

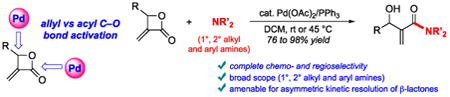
Abstract
A Pd-catalyzed ring-opening of β-lactones with various types of amines (primary, secondary and aryl) to provide β-hydroxy amides with excellent selectivity towards acyl C–O bond cleavage is reported. The utility of this protocol is demonstrated in an asymmetric kinetic resolution providing enantioenriched α-Methylene-β-lactones
Graphical abstract
β-lactones are important intermediates in organic synthesis.1 They can be readily accessed in high enantiomeric purity, and they undergo a broad range of transformations, providing highly functionalized products. As part of our interest in the utility of β-lactones or β-lactone-derived strained heterocycles in organic synthesis, we have reported several of their reactions in the presence of transition metal (TM) catalysts.2 In particular, we reported that α-Methylene-β-lactones 1 readily undergo cross-metathesis reactions2e and recently used this to access a focused library of 3,4-disubstituted β-lactones for proteomic profiling.2c,d A current interest is to develop further useful methods employing 1, especially applications involving ring-opening reactions.
The ring-opening of β-lactones with different nucleophiles has been utilized in the synthesis of biologically important synthetic and natural products. Nevertheless, a major problem in opening β-lactones can be the formation of two isomeric products due to competing alkyl C–O and acyl C–O bond cleavages (Figure 1A).1a In particular, the selective opening of β-lactones with amines has proven to be challenging.3 We hypothesized that α-Methylene-β-lactones 1 could undergo selective ring opening with amine nucleophiles under Pd catalysis (Figure 1B). These unsaturated β-lactones could be expected to undergo allyl C–O bond activation with Pd to provide palladacycle A4 (Figure 1B, path a). Alternatively, the olefin could act as a directing group5 to facilitate the oxidative addition of Pd into the acyl C–O bond to form palladacycle B (Figure 1B, path b). Herein the development of a Pd-catalyzed activation of α-Methylene-β-lactones to provide solely β-hydroxy-α-Methyleneamides in good to excellent yields is reported. The broad scope of the transformation using various β-lactones and amines is described.
Figure 1.

Alkyl vs acyl C–O bond cleavage in β-lactones.
As mentioned above, we postulated that selective opening of α-Methylene-β-lactones might be promoted by oxidative addition of a TM into either the alkyl or acyl C–O bond. There is direct precedent for alkyl C–O bond activation of β-lactones. Puddephatt reported the oxidative addition of oxetan-2-one with a stoichiometric amount of a Pt complex via alkyl C–O cleavage (Figure 2).6 Noels described a Pd-catalyzed opening of vinyl-substituted β-lactones to form butadiene acids.7 This transformation was proposed to involve allyl C–O bond activation to form a palladalactone which then undergoes β-hydride elimination. This mode of activation was utilized by Hattori with vinyl β-lactones generated in situ from the reaction of ketene with α,β-unsaturated aldehydes.8 These allylic systems would appear to be especially relevant to an expectation that Pd-catalysis might be used for alkyl C–O bond cleavage in α-Methylene-β-lactones.
Figure 2.

Transition metal activation of alkyl C–O bonds in β-lactones.
There are, to our knowledge, no reports of TM-catalyzed ring opening of β-lactones at the acyl C–O bond. Consequently we looked into TM-catalyzed coupling of esters with amines. Certain types of esters, activated with α-aromatic/heteroaromatic or CF3 substituents or certain alkoxy moieties, have been shown to undergo TM-catalyzed acyl C–O bond activation. The intermediates can be cross-coupled to form ketones9 or undergo decarbonylation10 before reductive coupling. Of potential direct relevance, Bao and coworkers recently developed a Pd-catalyzed amidation of activated esters that was believed to involve an acyl C–O insertion with a Pd catalyst.11 Also, the Garg group utilized a nickel catalyst for the activation of aromatic methyl esters for amide formation.12 We surmised that the α-Methylene could play the role of an activating group for C–O insertion.
