

Abstract
Schistosomiasis is a debilitating neglected tropical disease, caused by flatworms of Schistosoma genus. The treatment relies on a single drug, praziquantel (PZQ), making the discovery of new compounds extremely urgent. In this work, we integrated QSAR-based virtual screening (VS) of Schistosoma mansoni thioredoxin glutathione reductase (SmTGR) inhibitors and high content screening (HCS) aiming to discover new antischistosomal agents. Initially, binary QSAR models for inhibition of SmTGR were developed and validated using the Organization for Economic Co-operation and Development (OECD) guidance. Using these models, we prioritized 29 compounds for further testing in two HCS platforms based on image analysis of assay plates. Among them, 2-[2-(3-methyl-4-nitro-5-isoxazolyl)vinyl]pyridine and 2-(benzylsulfonyl)-1,3-benzothiazole, two compounds representing new chemical scaffolds have activity against schistosomula and adult worms at low micromolar concentrations and therefore represent promising antischistosomal hits for further hit-to-lead optimization.
Graphical Abstract
INTRODUCTION
Schistosomiasis is a neglected tropical disease caused by flatworms of the genus Schistosoma. These worms cause a chronic and often debilitating infection that impairs development and productivity, and exposure to these worms is strongly linked to extreme poverty.1–4 Recent estimates of World Health Organization suggest that around 258 million people are infected resulting up to 200000 deaths annually. Currently, schistosomiasis is endemic in 78 countries worldwide, mainly in sub-Saharan Africa, the Middle East, the Caribbean, and South America, where infections are mediated through poor knowledge about the disease, poor sanitation, and lack of effective health policies.5
In the absence of a vaccine, the control of schistosomiasis relies on a single drug, praziquantel (PZQ), which has been used in clinical practice for almost four decades.6 However, because of high incidence of reinfection, the widespread and repeated use of this drug in endemic areas raises concerns about the development of drug resistance by the parasite.7–11 This problem is further emphasized by the known lack of efficacy of PZQ against juvenile worms,12 which is a potential cause of treatment failure in endemic areas. Hence, there is an urgent need for new antischistosomal drugs with novel mechanisms of action.
The complete genome sequencing of Schistosoma mansoni,13,14 Schistosoma japonicum,15 and Schistosoma hematobium16 has provided new information on their biological pathways, identifying potentially relevant targets for therapeutic intervention.17 Thioredoxin glutathione reductase (TGR) is one of these targets; it plays a crucial role in the redox homeostasis of the parasite.18 TGR is a multifunctional enzyme that acts in the detoxification of reactive oxygen species (ROS) generated by digestion of red blood cells19,20 and by the host immune system.21,22 In mammalian cells, there are two major systems to detoxify ROS, one is based on glutathione (GSH) and the other is based on thioredoxin (Trx). In both systems, NADPH provides reducing equivalents via two specialized oxidoreductase flavoenzymes. Glutathione reductase (GR) reduces glutathione disulfide (GSSG) and drives the GSH-dependent systems, whereas Trx reductases (TR) are pivotal in the Trx-dependent system. On the other hand, in schistosomes, thiol redox homeostasis is completely dependent on TGR, which controls the NADPH reduction of GSSG and Trx in both systems.23–25 Given these characteristics, it is expected that the maintenance of the homeostatic levels of Trx and GSH in schistosomes play a key role in a variety of cellular processes such as defense against oxidative stress, DNA synthesis, detoxification, protein folding, and repair.26 Moreover, RNA interference studies have showed that inactivation of TGR of S. mansoni (SmTGR)18 and TGR of S. japonicum (SjTGR)27,28 has profound effects on worm survival rates both in culture medium and infected mice.
Due to the importance of TGR in parasite’s redox balance, we hypothesized that known SmTGR inhibitors listed on publicly available databases may serve as the chemical basis to discover new antischistosomal compounds by virtual screening (VS). Docking-based and pharmacophore-based approaches are the most popular VS strategies to identify putative hits in chemical libraries. However, in recent years, quantitative structure–activity relationships (QSAR) models have been used widely in VS applications as well.29–35
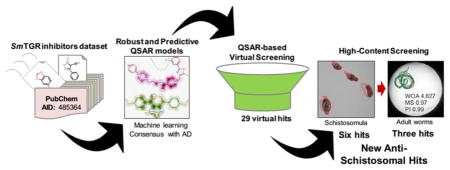
The main goal of this study was the identification of new structurally dissimilar compounds with high antischistosomal activity. To achieve this goal, we designed a study with the following steps: (i) collection, rigorously curation, and integration of the largest possible data set of SmTGR inhibitors, (ii) development of rigorously validated and mechanistically interpretable binary QSAR models, (iii) application of generated models for VS of three subsets from ChemBridge library (~150000 compounds), (iv) interpretation of developed models to derive structural rules useful for targeted design of new inhibitors, and (v) experimental validation of prioritized/designed hits on live schistosomula and adult worms in two distinct HCS platforms. As a result of this study, we found that the QSAR models were efficient for prediction of new SmTGR inhibitors and identified six novel antischistosomal hit compounds active against schistosomula and three hits active against adult worms. Among them, two hits, 2-[2-(3-methyl-4-nitro-5-isoxazolyl)vinyl]pyridine (3) and 2-(benzylsulfonyl)-1,3-benzothiazole (4), representing new chemical scaffolds structurally dissimilar to known inhibitors of S. mansoni, could be considered as promising antischistosomal agents.
RESULTS AND DISCUSSION
Data Set Balancing
Initially, thousands compounds with SmTGR inhibition data were retrieved from the PubChem Bioassay Database (AID: 485364) and used to build binary QSAR models. Further, uncurated chemical structures were standardized, duplicates were removed, and 2854 compounds with reproducible potency (IC50) ≤10 μM were considered as inhibitors, whereas the remaining 337327 compounds were considered as noninhibitors. Because the original data set was highly unbalanced, i.e., 2854 inhibitors and 337327 non-inhibitors (1:118 ratio), it is not recommended for building binary QSAR models for the entire data set. During model building, most machine learning methods need equal weighting of the classes in terms of both the number of instances and the level of importance (i.e., active class has the same importance as inactive class). Consequently, when trying to predict a minority class in an unbalanced data set, machine learning methods are prone to assign most samples to the majority class, resulting in a large number of erroneous predictions for minority class.36
To reduce the number of the noninhibitors and ideally maintain the “chemical space” of the original data set, we evaluated the optimal number of representative compounds. To accomplish this task, we developed an undersampling workflow based on k-nearest neighbors (kNN) distances of the each noninhibitor to all inhibitors using the public available 166 substructures MACCS keys. We tested different sizes of the data set by removing noninhibitors and changing the inhibitors-to-noninhibitors ratios of 1:1 (balanced), 1:2, and 1:3.
To visualize the structural diversity of our data set before and after balancing, we performed a principal component analysis (PCA). PCA transforms the original measured variables into new orthogonal variables called principal components, which are a linear combination of the original variables. Detailed results of structural diversity investigation are shown in Supporting Information, Figure S1. The top two principal components retained 20% of the original information. Supporting Information, Figure S1A, represents the PCA plot of 2854 inhibitors (blue dots) vs all 337327 noninhibitors (gray dots). As we can see, the inhibitors are widely distributed across chemical space, reflecting significant chemical diversity. Supporting Information, Figure S1B–D, shows the non-inhibitors selected with different ratios: 1:1 or 2854 non-inhibitors, Figure S1B; 1:2 or 5705 noninhibitors, Figure S1C; and 1:3 or 8562 noninhibitors, Figure S1D. As we can see from the distribution of these dots, the most representative compounds were chosen that allowed minimal reduction of the original chemical space.
Performance of Individual QSAR Models
The balanced (ratio of 1:1) and unbalanced data sets (ratios of 1:2 and 1:3) were modeled by a combination of AtomPair,37,38 molecular access system (MACCS),39–41 and Morgan fingerprints,38,42 chemistry development kit (CDK),43 and Dragon descriptors44,45 along with eight machine learning methods leading to 120 different binary QSAR models (Supporting Information, Tables S1, S2, and S3). According to the statistical results of a 5-fold external cross-validation procedure, we could draw three general conclusions: (i) random forest (RF), support vector machine (SVM), and gradient boosting machine (GBM) methods showed the best prediction ability among the eight tested machine learning methods; (ii) QSAR models built on balanced data sets are better than unbalanced (1:2 and 1:3 ratios) due to discrepant values between sensitivity (SE) and specificity (SP), the latter are prone to assign most samples as noninhibitors, resulting in a large number of erroneous predictions; and (iii) the QSAR models which were built from the balanced data set showed a high level of agreement between correct classification rate (CCR), SP, and SE values. Table 1 shows the detailed performances of the more predictive QSAR models derived from the balanced data set.
Table 1.
Summarized Statistical Characteristics of QSAR Models Developed with Balanced Dataseta
| model | CCR | k | SE | SP | coverage |
|---|---|---|---|---|---|
| Morgan–RF | 0.85 | 0.71 | 0.85 | 0.86 | 0.62 |
| MACCS–RF | 0.83 | 0.66 | 0.83 | 0.83 | 0.67 |
| AtomPair–SVM | 0.81 | 0.62 | 0.81 | 0.81 | 0.65 |
| AtomPair–GBM | 0.81 | 0.62 | 0.81 | 0.81 | 0.65 |
| Dragon–SVM | 0.85 | 0.70 | 0.85 | 0.84 | 0.69 |
| Dragon–GBM | 0.85 | 0.70 | 0.85 | 0.84 | 0.69 |
| CDK–SVM | 0.84 | 0.69 | 0.85 | 0.84 | 0.77 |
| consensus | 0.87 | 0.74 | 0.87 | 0.88 | 1.00 |
| consensus rigor | 0.91 | 0.81 | 0.96 | 0.87 | 0.38 |
RF, random forest; SVM, support vector machine; GBM, gradient boosting machine; CCR, correct classification rate; k, Cohen’s κ coefficient; SE, sensitivity; SP, specificity. Consensus and consensus rigor models were built by averaging the predicted values from the individual model for each machine learning technique (Morgan–RF, MACCS–RF, AtomPair–SVM, Dragon–SVM, and CDK–SVM).
