. Botanical Magazine, Tokyo 51: 645. [Google Scholar]
- Hubisz MJ, Falush D, Stephens M, Pritchard JK. 2009. Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources 9: 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi Y, Narukawa J, Kato T. 2005. Last 12,000 years vegetation history in and around the Furano Basin, central Hokkaido, Japan. Bulletin of the Tokyo University Forests 114: 115–132. [Google Scholar]
- Iwatsuki K, Yamazaki T, Boufford DE, Ohba H. 1995. Flora of Japan, Vol. IIIb: Angiospermae, Dicotyledoneae, Sympetalae (b). Tokyo: Kodansha. [Google Scholar]
- Jang CS, Yang SG, Oh BU. 2012. A taxonomical review of Solidago japonica and its relatives (Asteraceae). Korean Journal of Plant Taxonomy 42: 40–49. [Google Scholar]
- Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota Y. 2008. Solidago horieana (Asteraceae), a new species from Hokkaido, northern Japan. Journal of Japanese Botany 83: 233–238. [Google Scholar]
- Kawano S. 1988. Life history of Solidago virgaurea. Shokubutsu no Sekai 3. Tokyo: Kyoikusha. [Google Scholar]
- Kiełtyk P, Mirek Z. 2014. Taxonomy of the Solidago virgaurea Group (Asteraceae) in Poland, with special reference to variability along an altitudinal gradient. Folia Geobotanica 49: 259–282. [Google Scholar]
- Kikuchi R, Jae-Hong P, Takahashi H, Maki M. 2010. Disjunct distribution of chloroplast DNA haplotypes in the understory perennial Veratrum album ssp. oxysepalum (Melanthiaceae) in Japan as a result of ancient introgression. New Phytologist 188: 879–891. [DOI] [PubMed] [Google Scholar]
- Kimura M. 1996. Quaternary paleogeography of the Ryukyu Arc. Journal of Geography 105: 259–285. [Google Scholar]
- Kubota S, Iwasaki T, Hanada K et al. . 2015. A genome scan for genes underlying microgeographic-scale local adaptation in a wild Arabidopsis species. PLoS Genetics 11: e1005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn I, Durka W, Klotz S. 2004. BiolFlor: a new plant-trait database as a tool for plant invasion ecology. Diversity and Distributions 10: 363–365. [Google Scholar]
- Magri D, Vendramin GG, Comps B et al. . 2006. A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytologist 171: 199–221. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. 2011. The CIPRES science gateway: a community resource for phylogenetic analyses. In: Proceedings of the 2011 TeraGrid Conference: extreme digital discovery. ACM Digital Library, Article 41.
- El Mousadik A, Petit RJ. 1996. High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L) Skeels endemic to Morocco. Theoretical and Applied Genetics 92: 832–839. [DOI] [PubMed] [Google Scholar]
- Murray MG, Thompson WF. 1980. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research 8: 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. 1987. Molecular evolutionary genetics.New York: Columbia University Press. [Google Scholar]
- Nishizawa T, Kinoshita E, Yakura K, Shimizu T. 2001. Morphological variation of the heads characters in Solidago virgaurea L. inhabiting three mountains in central Honshu. Journal of Phytogeography and Taxonomy 49: 117–127. [Google Scholar]
- Nosil P. 2012. Ecological speciation. Oxford: Oxford University Press. [Google Scholar]
- Nosil P, Funk DJ, Ortiz-Barrientos D. 2009. Divergent selection and heterogeneous genomic divergence. Molecular Ecology 18: 375–402. [DOI] [PubMed] [Google Scholar]
- Ohba H. 1999. Family Araliacecae. In: Iwatsuki K, Boufford DE, Ohba H, eds. Flora of Japan. Tokyo: Kodan-sha. [Google Scholar]
- Okaura T, Quang ND, Ubukata M, Harada K. 2007. Phylogeographic structure and late Quaternary population history of the Japanese oak Quercus mongolica var. crispula and related species revealed by chloroplast DNA variation. Genes & Genetic Systems 82: 465–477. [DOI] [PubMed] [Google Scholar]
- Pante E, Abdelkrim J, Viricel A et al. . 2015. Use of RAD sequencing for delimiting species. Heredity 114: 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. 2006. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLOS ONE 7(5):e37135. doi:10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Wen X, Falush D. 2009. Documentation for structure software: Version 2.3 http://pritch.bsd.uchicago.edu/structure_software/release_versions/v2.3.2/structure_doc.pdf.