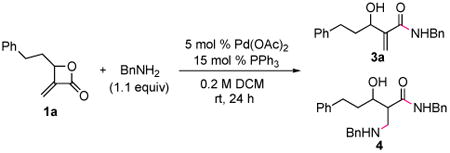
We initially probed the ring-opening of α-Methylene-β-lactone 1a with benzylamine in the presence of a catalytic amount of Pd(OAc)2 and PPh3 in DCM at rt (Table 1). The use of 2 equivalents of benzylamine provided a 4:1 mixture of β-hydroxy amides 3a and 4 (entry 1). The latter was believed to arise from aza-Michael addition of the excess amine to product 3a. After optimization of the concentration and of the molar ratio of benzylamine, 3a was isolated in 92% yield (entry 2). At 45 °C, complete conversion was achieved after 12 h, providing 3a in nearly quantitative yield. Other solvents, such as THF and CHCl3, as well as biphosphine ligands (BINAP and SEGPHOS), gave outcomes similar to entry 2. Other Pd sources (entries 4 to 6) also promoted the transformation. Notably, the use of catalytic Pd(PPh3)4 without exogenous phosphine ligand provided an efficient conversion, but 3a and 4 were formed in a 10:1 ratio. When the reaction was carried out in the absence of Pd catalyst or phosphine ligand (entries 7–9), no significant conversion was observed. It is also worth noting that the other possible product, β-amino acid 2 (Figure 1B, path a) was never observed. Consistent with our alternate hypothesis (Figure 1B, path b), these results indicate that the reaction is promoted by initial oxidative addition of Pd(0) to the acyl C–O bond of β-lactone 1.
Table 1. Initial Studies on the Pd-Catalyzed Amidation of β-Lactone 1a with Benzyl Aminea.
| |||
|---|---|---|---|
|
| |||
| entry | variation from general conditions | ratio 3a:4b | yield 3ac |
| 1 | 2 equiv of BnNH2, 0.5 M | 4:1 | 80% |
| 2 | none | >20:1 | 92% |
| 3 | 45 °C, 12 h | >20:1 | 98% |
| 4 | 2 mol % [Pd(allyl)Cl]2; 12 mol % PPh3 | >20:1 | 90%d |
| 5 | 2 mol % Pd2(dba)3; 12 mol % PPh3 | >20:1 | 95%d |
| 6 | 5 mol % Pd(PPh3)4; no PPh3 | 10:1 | 85%d |
| 7 | 5 mol % Pd2(dba)3; no PPh3 | - | n. r.e |
| 8 | no Pd(OAc)2 | - | <5% convb |
| 9 | no PPh3 | - | ∼10% convb |
General conditions: 0.1 mmol 1a, benzylamine (1.1 equiv), Pd catalyst (5 mol % Pd(OAc)2), 15 mol % PPh3 in DCM (0.2 M) atrt for 24 h.
Ratios and conversions were estimated by 1H NMR analysis of the crude reaction mixture.
Isolated yields except where noted.
1H NMR yields using 1,3,5-trimethoxybenzene as internal standard.
The starting material was recovered; Pd black was observed on the wall of the reaction tube.
The optimized conditions shown in the reaction scheme in Table 1 were utilized for the ring-opening of several α-Methylene-β-lactones with various types of amines. As highlighted in Scheme 1, primary, secondary and allyl amines provided β-hydroxy amides 3a-f in good to excellent yields. The outcome observed with morpholine (3e) is noteworthy. Adam and coworkers found that cyclic, secondary amines (such piperidine and pyrrolidine) reacted with α-Methylene-β-lactones in the absence of a Pd catalyst to give conjugate addition products.13
Scheme 1. Scope of Pd-Catalyzed Amidation of α-Methylene-β-lactonesa with Various Aminesb.

aFor the syntheses of the α-Methylene-β-lactones see the Supporting Information. bGeneral conditions: 0.1 to 0.2 mmol 1 (1 equiv), amine (1.1 equiv), Pd(OAc)2 (5 mol %), PPh3 (15 mol%) in DCM (0.2 M) at rt or 45 °C for 24 h. For aryl amines: 2-4 equiv of aryl amine was used in DCM (0.5 M) at 45 °C. c1.0 mmol scale.
Aryl amines were also successfully coupled to give exclusively the corresponding amides. When these amines were used in excess (2-4 equivalents), and the reaction was conducted at 45 °C β-hydroxy-α-Methylene arylamides 3g-j were obtained in high yields.
To extend the generality of this method, we next explored whether the Pd-catalyzed ring-opening can be used for simple β-lactones. As shown in Scheme 2, α-phenyl-β-lactone 4 underwent facile ring-opening with benzylamine, providing the ring-opened product in excellent yield at rt. Racemic trans-disubstituted β-lactone 5 also gave the desired product with complete selectivity, albeit in slightly lower yield. Notably, when β-lactone 4 or 5 was reacted with benzylamine in the absence of a Pd catalyst, the reaction was messier (based on 1H NMRs of crude reaction mixtures), and the isolated yield for 3k (65%) or 3l (52%) was lower. Homochiral β-lactone (R)-1a (99% ee) also underwent ring opening to yield β-hydroxy amide (R)-3a (99% ee) without erosion of stereochemical integrity. Likewise, α-alkylidene-β-lactone 6, prepared by the Ru-catalyzed cross-metathesis of its corresponding α-Methylene-β-lactone,2e provided α-alkylidene-β-hydroxy amide with complete retention of olefin geometry.