The combination of Morgan fingerprints with RF (Morgan–RF), MACCS key with RF (MACCS–RF), AtomPair finger-prints with SVM (AtomPair–SVM) and GBM (AtomPair–GBM), Dragon descriptors with SVM (Dragon–SVM) and GBM (Dragon–GBM), and CDK descriptors with SVM (CDK–SVM) led to more predictive QSAR models, with correct classification rate (CCR) ranging between 0.81 and 0.85 and coverage of 0.62–0.77 (Table 1). The best individual model was built using the combination of Morgan–RF (CCR = 0.85, SE = 0.85, and SP = 0.86).
To ensure that the accuracy of the models was not due to chance correlation, 10 rounds of Y-randomization were performed for each data set (Supporting Information, Table S4). The results from this analysis (CCR values around 0.50) indicate that our models built using balanced data set are statistically robust.
Performance of Consensus Models
Several individual QSAR models were generated using multiple machine learning algorithms and descriptors/fingerprints. However, our previous experience suggests that consensus models that combine individual QSAR models are advantageous46–49 and naturally minimize prediction errors during a VS campaign. Therefore, consensus models were built by averaging the predicted values obtained after combining the individual models built using the balanced data set. The detailed performances of 12 consensus models are given in Supporting Information, Table S5. Among them, the consensus model built by combining the Morgan–RF, MACCS–RF, AtomPair–SVM, Dragon–SVM, and CDK–SVM (Table 1 and Supporting Information, Table S5) showed the best performance among all constructed consensus models (CCR = 0.87, SE = 0.87, and SP = 0.88). This consensus model discriminates inhibitors and noninhibitors better than any of the individual QSAR models, with a 2% of increase in CCR, SE, and SP when compared with the best individual model (Morgan–RF).
In addition, the most rigorous consensus model (consensus rigor)46 was built by combining five individual models with more restrictive conditions. A consensus rigor model only considers the outcome to be reliable when a compound was inside the applicability domain (AD) for the five models. If the compound was outside the AD for any model, then the outcome was specified as unreliable. Expectedly, the combination of Morgan–RF, MACCS–RF, AtomPair–SVM, Dragon–SVM, and CDK–SVM models (Tables 1 and Supporting Information, Table S5) also showed the best performance among all built consensus rigor models (CCR = 0.91, SE = 0.96, and SP = 0.87). In summary, the best consensus rigor model demonstrated better statistical results, with a 5% of increase in CCR, and 11% of increase in SE when compared with the best individual model (Morgan–RF). Although the AD of consensus rigor is limited only for certain chemical classes (coverage of 0.38), it has very high predictive power (CCR = 0.91).
Model Interpretation
The Morgan–RF model exhibited the best predictive performance, and, consequently, it possesses the features that are best correlated with SmTGR inhibition activity. Therefore, we translated its features (fingerprints) into predicted probability maps (PPMs) and visualized the atomic and fragment contributions predicted by the QSAR model (Figures 1 and 2). Atoms and fragments promoting the inhibition are highlighted by green (Figure 1), atoms and fragments decreasing the inhibitory potential are highlighted by purple (Figure 2), and gray lines (Figures 1 and 2) delimit the region of split between the favorable and the unfavorable contributions.50
Figure 1.

Favorable fragments (green) for SmTGR inhibition predicted by the best individual QSAR model and their respective frequencies in inhibitors and noninhibitors sets.
Figure 2.

Unfavorable fragments (purple) for SmTGR inhibition predicted by the best individual QSAR model and their respective frequencies in inhibitors and noninhibitors sets.
Analyzing the fragments with favorable contributions highlighted by PPMs, we noticed that 14 fragments were more frequent in the inhibitors set and absent in the noninhibitors set (Figure 1). Examples of favorable fragments for SmTGR inhibition activity are nitrofuran, 2-ethenylfuran, (ethanesulfonyl) benzene, 2-(sulfonylmethyl) furan, carbonyl thiourea, and 4-methanesulfonyl-1,3-oxazole. By analyzing the fragments with unfavorable contribution into SmTGR inhibition activity (Figure 2), several fragments, such as benzylsulfonamide, methylurea, morpholine-4-carbonyl, piperidine-4-carboxamide, 1-methanesulfonylpiperidine, and cyclohexanecarbonyl, were more frequent in the noninhibitors set. Compounds that contain these fragments may show a decreased SmTGR inhibitory activity. This information could be useful for designing or optimizing new SmTGR inhibitors by replacing unfavorable fragments by favorable fragments.
Reaction Mechanism of SmTGR Inhibition
Although the inhibition mechanisms of most of the SmTGR inhibitors are not well understood at the molecular level, the reaction mechanisms by which oxadiazole-2-oxides and cephalosporins operate could be identified according to a graphical interpretation of PPMs. However, for the best understanding of molecular inhibition mechanisms, it is important to highlight that the active site of SmTGR is composed by a cysteine pair (Cys28/Cys31) in the glutharedoxin domain, a cysteine pair (Cys154/Cys159) in the thioredoxin domain, and a redox-active cysteine/selenocysteine pair (Cys596/Sec597) in the C-terminal tail. The latter should be highly mobile to accept electrons from the Cys154/Cys159 pair and to donate electron pairs to Cys28/Cys31 pair.24 These amino acids provide the perfect chemical environment for covalent inhibition. The higher nucleophilicity and low pKa of the selenol group of Sec are thought to confer Sec a catalytic advantage over Cys at the attacking position.51–53 Nonetheless, the thioredoxin domain contains His571 and Glu576, a catalytic dyad that can facilitate proton abstraction of Cys159, thus impacting the catalytic efficiency of the thioredoxin domain of SmTGR.24
We observed that the carbons 3 and 4 of the oxadiazole-2-oxide core presented the most important contributions for SmTGR inhibition activity (Figure 3A–C). With PPMs information for this chemotype, a mechanistic rationale for inhibition was initiated through nucleophilic attack (presumably by a thiolate or selenoate of Cys or Sec, respectively) at either the position 3 or 4 of the oxadiazole ring and subsequent rearrangement of the heterocycle in a manner that allows release of the nitroxyl anion. An enzymatic oxidation is posited to transform this agent to nitric oxide (Figure 3D). These pieces of information corroborate with mechanism of inhibition proposed by Rai and colleagues54 and mechanism of nitric oxide release in physiological solution under the action of thiols studied by Gasco and colleagues55 In addition, PPMs indicated that the presence of amine-oxide group in core and electron-withdrawing substituents, such as carbonyl, at R1 and R2 positions are favorable for SmTGR inhibition (Figure 3A), while removal of the amine-oxide group (Figure 3B) and presence of electron deficient substituents at R1 and R2 positions (Figure 3C) leads to modest potencies in terms of SmTGR inhibition. These pieces of information corroborate with structure–activity relationships rules established by Rai and colleagues.54
Figure 3.

Predicted probability maps generated for oxadiazoles (A, B, and C) and their proposed reaction mechanism in the SmTGR active site (D).
The reaction mechanism by which cephalosporins exert their SmTGR inhibition activity was also proposed using the PPMs information (Supporting Information, Figure S2A). For both compounds, the PPMs picked up the positive contributions of the basic core structure of cephalosporines, more specifically carbon 8 and nitrogen 5 of β-lactam ring, and partially positive contribution of 1-methyl-5-tetrazolethione core for inhibition of SmTGR. On the basis of these results, we suggest that inhibition of SmTGR by cephalosporins may occur via a mechanism similar to proposed by Triboulet and colleagues,56 i.e., a nucleophilic attack of Cys or Sec on β-lactam carbonyl carbon, with formation of a tetrahedral intermediate, which collapses with β-lactam ring opening by N5–C8 bond fission. Then, the acyl-enzyme intermediate could hydrolyze or react further, with expulsion of the 1-methyl-5-tetrazolethione from carbon 3 generating a reactive methylene that could be trapped by other thiolate or selenoate (Supporting Information, Figure S2B).
QSAR-Based Virtual Screening
The QSAR-based VS was carried out following the workflow presented in Figure 4. Initially, 150000 compounds available on PremiumSet, DIVERSet-CL, and DIVERSet-EXP libraries of ChemBridge were downloaded and prepared for VS. As drug-like ligands are highly desirable for the development of new leads with good oral bioavailability, we first filtered these libraries and excluded 1285 compounds that violated Veber57 and Lipinski’s rules.58 The remaining compounds were predicted by the consensus and consensus rigor models. To narrow down the compounds list and to obtain the highest level of confidence for each prediction, we took both the consensus score (average class prediction) and consensus model coverage into consideration. Consensus model coverage was defined as a fraction of individual models for which a compound was found to fall within the respective ADs. In that sense, introducing probability cutoffs can lead to predictions with higher confidence. Therefore, only putative hits with an average class number prediction of 1.0 and consensus model coverage over 50% were selected (470 putative hits). In addition, we removed compounds with previous bioactivity data reported against SmTGR or S. mansoni and pan-assay interference compounds (PAINs)59,60 so that selected compounds would be novel SmTGR inhibitors and contain no PAINs structures. Finally, the compounds were evaluated by predicting a panel of properties including high aqueous solubility (CIQPlogS),61 acceptable binding to human serum albumin (QPlogKhsa),61 acceptable brain/blood partition coefficient (QPlogBB),61 nonblocking or weak blocking of hERG channel,46,47 and absence of carcinogenicity and hepatotoxicity.32 At the end of the VS workflow, 29 putative hits were visually inspected and acquired for biological evaluation (Supporting Information, Table S6).