- Purcell S, Neale B, Todd-Brown K et al. . 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X-S, Chen C, Comes HP et al. . 2012. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytologist, 196: 617–630. [DOI] [PubMed] [Google Scholar]
- Quesada H, Posada D, Caballero A, Morán P, Rolán‐Alvarez E. 2007. Phylogenetic evidence for multiple sympatric ecological diversification in a marine snail. Evolution 61: 1600–1612. [DOI] [PubMed] [Google Scholar]
- Räsänen K, Hendry AP. 2008. Disentangling interactions between adaptive divergence and gene flow when ecology drives diversification. Ecology Letters 11: 624–636. [DOI] [PubMed] [Google Scholar]
- Rambaut A. 2009. FigTree, ver. 1.3.1. http://tree.bio.ed.ac.uk/software/figtree/. [Google Scholar]
- Rieseberg LH, Willis JH. 2007. Plant speciation. Science 317: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundle HD, Nosil P. 2005. Ecological speciation. Ecology Letters 8: 336–352. [Google Scholar]
- Sakaguchi S, Ito M. 2014. Development and characterization of EST-SSR markers for the Solidago virgaurea complex (Asteraceae) in the Japanese Archipelago. Applications in Plant Sciences 2: 1400035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Takeuchi Y, Yamasaki M, Sakurai S, Isagi Y. 2011. Lineage admixture during postglacial range expansion is responsible for the increased gene diversity of Kalopanax septemlobus in a recently colonised territory. Heredity 107: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Qiu Y-X, Liu Y-H et al. . 2012. Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Molecular Ecology 21: 3823–3838. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Bowman DM, Prior LD, Crisp MD, Linde CC, Tsumura Y, Isagi, Y. 2013. Climate, not Aboriginal landscape burning, controlled the historical demography and distribution of fire-sensitive conifer populations across Australia. Proceedings of the Royal Society of London B: Biological Sciences 280(1773): 20132182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sugino T, Tsumura Y et al. . 2015. High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genetics & Genomes 11: 1–12. [Google Scholar]
- Sakaguchi S, Horie K, Ishikawa N, Nagano AJ, Yasugi M, Kudoh H, Ito M. 2017. Simultaneous evaluation of the effects of geographic, environmental and temporal isolation in ecotypic populations of Solidago virgaurea. New Phytologist 216(4): 1268–1280. [DOI] [PubMed] [Google Scholar]
- Sakata Y, Kaneko S, Hayano A, Inoue-Murayama M, Ohgushi T, Isagi Y. 2013. Isolation and characterization of microsatellite loci in the invasive herb Solidago altissima (Asteraceae). Applications in Plant Sciences 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata Y, Yamasaki M, Isagi Y, Ohgushi T. 2014. An exotic herbivorous insect drives the evolution of resistance in the exotic perennial herb Solidago altissima. Ecology 95: 2569–2578. [Google Scholar]
- Schluter D, Nagel LM. 1995. Parallel speciation by natural selection. American Naturalist 146: 292–301. [Google Scholar]
- Schmid B, Bazzaz FA. 1990. Plasticity in plant size and architecture in rhizome-derived vs. seed-derived Solidago and Aster. Ecology 71: 523–535. [Google Scholar]
- Semple JC. 2013. On the name Solidago mirabilis (Asteraceae: Astereae) and a new name for a Japanese species of goldenrod. Phytoneuron 24: 1–9. doi:10.1890/13-1455.1. [Google Scholar]
- Semple JC. 2016. An intuitive phylogeny and summary of chromosome number variation in the goldenrod genus Solidago. (Asteraceae: Astereae). Phytoneuron 32: 1–9. [Google Scholar]
- Semple JC, Cook RE. 2006. Solidago Linnaeus. In: Flora North America Editorial Committee. ed. Flora of North America. Vol. 20. Asteraceae. Part 2. Astereae and Senecioneae. New York: Oxford University Press, 107–166. http://www.efloras.org/florataxon.aspx?flora_id=1&taxon_id=130659. [Google Scholar]
- Semple JC, Ringius GS, Leeder C, Morton G. 1984. Chromosome numbers of goldenrods, Euthamia and Solidago (Compositae: Astereae). II. Additional counts with comments on cytogeography. Brittonia 36: 280–292. [Google Scholar]
- Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J. 2005. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany 92: 142–166. [DOI] [PubMed] [Google Scholar]
- Slavík B. 2004. Solidago L. – zlatobýl. In: Slavík B, Š t ě pánková J eds. Flora of the Czech Republic 7. Praha: Academia, 114–123. [Google Scholar]
- Sork VL, Gugger PF, Chen J-M, Werth S. 2016. Evolutionary lessons from California plant phylogeography. Proceedings of the National Academy of Sciences of the USA 113: 8064–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitze K. 1993. Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K, Kaneko Y, Ito S et al. . 2011. Phylogeography of Japanese horse chestnut (Aesculus turbinata) in the Japanese Archipelago based on chloroplast DNA haplotypes. Journal of Plant Research 124: 75–83. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Matsuki S. 2016. Morphological variations of the Solidago virgaurea L. complex along an elevational gradient on Mt Norikura, central Japan. Plant Species Biology 32: 238−246. [Google Scholar]
- Takasu H. 1978. Variation and taxonomy of Solidago virgaurea. Shu Seibutsu Kenkyu 2: 54–64. [Google Scholar]
- Takasu H. 1982. A study on the variation in gross morphology of Solidago virgaurea L. sensu lato from Kamtschatka and East Siberia. Journal of Phytogeography and Taxonomy 30: 98–103. [Google Scholar]
- Tono A, Iwasaki T, Seo A, Murakami N. 2015. Environmental factors contribute to the formation and maintenance of the contact zone observed in deciduous broad-leaved tree species in Japan. Journal of Plant Research 128: 535–551. [DOI] [PubMed] [Google Scholar]
- Turesson G. 1925. The plant species in relation to habitat and climate. Hereditas 6: 147–236. [Google Scholar]
- Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV. 2010. Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nature Genetics 42: 260–263. [DOI] [PubMed] [Google Scholar]
- Watanabe K. 2017. Index to chromosome numbers in Asteraceae. Kobe University. http://www.lib.kobe-u.ac.jp/infolib/meta_pub/G0000003asteraceae_e. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.