Scheme 2. Scope of Pd-Catalyzed Amidation of Various Types of β-lactonesa.

aFor the syntheses of the β-lactones see the Supporting Information. General conditions: 0.2 mmol 1 (1 equiv), amine (1.1 equiv), Pd(OAc)2 (5 mol %), PPh3 (15 mol %) in DCM (0.2 M) at rt or 45 °C for 24 h.
To date, enantioenriched α-Methylene-β-lactones 1 have only been accessed via enzymatic kinetic resolution.14 Our interest in α-Methylene-β-lactones 1 as privileged intermediates in organic synthesis led us to explore the Pd-catalyzed amidation for potential resolution of racemic β-lactones. Several chiral phosphine ligands typically used in asymmetric Pd-catalyzed C–N bond coupling reactions were evaluated (Scheme 3). Racemic β-lactone 1a underwent efficient amidation. Reactions were monitored by 1H NMR analysis and were quenched after obtaining ∼50-55% conversions, typically after 16 to 20 h. (R)-BINAP and (R)-SEGPhos (not shown) did not provide any selectivity. When chiral Trost ligands 15 L2 and L3 were utilized, 5–38% ee's were obtained. The use of chiral spiroketal phosphine (SKP) ligands, recently developed by Ding and co-workers,16 provided improved resolution, up to 68% ee (using SKP-L4). Further optimization (such as the use of various Pd sources, solvents, type and amounts of amine, reaction concentration, and temperature) did not improve the enantioselectivities. The conditions developed above for Pd-catalyzed asymmetric kinetic resolution were utilized for other β-lactones. With the exception of α-phenyl-β-lactone 4, good yields and moderate enantioselectivities were obtained.
Scheme 3. Pd-Catalyzed Asymmetric Kinetic Resolution of β-lactonesa.

aGeneral conditions: 0.1 to 0.2 mmol 1 (1 equiv), amine (1 equiv), Pd(OAc)2 (2 mol %), chiral ligand (5 mol %) in CDCl3 (0.2 M) at rt for 16 to 20 h.
In conclusion, we have developed a highly selective Pd-catalyzed ring opening of α-Methylene-β-lactones and β-lactones with various types of amines (primary, secondary, and aryl) to give amides via acyl C–O activation. The complete chemoselectivity and efficiency of the transformation are remarkable. Moreover, enantioenriched α-Methylene-β-lactones can be obtained through kinetic resolution by using chiral phosphine ligands. The kinetic resolution of α-Methylene-β-lactones has previously only been achieved by an enzymatic process.
Supplementary Material
Acknowledgments
This paper is based upon work partially supported by the National Institutes of Health (NIH) under Grant: R01 CA193994. Boehringer Ingelheim (BI Fellowship for C.A.M.) is acknowledged for financial support. S.M.acknowledges support from the University of Connecticut McNair Scholars program.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, analytical and spectral data for all new compounds and HPLC traces for kinetic resolution experiments (PDF).
References
- 1.For selected reviews see: Nelson SG, Dura RD, Peelen TJ. Org React. 2013;82:471.; (b) Wang Y, Tennyson RL, Romo D. Heterocycles. 2004;64:605. [Google Scholar]; (c) Calter MA, Orr RK. Tetrahedron. 2003;59:3545. [Google Scholar]; (d) Schneider C. Angew Chem Int Ed. 2002;41:744. doi: 10.1002/1521-3773(20020301)41:5<744::aid-anie744>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (e) Yang HW, Romo D. Tetrahedron. 1999;55:6403. [Google Scholar]
- 2.(a) Malapit CA, Howell AR. J Org Chem. 2015;80:8489. doi: 10.1021/acs.joc.5b01255. [DOI] [PubMed] [Google Scholar]; (b) Malapit CA, Chitale SM, Thakur MS, Taboada R, Howell AR. J Org Chem. 2015;80:5196. doi: 10.1021/acs.joc.5b00604. [DOI] [PubMed] [Google Scholar]; (c) Camara K, Lasota CC, Kamat SS, Cravatt BF, Howell AR. Bioorg Med Chem Lett. 2015;25:317. doi: 10.1016/j.bmcl.2014.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kamat SS, Camara K, Parsons WH, Chen DH, Dix MM, Bird TD, Howell AR, Cravatt BF. Nat Chem Biol. 2015;11:164. doi: 10.1038/nchembio.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Raju R, Howell AR. Org Lett. 2006;8:2139. doi: 10.1021/ol060624v. [DOI] [PubMed] [Google Scholar]
- 3.For selected examples see: Wiedemann EH, Mandl FA, Blank ID, Ochsenfeld C, Ofial AR, Sieber SA. Chem Plus Chem. 2015;80:1673. doi: 10.1002/cplu.201500246.; (b) Noel A, Delpech B, Crich D. Org Biomol Chem. 2012;10:6480. doi: 10.1039/c2ob25640a. [DOI] [PubMed] [Google Scholar]; (c) Calter MA, Guo X. J Org Chem. 1998;63:5308. [Google Scholar]; (d) Ratemi ES, Vederas JC. Tetrahedron Lett. 1994;35:7605. [Google Scholar]; (e) Griesbeck A, Seebach D. Helv Chim Acta. 1987;70:1326. [Google Scholar]; (f) Arnold LD, Kalantar TH, Vederas JC. J Am Chem Soc. 1985;107:7105. [Google Scholar]
- 4.For the intermediacy of an α-alkylidenepallada-γ-lactone see: Choi JC, Shiraishi K, Takenaka Y, Yasuda H, Sakakura T. Organometallics. 2013;32:3411.