Figure 4.

QSAR-based VS workflow used for identifying new compounds active against S. mansoni.
Ex Vivo Activity Against Schistosomula
Compared to target-based VS approaches, the traditional whole-organism schistosome screening approach (phenotypic screening) is an old but indispensable method to discover new antischistosomal agents. This phenotypic approach may be used to validate if the predicted SmTGR-inhibitor interaction has antischistosomal activity. Moreover, a validated compound from a phenotypic assay must have been able to reach its target within the assayed organism only after crossing several biological membranes and resisting degradation by detoxification enzymes. Hence, a hit coming from a phenotypic screen has much more biological value than one coming from a simple biochemical assay. Advances in automated microscopes, liquid handling systems, and computer-based image analysis programs have enabled the development of high-throughput phenotypic assays with cells or small whole organisms, a technique known as high-content screening (HCS).62,63 HCS microscopes are able to capture high resolution images of live organisms in quick succession, a feature that has been explored to evaluate phenotypic and motility changes in schistosomula64 or adult worms.65,66
Therefore, we employed a HCS assay to evaluate the biological activity of the selected compounds from virtual screening against the S. mansoni schistosomula. Assaying against this larval stage is commonly used as an initial screening step in antischistosomal drug discovery campaigns67–72 because schistosomula are easier to obtain in larger numbers than adult worms. Of the 29 compounds tested against schistosomula, six were declared confirmed actives based on motility and phenotype scores at 20 μM after 48 h of exposure (Supporting Information, Table S6). The chemical structures of the six primary hits are shown in Figure 5.
Figure 5.

Chemical structures of six priority hits selected for further follow up.
Following the initial screening on schistosomula, the six primary hits were selected for determining half-maximal motility concentration (EC50) at 0.31–20 μM range (Table 2 and Supporting Information, Figure S3). Among primary hits, 1,2-dimethoxy-4-(2-nitrovinyl)benzene (1), 1-(4-iodophenyl)-3-(4H-1,2,4-triazol-3-ylthio)-2,5-pyrrolidinedione (2), 3-[(4-acetylphenyl)amino]-1-(2-thienyl)-1-propanone (5), and 3-(2-furyl)-1-phenyl-1H-pyrazole-4-carbonitrile (6) only showed inhibition activity at the highest tested concentration (>20 μM). On the other hand, 2-[2-(3-methyl-4-nitro-5-isoxazolyl)-vinyl]pyridine (3) and 2-(benzylsulfonyl)-1,3-benzothiazole (4) showed efficacy in the same range of activity of the reference drug PZQ (EC50 = 1.90 μM), with EC50 values of 3.23 and 2.62 μM, respectively. This is an important feature for a new antischistosomal drug because modern lead discovery pipelines prioritize compounds that possess bioactivity across the entire developmental cycle of the parasite in the mammalian host.73,74
Table 2.
Biological Activity Data for Hits of Interest
| compd | schistosomula EC50 (μM) | adult EC50 (μM)
|
WSS-1 CC50 (μM) | papain IC50 (μM) | |
|---|---|---|---|---|---|
| male | female | ||||
| 1 | >20 | 29.8 | 5.77 | 17.48 | >100 |
| 2 | >20 | 10.2 | 17.9 | 133.40 | >100 |
| 3 | 3.23 | 6.43 | 5.68 | 16.38 | >100 |
| 4 | 2.62 | 21.1 | 4.91 | 28.49 | >100 |
| 5 | >20 | NDa | ND | ND | ND |
| 6 | >20 | ND | ND | ND | ND |
| PZQ | 1.90 | 0.22b | 0.64 | >400 | ND |
ND: not determined.
EC50 values produced for adult male after 72 h of exposure. WSS-1 human kidney epithelial cells were used to evaluate cytotoxicity.
Analysis of Phenotypic Profile
Compounds 3 and 4 promoted the internal disruption of larvae as evidenced by the appearance of multiple vacuoles as well as the rounding and darkening of the schistosomula (Figure 6). To evaluate if schistosomula response profiles toward hits resemble those observed in the presence of known antischistosomal drugs (OLT, PZQ, dihydro-artemisinin, methylclonazepam, Ro15-5458, and oxamniquine), we applied a Bayesian treatment class model using phenotype scores.64 This analysis indicated a shared target and/or mechanism of action between OLT and hits, and therefore, all six hits were classified as OLT-like compounds. At least in part, these results could be related to SmTGR inhibition because OLT has already been identified as a noncompetitive inhibitor of this enzyme. It is also important to note that these phenotypic profile has been also observed after SmTGR gene knockout.18
Figure 6.

Phenotypes of schistosomula exposed for 48 h to 0.625% DMSO (control, A), 20 μM of 4 (B), and 10 μM of PZQ (C) and OLT (D). The outlines represent the position of each parasite over 5 time points (11 s interval).
Ex Vivo Activity on Adult Worms
Our next step was to investigate if the compounds identified as hits for schistosomula also had an effect on adult S. mansoni worms.65,66 Therefore, we employed a new HCS platform recently developed by our group that allows for systematic evaluation of gender-, dose-, and time-dependent drug effects on individual male and female parasites by measuring over 100 image features related to worm motility and morphology. Previously, we have demonstrated the successful application of this platform in identification of potent antischistosomal hit compounds.65,66 In this study, four compounds (1–4) were screened at 0.1–100 μM concentrations for incubation times varying from 0 h (immediately after compound addition to culture medium) to 72 h.
Inspection of the measured features suggested that at least three features were able to distinguish active from inactive compound concentrations or the DMSO control: the Overlap_RandIndex feature, which is related to motility, the intensity, and the area of the identified worm object. Figure 7 shows a 3D plot of these relevant features for individual female worms exposed to the investigated compounds at 20 μM concentration as well as for the PZQ and negative control (treated with 0.1% DMSO) after 48 h incubation. The sample images are shown to exemplify the phenotypes that can be captured by these features. In general, the feature most correlated to the antischistosomal activity of these compounds was the Overlap_RandIndex, which roughly measures the difference in worm position from one time-lapse frame to the next and is inversely proportional to worm motility in a scale varying from 0 to 1. For simplicity, we hereafter refer to this feature as the “motility score”.
Figure 7.

3D scatter plot of the top three image features correlated to antischistosomal activity of the investigated compounds on female S. mansoni worms after 48 h drug exposure. Each point in the graph represents a well/condition in the assay. Sample images are shown for selected wells to illustrate the different phenotypes captured by these three parameters (OA, object area; MS, mobility score; PI, pixel intensity of the worm object). The green outlines represent the position of each parasite over five time points (3 s interval) overlaid on the initial position (red outline).
To determine the potency of the hit compounds against adult worms with the reference drug PZQ, we have determined EC50 values from dose response curves against male and female worms with varying incubation times (Supporting Information, Table S7 and Figure S7). Compounds showed motility inhibition potencies against adult worms ranging from 4.91 to 35 μM, depending on incubation time and gender (Table 2). Overall, inhibition was fully achieved after 48 h of incubation (Table 2). Compound 3 was the most active, with EC50 around 6.00 μM for both genders. Compounds 1 and 4 showed satisfactory potencies (<10 μM) for female worms, with EC50 = 5.77 and 4.91 μM, respectively, but not for male worms. Compound 2 was the less potent, with EC50 values of 10.2 and 17.9 for male and female, respectively. Despite the satisfactory potencies displayed, all compounds had a less pronounced effect on adult worms than PZQ at all incubation times (EC50 values ≤0.66 μM, see Supporting Information, Table S2). Results also indicated that female worms and schistosomula are slightly more sensitive to compounds action because they showed EC50 values up to 5–8 times lower than those determined in males. In part, this could be due to a gender-specific expression pattern of SmTGR and immature antioxidant system of the schistosomula. In fact, schistosomula express lower levels of SmTGR than adults, which make them more susceptible to oxidative damage caused by inhibitors.75,76
Cytotoxicity Against Human Cells
Compounds 1–4 and PZQ were further evaluated for its cytotoxicity against human epithelial cells (WSS-1) from human kidney using a resazurin-based viability assay (Table 2). PZQ showed the lowest cytotoxicity, exhibiting half-maximal cytotoxic concentration (CC50) above 400 μM. Compounds 2, 3, and 4 only were cytotoxic in concentrations higher than those necessary for antischistosomal activity. Compound 2 was the least cytotoxic compound (CC50 = 133.40 μM), followed by 4 (CC50 = 28.49 μM), 1 (CC50 = 17.48 μM), and 3 (CC50 = 16.38 μM).
Controls for Nonspecific Inhibition and Off-Target Effects
Colloidal aggregates have long plagued early drug discovery. When a colloid is formed, membrane and soluble proteins adsorb to its surface and are partially denatured, leading to nonspecific inhibition and occasionally activation.77,78 Therefore, adult worms were coincubated with investigated compounds (at 20 and 100 μM) and detergent Triton X-100 (0.01%) and their antischistosomal effect was compared with activities obtained without detergent for excluding a possible promiscuous colloidal aggregate effect. No significant differences were observed after comparison of inhibition activities of both groups, showing that antischistosomal activity of the hit compounds is related to specific inhibition (Supporting Information, Figure S5). Further, we also investigated possible off-target effects of the hit compounds toward nucleophilic thiols in a papain inhibition assay. Again, none of the antischistosomal hits showed significant inhibition of papain at 100 μM while positive control E-64 fully inhibits this enzyme at 20 μM concentration (Table 2 and Supporting Information, Figure S6).
CONCLUSIONS
To the best of our knowledge, this is the first study integrating QSAR-based VS and HCS methods to discover new antischistosomal agents. We have developed robust and predictive QSAR models for antischistosomal activity. Developed models were used in the most conservative way, i.e., in consensus fashion with the strictest AD criteria, for VS of three ChemBrigde data sets: DIVERSet-CL, DIVERSet-EXP, and PremiumSet. As a result, 470 putative SmTGR inhibitors were identified. Then, 29 compounds were selected and tested against S. mansoni schistosomula using a HCS platform and six of them showed significant inhibition activities at 20 μM. Among them, compounds 3 and 4 showed inhibitory effect equivalent to PZQ, with EC50 values around 2.50 μM. Both hits were also classified as OLT-like compounds, indicating a shared target with OLT, which has already been identified as an inhibitor of SmTGR.79 The results of gender-, dose-, and time-dependent inhibitory effect indicated that adult female worms of S. mansoni are slightly more sensitive than males to compounds action. Compounds 3 and 4 showed satisfactory potencies for female worms, with EC50 values around 6.00 μM. Both compounds also demonstrated low cytotoxicity to WSS-1 mammalian cells (CC50 > 16 μM) and inhibition of papain only in concentrations >100 μM. Finally, both compounds represent new chemical scaffolds which are structurally dissimilar to known inhibitors of S. mansoni and thus can be considered as new hit compounds for further chemical optimization.
EXPERIMENTAL SECTION
Computational
Data Set
The QSAR models were developed according to best practices of predictive QSAR modeling,80,81 which is fully compliant to Organization for Economic Co-operation and Development (OECD) guidance on development and validation of QSAR models such as (i) a defined end point, (ii) an unambiguous algorithm, (iii) a defined domain of applicability, (iv) appropriate measures of goodness-of-fit, robustness, and predictivity, and (v) mechanistic interpretation.82 All in silico steps developed in this study were implemented in a publicly available KSAR workflow (http://labmol.farmacia.ufg.br/ksar). The KSAR workflow is tightly integrated with R and KNIME and includes many modules such as the module for preparing the data, PCA, building of QSAR models, and VS.46,83 We first retrieved 359841 compounds containing half-maximal inhibitory concentration (IC50) data for the SmTGR enzyme from the PubChem BioAssay database (AID: 485364). Compounds with inconclusive IC50 results were considered experimental errors and thus were not included in this study to avoid noise in model building. A total of 2854 out of these 359841 compounds had reproducible potency (IC50 ≤ 10 μM) and were considered as inhibitors, whereas the remaining 356987 compounds were considered as noninhibitors.
Data Set Curation
Each compound of data set was carefully standardized according to the protocol proposed by Fourches and colleagues.84,85 Briefly, explicit hydrogens were added and salts were removed, whereas specific chemotypes such as aromatic and nitro groups were normalized using ChemAxon Standardizer (v.6.1.2, ChemAxon, Budapest, Hungary, http://www.chemaxon.com). Polymers, inorganic salts, organometallic compounds, and mixtures were also removed. In addition, 4437 compounds with multiple SmTGR measurements were identified during analyses of duplicates. Further analysis showed high concordance (99.9%) of duplicated records. In addition, 345 compounds with molecular weight greater than 700 Da were removed. In the end, the prepared data set contained 2854 inhibitors and 337327 noninhibitors.
Molecular Fingerprints and Descriptors
Three different types of fingerprints were used in this study: the Morgan fingerprint, a RDKit implementation38 of the extended-connectivity fingerprints,42 with radius of 2 and bit vector of 1024 bits; the MACCS structural key fingerprints;39–41 and the AtomPair fingerprints (RDKit implementation38 of the Carhart’s atom pairs)37 with bit vector of 1024 bits. All the fingerprints were calculated by the open-source cheminformatics toolkit RDKit v.2.4.0.86 A brief description of Morgan, AtomPair, and MACCS fingerprints is available in Supporting Information.
The Chemistry Development Kit (CDK, v.1.4.19, GNU Lesser General Public License) descriptors and 0–2D descriptors were calculated using the PaDEL-Descriptor program43 and DRAGON (v.5.5, Talete SRL, Milan, Italy), respectively. The complete list of CDK descriptors and a detailed discussion for DRAGON descriptors can be found elsewhere.44,45 The descriptors matrix was then normalized and constant/near constant and highly correlated (r ≥ 0.9) descriptors were removed.
Data Set Analysis and Undersampling
Because the original library was highly unbalanced (2854 inhibitors and 337327 non-inhibitors), it is not recommended for building binary QSAR models for the entire data set. Thus, we decided to balance the data set. Unlike the traditional undersampling methods which randomly balance the data set, our linear undersampling strategy retains most of the representative structures of the noninhibitors set, thus ensuring as high as possible coverage of original chemical space. The basic principle here is to measure the whole inhibitors matrix represented by the MACCS key fingerprints evaluating the Euclidean distance to the MACCS key fingerprints of each noninhibitor using a kNN method,87 implemented in R software v.3.0.3.88 Then, the samples on noninhibitors set were linearly extracted over the whole set by using k-distances and were used to generate balanced and partially balanced data sets. Finally, we generated three undersampled data sets with inhibitor-to-noninhibitor ratios of 1:1 (2854 inhibitors and 2854 noninhibitors), 1:2 (2854 inhibitors and 5705 noninhibitors), and 1:3 (2854 inhibitors and 8562 noninhibitors).
Machine Learning Implementation
The building and optimization of statistically acceptable QSAR models requires a close combination between chemical information (i.e., fingerprints or descriptors) and several machine learning classifiers. For this reason, eight different machine learning classifiers, including the SVM with the radial basis Kernel function,89 the RF,90 GBM,91 and partial least-squares discriminant analysis (PLS-DA)92 approaches, classification and regression trees (CART),93 kNN with Euclidean distance,87 multilayer perceptron (MLP),94 and multivariate adaptive regression splines (MARS)95 were used. All machine learning classifiers were implemented using the R v.3.0.3.88 A brief description about the theory of each machine learning method is described in Supporting Information.
5-Fold External Cross-Validation
The full data set of compounds with known inhibition activities is randomly divided into five subsets of equal size; then one of these subsets (20% of all compounds) is set aside as an external validation set and the remaining four sets together form the modeling set (80% of the full set). This procedure is repeated five times, allowing each of the five subsets to be used as external validation set. Models are built using the modeling set only, and it is important to emphasize that the compounds in momentary external set (fold) are not employed either to build or select the models.
Applicability Domain
The AD for each descriptor or fingerprint type was estimated based on the Euclidean distances among the training set of each QSAR model generated in the external 5-fold cross-validation procedure. The distance of a test compound to its nearest neighbor in the training set was compared to the predefined AD threshold level. If the distance was greater than this threshold level, the prediction was considered to be less trustworthy.96 In this study, we defined AD as a distance threshold DT between a compound under prediction and its closest nearest neighbors of the training set. It was calculated as follows:
| (1) |
Here, ȳ is the average Euclidean distance of the k nearest neighbors of each compound within the training set, σ is the standard deviation of these Euclidean distances, and Z is an arbitrary parameter to control the significance level. We set the default value of this parameter Z at 0.5. Thus, if the distance of the external compound from all of its nearest neighbors in the training set exceeds this threshold, the prediction is considered unreliable.
Evaluation of Performance and Robustness
To access the predictive performance of the binary QSAR models, SE, SP, and CCR were used. These statistic metrics are calculated by the following equations:
| (2) |
| (3) |
| (4) |
Here, TP and TN represent the number of true positives (correct classifications of inhibitors), and true negatives (correct classifications of noninhibitors), respectively, while FP and FN represent the number of false positives (incorrect classifications of inhibitors) and false negatives (incorrect classifications of noninhibitors), respectively.
In addition to the above model evaluation metrics, Cohen’s κ (k) was used to measure the agreement between model predictions and experimental data.97 This statistical parameter is calculated by the following equations:
| (5) |
| (6) |
| (7) |
Here, N denotes the total number of compounds, Pr(a) represents the relative observed agreement between the predicted classification of the model and the known classification, and Pr(e) is the hypothetical probability of chance agreement. In the end, k analysis returns values between −1.0 (no agreement) and 1.0 (complete agreement), but values between 0.6 and 1.0 denote that the model is predictive. Finally, to further ensure that the robustness of the models was not due to chance correlation, 10 rounds of Y-randomization were performed for each constructed model.
Consensus Modeling
After the building of QSAR models using all pairwise combinations of different types of chemical descriptors/fingerprints and various machine learning methods, the best models were used for consensus modeling, which can be derived by calculating an average for individual models. In consensus modeling, the final predicted value for each compound is estimated by including an average of the predicted values from the set of QSAR models. Thus, the averaged predicted activity for each compound is in the [0, 1] range. Formally, compounds with the predicted activity higher than 0.5 are classified as inhibitors, and those with the predicted activity lower than 0.5 are classified as noninhibitors. Obviously, the closer the average predicted value is to 1 or 0, the higher the concordance among all models and the higher our confidence is in the classification of compounds as inhibitors or noninhibitors, respectively.