- 5.(a) Gandeepan P, Cheng CH. Org Lett. 2013;15:2084. doi: 10.1021/ol400792y. [DOI] [PubMed] [Google Scholar]; (b) Gandeepan P, Cheng CH. J Am Chem Soc. 2012;134:5738. doi: 10.1021/ja300168m. [DOI] [PubMed] [Google Scholar]; (c) Yamada S, Obora Y, Sakaguchi S, Ishii Y. Bull Chem Soc Jpn. 2007;80:1194. [Google Scholar]
- 6.(b) Aye KT, Colpitts D, Ferguson G, Puddephatt RJ. Organometallics. 1988;7:1454. [Google Scholar]; (b) Aye KT, Gelmini L, Payne NC, Vittal JJ, Puddephatt RJ. J Am Chem Soc. 1990;112:2465. [Google Scholar]
- 7.Noels AF, Herman JJ, Teyssie P. J Org Chem. 1976;41:2527. [Google Scholar]
- 8.(a) Hattori T, Suzuki Y, Uesugi O, Oi S, Miyano S. Chem Commun. 2000:73. [Google Scholar]; (b) Hattori T, Suzuki Y, Ito Y, Hotta D, Miyano S. Tetrahedron. 2002;58:5215. [Google Scholar]
- 9.For selected examples of TM-catalyzed acyl C–O bond activation of esters see: Halima TB, Zhang W, Yalaoui I, Hong X, Yang YF, Houk KN, Newman SG. J Am Chem Soc. 2017;139:13ll. doi: 10.1021/jacs.6b12329.; Tatamidani H, Yokota K, Kakiuchi F, Chatani N. J Org Chem. 2004;69:5615. doi: 10.1021/jo0492719. [DOI] [PubMed] [Google Scholar]
- 10.For selected examples of TM-catalyzed activation of esters with decarbonylation see: Muto K, Hatakeyama T, Itami K, Yamaguchi J. Org Lett. 2016;18:5106. doi: 10.1021/acs.orglett.6b02556.; (b) Pu X, Hu J, Zhao Y, Shi Z. ACS Catal. 2016;6:6692. [Google Scholar]; (c) Muto K, Yamaguchi J, Musaev DG, Itami K. Nat Commun. 2015;6:7508. doi: 10.1038/ncomms8508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) La Berge NA, Love JA. Eur J Org Chem. 2015:5546. [Google Scholar]; (e) Kakino R, Shimizu I, Yamamoto A. Bull Chem Soc Jpn. 2001;74:371. [Google Scholar]
- 11.Bao YS, Bao Z, Bao A, Baiyin MH, Jia ML. J Org Chem. 2014;79:803. doi: 10.1021/jo4023974. [DOI] [PubMed] [Google Scholar]
- 12.Hie L, Fine Nathel NF, Hong X, Yang YF, Houk KN, Garg NK. Angew Chem Int Ed. 2016;55:2810. doi: 10.1002/anie.201511486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adam W, Nava-Salgado VO. J Org Chem. 1995;60:578. [Google Scholar]
- 14.Adam W, Groer P, Saha-Moller CR. Tetrahedron Asymm. 1997;8:833. [Google Scholar]
- 15.(a) Trost BM, Van Vranken DL, Bingel C. J Am Chem Soc. 1992;114:9327. [Google Scholar]; (b) Trost BM, Bunt RC, Lemoine RC, Calkins TL. J Am Chem Soc. 2000;122:5968. [Google Scholar]
- 16.(a) Wang X, Guo P, Han Z, Wang X, Ding K. J Am Chem Soc. 2014;136:405. doi: 10.1021/ja410707q. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Meng F, Wang Y, Han Z, Chen Y, Liu L, Wang Z, Ding K. Angew Chem Int Ed. 2012;51:9276. doi: 10.1002/anie.201204925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.