Mechanistic Interpretation
To explore favorable or unfavorable structural fragments for SmTGR inhibition, the PPMs were generated to visualize the atomic and fragment contributions predicted by the best QSAR model.50
Virtual Screening
The purpose of VS is to identify in a library of chemicals a subset of compounds with the desired properties based on computational calculations. Here the DIVERSet-CL, DIVERSet-EXP, and PremiumSet diversity data sets taken from the ChemBridge database were screened to identify inhibitors of SmTGR. Prior to screening, the data sets were curated in the same way as modeling set (see Data Set Curation section) and filtered using the Veber57 and Lipinski’s rules58 to obtain drug-like compounds. Fingerprints and molecular descriptors were generated for all compounds and normalized (except fingerprints) based on the minimum and maximum values of each descriptor of the modeling set. Then, best consensus and consensus rigor models were used to predict the SmTGR inhibition activity of compounds. The prediction results were accepted only when the compound was found within the applicability domains of more than 50% of all models used in consensus prediction. In addition, to estimate the structural novelty of putative hits, we calculated the pairwise Tanimoto coefficients (using MACCS key fingerprints) between each screened putative hit and compounds in the full data set of SmTGR inhibitors. Then, putative hits with previous bioactivity data against SmTGR or S. mansoni were identified and PAINS were removed using a workflow developed by Saubern and colleagues.98 Finally, hits were imported into Maestro workspace v.9.3 and their aqueous solubility (CIQPlogS), binding to human serum albumin (QPlogKhsa), and brain/blood partition coefficient (QPlogBB) properties were predicted using QikProp v.3.4,61 and hERG inhibition, carcinogenicity, and hepatotoxicity were predicted using the Pred-hERG server,46,47,99 admetSAR server,100,101 and PaDEL-DDPredictor program,102,103 respectively.
Experimental
Materials
Investigated compounds were purchased from ChemBridge (San Diego-CA, USA), resuspended in 100% DMSO, and used immediately in the assays. It is important to mention that all chemical structures were confirmed using proton (1H) NMR spectra at 300/400 MHz and liquid chromatography–mass spectrometry (LC-MS) analysis with evaporative light scattering and ultraviolet detectors confirmed a minimum purity of 95% for all compounds (spectra of compounds are listed in Supporting Information). DMEM and M169 media were purchased from Vitrocell Embriolife (Campinas-SP, Brazil). All other reagents were purchased from Sigma-Aldrich (St. Louis-MO, USA).
Automated ex Vivo Larval S. mansoni HCS Assay
Cercarie (S. mansoni, BH strain) were vortexed at maximum speed for 5 min for tail shedding and transformation into schistosomula by an adapted method from literature.104,105 Briefly, schistosomula were resuspended in Medium 169, placed in 384-well plates (120 per well), and maintained in an incubator with 5% CO2 overnight before compound addition. The worms were then incubated with investigated compounds and PZQ at 0.31–20 μM concentrations or DMSO (0.625%). The effect of the compounds on schistosomula motility and phenotypes was assessed at 48 h after compound addition using an automated bright-field ImageXpressMicro HCS microscope (IXM; Molecular Devices, Wokingham, UK). For motility analysis 5 × 11 s interval time-lapse images were collected using a 4× objective. For detailed morphology, a 10× objective was used to collect four adjacent images fields from within a well in order to increase the number of schistosomula for phenotype analysis. Analysis of both the larval phenotype and motility was then carried out in Pipeline Pilot 9 as described by Paveley and colleagues.64 Phenotype analysis of individual parasites was carried out by a two class Laplacian-modified Bayesian categorization analysis of 80 image descriptors which constituted shape, size, image intensity, and texture statistics and compared to a training set of data comprising 20000 parasites. Motility analysis of individual parasites was also carried out by the average object displacement from the origin point in subsequent 4× image across the time frame series. Both the Bayesian phenotype and motility scores were subsequently adjusted to the control wells (DMSO treated) on each plate.64
Automated ex Vivo Adult S. mansoni HCS Assay
After 42–49 days post percutaneously infection of infant Swiss mice with 150 ± 10 S. mansoni cercariae (BH strain), animals were euthanized, and worms perfused from portal hepatic and mesenteric veins. Male and female parasites were rinsed and individually transferred into 96-well plates with complete DMEM media (i.e., DMEM plus 10% fetal calf serum, 2 mM L-glutamine, 100 μM/mL penicillin, 100 μg/mL streptomycin). The plates were maintained overnight at 37 °C in a humidified atmosphere of 5% CO2. Further, worms were then incubated up to 72 h with 0.10–100 μM of selected compounds and PZQ or negative control DMSO at 0.1%. The effect of the compounds on adult worm motility or phenotype was assessed either immediately 24, 48, or 72 h after compound addition using a newly developed HCS assay. Our method uses 100 time-lapse images captured every 250–300 ms with an automated bright-field microscope using a 2× objective lens (ImageXpress Micro XLS, Molecular Devices, CA). Subsequent quantitative image analysis used a custom-developed pipeline for detecting changes in parasite motility and morphology using the open-source CellProfiler software v. 2.1.2.106 The pipeline along with its validation will be thoroughly described in a subsequent publication, and the pipeline itself is freely available (www.cellprofiler.org/published_pipelines.shtml). Briefly, our strategy for motility measurement was based on sequential pairwise comparison of the 100 captured time-lapse images. The motility measurement called “AdjustedRandIndex” is calculated by comparing worm objects identified on images captured at times tn and tn−1 with CellProfiler’s CalculateImageOverlap module. This measure ranges from 0 to 1, with 1 meaning two objects are perfectly aligned (no movement). In addition to the “Overlap” mobility score, over 100 features related to size, shape, intensity, texture, and granularity are calculated for worm objects identified in the image analysis pipeline and saved in a database. These features are expected to describe different parasite phenotypes in response to drug exposure.
Cytotoxicity Assay
WSS-1 [WS-1](ATCCCRL-2029) epithelial cells derived from human kidney were grown in DMEM medium, supplemented with 4.5 g/L glucose, 50 μg/mL gentamicin, and 10% fetal bovine serum, and seeded into 96-well microplates at 5 × 104 cells/mL. Twenty hours later, cells were exposed to 0.2–400 μM of PZQ, OLT, and LabMol compounds and kept under a humidified atmosphere (37 °C, 5% CO2) for 48 h. To evaluate the cytotoxic effects of the compounds, the fluorescent viability dye resazurin was added to each well at a final concentration of 0.01 mg/mL 4 h before the end of the incubation. Resorufin fluorescence readings (λex = 560 nm, λem = 590 nm) were performed immediately and 4 h after resazurin addition in a FlexStation 3 Benchtop multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). The percentage of viable cell was calculated using cells treated only with DMSO (0.2–0.8%) as controls.
Colloidal Aggregation Assay
Adult worms were coincubated with compounds (at 20 and 100 μM) and detergent Triton X-100 (0.01%). The, motility measurements were performed after 48 h and 72 h, and their antischistosomal effect was compared with activities obtained without detergent.
Papain Inhibition Assay
Enzymatic assay was performed at 37 °C in 100 mM sodium acetate buffer, pH 3.5. Positive control E-64 and compounds were incubated at 20 and 100 μM concentrations for 5 min with papain (5 μg/mL), and the reaction was initiated with the addition of 50 μM Z-FR-AMC fluorogenic peptide substrate.
Statistical Analysis
One-way ANOVA followed by Tukey’s multiple comparisons test was performed using GraphPad Prism v.5.00 (GraphPad Software, La Jolla California USA, www.graphpad.com). The EC50 and CC50 values were determined by four-parameter logist curve function using the same software. EC50 values obtained for adult worms were calculated using TIBCO Spotfire software (Boston, MA).
Ethics Statement
Animal’s maintenance and experiments were carried out in accordance with the Institutional Ethics Committee for Laboratory Animal Use at the Oswaldo Cruz Foundation (CEUA/FIOCRUZ, Brazil; license no. L-044/15).
Supplementary Material
Acknowledgments
We thank Brazilian funding agencies, CNPq, CAPES, FAPEG, FAPERJ, and FIOCRUZ for financial support and fellowships. We are grateful to ChemAxon for providing academic license of their program. We also thank the Malacology Laboratory (Dr. Silvana C. Thiengo) from IOC/FIOCRUZ for providing S. mansoni cercariae, and the Bioassays and Drug Screening Platform (FIOCRUZ RPT11-I subunit), and NIH R01 GM095672 (Automated image analysis for high-throughput phenotypic screening in Caenorhabditis elegans) for technological support. B.J.N. was supported by a fellowship from the Coordination for the Improvement of Higher Education Personnel (CAPES). This work has been funded by the National Counsel of Technological and Scientific Development (CNPq), the State of Goiás Research Foundation (FAPEG), and State of Rio de Janeiro Research Foundation (FAPERJ). C.H.A. and F.P.S.Jr are CNPq productivity fellows. L.E.K. and A.E.C. were supported by a grant from the U.S. National Institutes of Health (GM095672). E.M. also acknowledges NIH (grants GM66940 and GM096967), CNPq (grant 400760/2014–2), and UNC for Junior Faculty Development Award for partial financial support. Authors also thank Molecular Devices for providing access to the HCS equipment. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ABBREVIATIONS USED
- AD
applicability domain
- CART
classification and regression trees
- CC50
half-maximal cytotoxic concentration
- CDK
chemistry development kit
- EC50
half-maximal motility concentration
- FN
false negatives
- FP
false positives
- GBM
gradient boosting machine
- GR
glutathione reductase
- GSH
glutathione
- GSSG
glutathione disulflde
- HCS
high content screening
- IC50
half-maximal inhibitory concentration
- kNN
k-nearest neighbors
- MACCS
Molecular ACCess System (MACCS) keys
- MARS
multivariate adaptive regression splines
- MLP
multilayer perceptron
- NADPH
nicotinamide adenine dinucleotide phosphate
- OECD
Organization for Economic Cooperation and Development
- OLT
oltipraz
- PCA
principal component analysis
- PLS-DA
partial least-squares discriminant analysis
- PPMs
predicted probability maps
- PZQ
praziquantel
- QSAR
quantitative structure–activity relationships
- RF
random forest
- S. mansoni
Schistosoma mansoni
- SAR
structure–activity relationships
- SE
sensitivity
- SMARTS
SMILES arbitrary target specification
- SmTGR
S. mansoni TGR
- SP
specificity
- SVM
support vector machine
- TGR
thioredoxin glutathione reductase
- TN
true negatives
- TP
true positives
- TR
thioredoxin reductase
- Trx
thioredoxin
- VS
virtual screening
Footnotes
Author Contributions
B.J.N., R.F.D., and M.R.S. contributed equally.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b02038.
More computational details regarding molecular finger-prints calculation and QSAR model development, as well as additional tables and figures of experimental results (PDF)
Molecular formula strings (CSV)
References
- 1.Colley DG, Bustinduy AL, Secor WE, King CH. Human Schistosomiasis. Lancet. 2014;383(9936):2253–2264. doi: 10.1016/S0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross AGP, Bartley PB, Sleigh AC, Olds GR, Li Y, Williams GM, McManus DP. Schistosomiasis. N Engl J Med. 2002;346(16):1212–1220. doi: 10.1056/NEJMra012396. [DOI] [PubMed] [Google Scholar]
- 3.King CH. Toward the Elimination of Schistosomiasis. N Engl J Med. 2009;360(2):106–109. doi: 10.1056/NEJMp0808041. [DOI] [PubMed] [Google Scholar]
- 4.Gryseels B, Polman K, Clerinx J, Kestens L. Human Schistosomiasis. Lancet. 2006;368(9541):1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 5.Schistosomiasis. World Health Organization; Geneva: Feb, 2016. [accessed June 13, 2016]. http://www.who.int/mediacentre/factsheets/fs115/en. [Google Scholar]
- 6.Gönnert R, Andrews P. Praziquantel, a New Board-Spectrum Antischistosomal Agent. Z Parasitenkd. 1977;52(2):129–150. doi: 10.1007/BF00389899. [DOI] [PubMed] [Google Scholar]
- 7.Ismail M, Metwally A, Farghaly A, Bruce J, Tao LF, Bennett JL. Characterization of Isolates of Schistosoma Mansoni from Egyptian Villagers That Tolerate High Doses of Praziquantel. Am J Trop Med Hyg. 1996;55(2):214–218. doi: 10.4269/ajtmh.1996.55.214. [DOI] [PubMed] [Google Scholar]
- 8.Melman SD, Steinauer ML, Cunningham C, Kubatko LS, Mwangi IN, Wynn NB, Mutuku MW, Karanja DMS, Colley DG, Black CL, Secor WE, Mkoji GM, Loker ES. Reduced Susceptibility to Praziquantel among Naturally Occurring Kenyan Isolates of Schistosoma Mansoni. PLoS Neglected Trop Dis. 2009;3(8):e504. doi: 10.1371/journal.pntd.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fallon PG, Sturrock RF, Niang AC, Doenhoff MJ. Short Report: Diminished Susceptibility to Praziquantel in a Senegal Isolate of Schistosoma Mansoni. Am J Trop Med Hyg. 1995;53(1):61–62. [PubMed] [Google Scholar]
- 10.Hagan P, Appleton CC, Coles GC, Kusel JR, Tchuem-Tchuenté LA. Schistosomiasis Control: Keep Taking the Tablets. Trends Parasitol. 2004;20(2):92–97. doi: 10.1016/j.pt.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Loukas A, Bethony JM. New Drugs for an Ancient Parasite. Nat Med. 2008;14(4):365–367. doi: 10.1038/nm0408-365. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Wang L, Liang Y. Susceptibility or Resistance of Praziquantel in Human Schistosomiasis: A Review. Parasitol Res. 2012;111(5):1871–1877. doi: 10.1007/s00436-012-3151-z. [DOI] [PubMed] [Google Scholar]
- 13.Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, Mashiyama ST, Al-Lazikani B, Andrade LF, Ashton PD, Aslett Ma, Bartholomeu DC, Blandin G, Caffrey CR, Coghlan A, Coulson R, Day Ta, Delcher A, DeMarco R, Djikeng A, Eyre T, Gamble Ja, Ghedin E, Gu Y, Hertz-Fowler C, Hirai H, Hirai Y, Houston R, Ivens A, Johnston Da, Lacerda D, Macedo CD, McVeigh P, Ning Z, Oliveira G, Overington JP, Parkhill J, Pertea M, Pierce RJ, Protasio AV, Quail Ma, Rajandream M-A, Rogers J, Sajid M, Salzberg SL, Stanke M, Tivey AR, White O, Williams DL, Wortman J, Wu W, Zamanian M, Zerlotini A, Fraser-Liggett CM, Barrell BG, El-Sayed NM. The Genome of the Blood Fluke Schistosoma Mansoni. Nature. 2009;460(7253):352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett Ma, De Silva N, Velarde GS, Anderson TJC, Clark RC, Davidson C, Dillon GP, Holroyd NE, LoVerde PT, Lloyd C, McQuillan J, Oliveira G, Otto TD, Parker-Manuel SJ, Quail Ma, Wilson RA, Zerlotini A, Dunne DW, Berriman M. A Systematically Improved High Quality Genome and Transcriptome of the Human Blood Fluke Schistosoma Mansoni. PLoS Neglected Trop Dis. 2012;6(1):e1455. doi: 10.1371/journal.pntd.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y, Zheng H, Chen Y, Zhang L, Wang K, Guo J, Huang Z, Zhang B, Huang W, Jin K, Dou T, Hasegawa M, Wang L, Zhang Y, Zhou J, Tao L, Cao Z, Li Y, Vinar T, Brejova B, Brown D, Li M, Miller DJ, Blair D, Zhong Y, Chen Z, Liu F, Hu W, Wang ZQ, Zhang QH, Song HD, Chen S, Xu X, Xu B, Ju C, Huang Y, Brindley PJ, McManus DP, Feng Z, Han ZG, Lu G, Ren S, Wang Y, Gu W, Kang H, Chen J, Chen X, Chen S, Wang L, Yan J, Wang B, Lv X, Jin L, Wang B, Pu S, Zhang X, Zhang W, Hu Q, Zhu G, Wang J, Yu J, Wang J, Yang H, Ning Z, Beriman M, Wei CL, Ruan Y, Zhao G, Wang S, Liu F, Zhou Y, Wang ZQ, Lu G, Zheng H, Brindley PJ, McManus DP, Blair D, Zhang Q, Zhong Y, Wang S, Han ZG, Chen Z, Wang S, Han ZG, Chen Z. The Schistosoma Japonicum Genome Reveals Features of Host–parasite Interplay. Nature. 2009;460(7253):345–351. doi: 10.1038/nature08140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young ND, Jex AR, Li B, Liu S, Yang L, Xiong Z, Li Y, Cantacessi C, Hall RS, Xu X, Chen F, Wu X, Zerlotini A, Oliveira G, Hofmann A, Zhang G, Fang X, Kang Y, Campbell BE, Loukas A, Ranganathan S, Rollinson D, Rinaldi G, Brindley PJ, Yang H, Wang J, Wang J, Gasser RB. Whole-Genome Sequence of Schistosoma Haematobium. Nat Genet. 2012;44(2):221–225. doi: 10.1038/ng.1065. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira LG, Oliva G, Andricopulo AD. Target-Based Molecular Modeling Strategies for Schistosomiasis Drug Discovery. Future Med Chem. 2015;7(6):753–764. doi: 10.4155/fmc.15.21. [DOI] [PubMed] [Google Scholar]
- 18.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ESJ, Williams DL. Thioredoxin Glutathione Reductase from Schistosoma Mansoni: An Essential Parasite Enzyme and a Key Drug Target. PLoS Med. 2007;4(6):e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.TIMMS AR, BUEDING E. Studies of a Proteolytic Enzyme from Schistosoma Mansoni. Br J Pharmacol Chemother. 1959;14(1):68–73. doi: 10.1111/j.1476-5381.1959.tb00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall SL, Braschi S, Truscott M, Mathieson W, Cesari IM, Wilson RA. Insights into Blood Feeding by Schistosomes from a Proteomic Analysis of Worm Vomitus. Mol Biochem Parasitol. 2011;179(1):18–29. doi: 10.1016/j.molbiopara.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 21.LoVerde P. Do Antioxidants Play a Role in Schistosome Host–Parasite Interactions? Parasitol Today. 1998;14(7):284–289. doi: 10.1016/s0169-4758(98)01261-7. [DOI] [PubMed] [Google Scholar]
- 22.Callahan HL, Crouch RK, James ER. Helminth Anti-Oxidant Enzymes: A Protective Mechanism against Host Oxidants? Parasitol Today. 1988;4(8):218–225. doi: 10.1016/0169-4758(88)90162-7. [DOI] [PubMed] [Google Scholar]
- 23.Alger HM, Williams DL. The Disulfide Redox System of Schistosoma Mansoni and the Importance of a Multifunctional Enzyme, Thioredoxin Glutathione Reductase. Mol Biochem Parasitol. 2002;121(1):129–139. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 24.Huang H, Day L, Cass CL, Ballou DP, Williams CH, Williams DL. Investigations of the Catalytic Mechanism of Thioredoxin Glutathione Reductase from Schistosoma Mansoni. Biochemistry. 2011;50(26):5870–5882. doi: 10.1021/bi200107n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonilla M, Denicola A, Marino SM, Gladyshev VN, Salinas G. Linked Thioredoxin-Glutathione Systems in Platyhelminth Parasites: Alternative Pathways for Glutathione Reduction and Deglutathionylation. J Biol Chem. 2011;286(7):4959–4967. doi: 10.1074/jbc.M110.170761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams DL, Bonilla M, Gladyshev VN, Salinas G. Thioredoxin Glutathione Reductase-Dependent Redox Networks in Platyhelminth Parasites. Antioxid Redox Signaling. 2013;19(7):735–745. doi: 10.1089/ars.2012.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song L, Li J, Xie S, Qian C, Wang J, Zhang W, Yin X, Hua Z, Yu C. Thioredoxin Glutathione Reductase as a Novel Drug Target: Evidence from Schistosoma Japonicum. PLoS One. 2012;7(2):e31456. doi: 10.1371/journal.pone.0031456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y, Fu Z, Hong Y, Zhang M, Han H, Lu K, Yang J, Li X, Lin J. Inhibitory Effects and Analysis of RNA Interference on Thioredoxin Glutathione Reductase Expression in Schistosoma Japonicum. J Parasitol. 2014;100(4):463–469. doi: 10.1645/13-397.1. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, Clark J, Connelly MC, Sigal M, Hodges D, Guiguemde A, Guy RK, Tropsha A. Discovery of Novel Antimalarial Compounds Enabled by QSAR-Based Virtual Screening. J Chem Inf Model. 2013;53(2):475–492. doi: 10.1021/ci300421n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neves BJ, Bueno RV, Braga RC, Andrade CH. Discovery of New Potential Hits of Plasmodium Falciparum Enoyl-ACP Reductase through Ligand- and Structure-Based Drug Design Approaches. Bioorg Med Chem Lett. 2013;23(8):2436–2441. doi: 10.1016/j.bmcl.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Bueno RV, Toledo NR, Neves BJ, Braga RC, Andrade CH. Structural and Chemical Basis for Enhanced Affinity to a Series of Mycobacterial Thymidine Monophosphate Kinase Inhibitors: Fragment-Based QSAR and QM/MM Docking Studies. J Mol Model. 2013;19(1):179–192. doi: 10.1007/s00894-012-1527-8. [DOI] [PubMed] [Google Scholar]
- 32.Braga RC, Alves VM, Silva AC, Nascimento MN, Silva FC, Liao LM, Andrade CH. Virtual Screening Strategies in Medicinal Chemistry: The State of the Art and Current Challenges. Curr Top Med Chem. 2014;14(16):1899–1912. doi: 10.2174/1568026614666140929120749. [DOI] [PubMed] [Google Scholar]
- 33.Melo-Filho CC, Braga RC, Andrade CH. 3D-QSAR Approaches in Drug Design: Perspectives to Generate Reliable CoMFA Models. Curr Comput -Aided Drug Des. 2014;10(2):148–159. doi: 10.2174/1573409910666140410111043. [DOI] [PubMed] [Google Scholar]
- 34.Artemenko AG, Muratov EN, Kuz’min VE, Kovdienko NA, Hromov AI, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. Identification of Individual Structural Fragments of N,N′-(bis-5-Nitropyrimidyl)dispirotripiperazine Derivatives for Cytotoxicity and Antiherpetic Activity Allows the Prediction of New Highly Active Compounds. J Antimicrob Chemother. 2007;60(1):68–77. doi: 10.1093/jac/dkm172. [DOI] [PubMed] [Google Scholar]
- 35.Kuz’min VE, Artemenko AG, Muratov EN, Volineckaya IL, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. Quantitative Structure-Activity Relationship Studies of [(Biphenyloxy)propyl]isoxazole Derivatives. Inhibitors of Human Rhinovirus 2 Replication. J Med Chem. 2007;50(17):4205–4213. doi: 10.1021/jm0704806. [DOI] [PubMed] [Google Scholar]
- 36.Zakharov AV, Peach ML, Sitzmann M, Nicklaus MC. QSAR Modeling of Imbalanced High-Throughput Screening Data in PubChem. J Chem Inf Model. 2014;54(3):705–712. doi: 10.1021/ci400737s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carhart RE, Smith DH, Venkataraghavan R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J Chem Inf Model. 1985;25(4):64–73. [Google Scholar]
- 38.Riniker S, Landrum Ga. Open-Source Platform to Benchmark Fingerprints for Ligand-Based Virtual Screening. J Cheminf. 2013;5(1):26. doi: 10.1186/1758-2946-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of MDL Keys for Use in Drug Discovery. J Chem Inf Comput Sci. 2002;42(6):1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- 40.Dill JD, Hounshell WD, Marson S, Peacock S, Wipke WT. Search and Retrieval Using an Automated Molecular Access System. 182nd National Meeting of the American Chemical Society; New York. Washington, DC: American Chemical Society; 1981. pp. 23–28. [Google Scholar]
- 41.Anderson S. Graphical Representation of Molecules and Substructure-Search Queries in MACCStm. J Mol Graphics. 1984;2:83–90. [Google Scholar]
- 42.Rogers D, Hahn M. Extended-Connectivity Fingerprints. J Chem Inf Model. 2010;50(5):742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 43.Yap CW. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J Comput Chem. 2011;32(7):1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 44. [accessed January 5, 2015];PaDEL-Descriptor. http://padel.nus.edu.sg/software/padeldescriptor/
- 45.Todeschini R, Consonni V. Handbook of Molecular Descriptors. In: Todeschini R, Consonni V, editors. Methods and Principles in Medicinal Chemistry. 1. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2000. [Google Scholar]
- 46.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Tropsha A, Andrade CH. Tuning HERG out: Antitarget QSAR Models for Drug Development. Curr Top Med Chem. 2014;14(11):1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Lião LM, Tropsha A, Andrade CH. Pred-hERG: A Novel Web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol Inf. 2015;34(10):698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alves VM, Muratov E, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Predicting Chemically-Induced Skin Reactions. Part I: QSAR Models of Skin Sensitization and Their Application to Identify Potentially Hazardous Compounds. Toxicol Appl Pharmacol. 2015;284(2):262–272. doi: 10.1016/j.taap.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alves VM, Muratov E, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Predicting Chemically-Induced Skin Reactions. Part II: QSAR Models of Skin Permeability and the Relationships between Skin Permeability and Skin Sensitization. Toxicol Appl Pharmacol. 2015;284(2):273–280. doi: 10.1016/j.taap.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riniker S, Landrum Ga. Similarity Maps - A Visualization Strategy for Molecular Fingerprints and Machine-Learning Methods. J Cheminf. 2013;5(9):42. doi: 10.1186/1758-2946-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonilla M, Denicola A, Novoselov SV, Turanov AA, Protasio A, Izmendi D, Gladyshev VN, Salinas G. Platyhelminth Mitochondrial and Cytosolic Redox Homeostasis Is Controlled by a Single Thioredoxin Glutathione Reductase and Dependent on Selenium and Glutathione. J Biol Chem. 2008;283(26):17898–17907. doi: 10.1074/jbc.M710609200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nauser T, Steinmann D, Koppenol WH. Why Do Proteins Use Selenocysteine instead of Cysteine? Amino Acids. 2012;42(1):39–44. doi: 10.1007/s00726-010-0602-7. [DOI] [PubMed] [Google Scholar]
- 53.Johansson L, Gafvelin G, Arnér ESJ. Selenocysteine in Proteins-Properties and Biotechnological Use. Biochim Biophys Acta, Gen Subj. 2005;1726(1):1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 54.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arnér ESJ, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure Mechanism Insights and the Role of Nitric Oxide Donation Guide the Development of Oxadiazole-2-Oxides as Therapeutic Agents against Schistosomiasis. J Med Chem. 2009;52(20):6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasco A, Fruttero R, Sorba G, Di Stilo A, Calvino R. NO Donors: Focus on Furoxan Derivatives. Pure Appl Chem. 2004;76(5):973–981. [Google Scholar]
- 56.Triboulet S, Dubée V, Lecoq L, Bougault C, Mainardi JL, Rice LB, Ethève-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet JE, Simorre JP, Arthur M. Kinetic Features of L,D-Transpeptidase Inactivation Critical for β-Lactam Antibacterial Activity. PLoS One. 2013;8(7):e67831. doi: 10.1371/journal.pone.0067831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 58.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Delivery Rev. 1997;23(1–3):3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 59.Baell J, Walters MA. Chemistry: Chemical Con Artists Foil Drug Discovery. Nature. 2014;513(7519):481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 60.Baell JB, Holloway Ga. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J Med Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 61.QikProp, version 34. Schrödinger Inc; New York: 2011. http://www.schrodinger.com/ [Google Scholar]
- 62.Zanella F, Lorens JB, Link W. High Content Screening: Seeing Is Believing. Trends Biotechnol. 2010;28(5):237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 63.Neves BJ, Muratov E, Machado RB, Andrade CH, Cravo PV. L Modern Approaches to Accelerate Discovery of New Antischistosomal Drugs. Expert Opin Drug Discovery. 2016;11(6):557–567. doi: 10.1080/17460441.2016.1178230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paveley R, Mansour NR, Hallyburton I, Bleicher LS, Benn AE, Mikic I, Guidi A, Gilbert IH, Hopkins AL, Bickle QD. Whole Organism High-Content Screening by Label-Free, Image-Based Bayesian Classification for Parasitic Diseases. PLoS Neglected Trop Dis. 2012;6(7):e1762. doi: 10.1371/journal.pntd.0001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melo-Filho CC, Dantas RF, Braga RC, Neves BJ, Senger MR, Valente WCG, Rezende-Neto JM, Chaves WT, Muratov EN, Paveley RA, Furnham N, Kamentsky L, Carpenter AE, Silva-Junior FPS, Andrade CH. QSAR-Driven Discovery of Novel Chemical Scaffolds Active Against Schistosoma Mansoni. J Chem Inf Model. 2016 doi: 10.1021/acs.jcim.6b00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neves BJ, Dantas RF, Senger MR, Valente WCG, Rezende-Neto JM, Chaves WT, Kamentsky L, Carpenter A, Silva-Junior FP, Andrade CH. The Antidepressant Drug Paroxetine as a New Lead Candidate in Schistosome Drug Discovery. Med Chem Comm. 2016;7:1176–1182. [Google Scholar]
- 67.Panic G, Flores D, Ingram-Sieber K, Keiser J. Fluorescence/luminescence-Based Markers for the Assessment of Schistosoma Mansoni Schistosomula Drug Assays. Parasites Vectors. 2015;8(8):624. doi: 10.1186/s13071-015-1233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cowan N, Keiser J. Repurposing of Anticancer Drugs: In Vitro and in Vivo Activities against Schistosoma Mansoni. Parasites Vectors. 2015;8:417. doi: 10.1186/s13071-015-1023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Panic G, Vargas M, Scandale I, Keiser J. Activity Profile of an FDA-Approved Compound Library against Schistosoma Mansoni. PLoS Neglected Trop Dis. 2015;9(7):e0003962. doi: 10.1371/journal.pntd.0003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ingram-Sieber K, Cowan N, Panic G, Vargas M, Mansour NR, Bickle QD, Wells TNC, Spangenberg T, Keiser J. Orally Active Antischistosomal Early Leads Identified from the Open Access Malaria Box. PLoS Neglected Trop Dis. 2014;8(1):e2610. doi: 10.1371/journal.pntd.0002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paveley RA, Bickle QD. Automated Imaging and Other Developments in Whole-Organism Anthelmintic Screening. Parasite Immunol. 2013;35(9–10):302–313. doi: 10.1111/pim.12037. [DOI] [PubMed] [Google Scholar]
- 72.Abdulla MH, Ruelas DS, Wolff B, Snedecor J, Lim KC, Xu F, Renslo AR, Williams J, McKerrow JH, Caffrey CR. Drug Discovery for Schistosomiasis: Hit and Lead Compounds Identified in a Library of Known Drugs by Medium-Throughput Phenotypic Screening. PLoS Neglected Trop Dis. 2009;3(7):e478. doi: 10.1371/journal.pntd.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mansour NR, Paveley R, Gardner JMF, Bell AS, Parkinson T, Bickle Q. High Throughput Screening Identifies Novel Lead Compounds with Activity against Larval, Juvenile and Adult Schistosoma Mansoni. PLoS Neglected Trop Dis. 2016;10(4):e0004659. doi: 10.1371/journal.pntd.0004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long T, Neitz RJ, Beasley R, Kalyanaraman C, Suzuki BM, Jacobson MP, Dissous C, McKerrow JH, Drewry DH, Zuercher WJ, Singh R, Caffrey CR. Structure-Bioactivity Relationship for Benzimidazole Thiophene Inhibitors of Polo-Like Kinase 1 (PLK1), a Potential Drug Target in Schistosoma Mansoni. PLoS Neglected Trop Dis. 2016;10(1):e0004356. doi: 10.1371/journal.pntd.0004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mkoji GM, Smith JM, Prichard RK. Antioxidant Systems in Schistosoma Mansoni: Evidence for Their Role in Protection of the Adult Worms against Oxidant Killing. Int J Parasitol. 1988;18(5):667–673. doi: 10.1016/0020-7519(88)90102-6. [DOI] [PubMed] [Google Scholar]
- 76.Nare B, Smith JM, Prichard RK. Schistosoma Mansoni: Levels of Antioxidants and Resistance to Oxidants Increase during Development. Exp Parasitol. 1990;70(4):389–397. doi: 10.1016/0014-4894(90)90122-s. [DOI] [PubMed] [Google Scholar]
- 77.Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK. An Aggregation Advisor for Ligand Discovery. J Med Chem. 2015;58(17):7076–7087. doi: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK. Colloidal Aggregation Affects the Efficacy of Anticancer Drugs in Cell Culture. ACS Chem Biol. 2012;7(8):1429–1435. doi: 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ESJ, Williams DL. Thioredoxin Glutathione Reductase from Schistosoma Mansoni: An Essential Parasite Enzyme and a Key Drug Target. PLoS Med. 2007;4(6):e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol Inf. 2010;29(6–7):476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 81.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR Modeling: Where Have You Been? Where Are You Going To? J Med Chem. 2014;57(12):4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.OECD Principles for the Validation, For Regulatory Purposes, of (Quantitative) Structure–Activity Relationship Models. Organisation for Economic Co-operation and Development; Paris: 2004. [accessed June 13, 2016]. http://www.oecd.org/chemicalsafety/risk-assessment/37849783.pdf. [Google Scholar]
- 83.Neves BJ, Braga RC, Bezerra JCB, Cravo PVL, Andrade CH. In Silico Repositioning-Chemogenomics Strategy Identifies New Drugs with Potential Activity against Multiple Life Stages of Schistosoma Mansoni. PLoS Neglected Trop Dis. 2015;9(1):e3435. doi: 10.1371/journal.pntd.0003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fourches D, Muratov E, Tropsha A. Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J Chem Inf Model. 2010;50(7):1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fourches D, Muratov E, Tropsha A. Curation of Chemogenomics Data. Nat Chem Biol. 2015;11(8):535. doi: 10.1038/nchembio.1881. [DOI] [PubMed] [Google Scholar]
- 86.Landrum G. RDKit: Open-Source Cheminformatics Software. SourceForge; 2014. http://www.rdkit.org/ [Google Scholar]
- 87.Altman N. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am Stat. 1992;46(3):175–185. [Google Scholar]
- 88.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- 89.Vapnik V. The Nature of Statistical Learning Theory. 2. Springer; New York: 2000. [Google Scholar]
- 90.Breiman L. Random Forests. Mach Learn. 2001;45(1):5–32. [Google Scholar]
- 91.Friedman JH. Greedy Function Approximation: A Gradient Boosting Machine. Ann Stat. 2001;29(5):1189–1232. [Google Scholar]
- 92.Barker M, Rayens W. Partial Least Squares for Discrimination. J Chemom. 2003;17:166–173. [Google Scholar]
- 93.Breiman L, Friedman J, Olshen RA, Charles JS. In: Classification and Regression Trees. 1. Breiman L, editor. Wadsworth & Brooks/Cole Advanced Books & Software; Monterey, CA: 1984. [Google Scholar]
- 94.Rosenblatt F. In: Principles of Neurodynamics; Perceptrons and the Theory of Brain Mechanisms. 1. Rosenblatt F, editor. Spartan Books; Washington, DC: 1962. [Google Scholar]
- 95.Friedman JH. Multivariate Adaptive Regression Splines. Ann Stat. 1991;19(1):1–67. doi: 10.1177/096228029500400303. [DOI] [PubMed] [Google Scholar]
- 96.Zhang S, Golbraikh A, Oloff S, Kohn H, Tropsha A. A Novel Automated Lazy Learning QSAR (ALL-QSAR) Approach: Method Development, Applications, and Virtual Screening of Chemical Databases Using Validated ALL-QSAR Models. J Chem Inf Model. 2006;46(5):1984–1995. doi: 10.1021/ci060132x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cohen J. A Coefficient of Agreement of Nominal Scales. Educ Psychol Meas. 1960;20:37–46. [Google Scholar]
- 98.Saubern S, Guha R, Baell JB. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol Inf. 2011;30(10):847–850. doi: 10.1002/minf.201100076. [DOI] [PubMed] [Google Scholar]
- 99.Alves VM, Braga RC, Silva MB, Muratov E, Fourches D, Tropsha A, Andrade CH. Pred-hERG: A Novel Web-Accessible Computational Tool for Predicting Cardiac Toxicity of Drug Candidates. Abstracts of Papers, 248th ACS National Meeting & Exposition; San Francisco, CA, United States. August 10–14, 2014; Washington, DC: American Chemical Society; 2014. p. CINF-40. [Google Scholar]
- 100.Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J Chem Inf Model. 2012;52(11):3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 101.Lagunin A, Filimonov D, Zakharov A, Xie W, Huang Y, Zhu F, Shen T, Yao J, Poroikov V. Computer-Aided Prediction of Rodent Carcinogenicity by PASS and CISOC-PSCT. QSAR Comb Sci. 2009;28(8):806–810. [Google Scholar]
- 102.He Y, Liew CY, Sharma N, Woo SK, Chau YT, Yap CW. PaDEL-DDPredictor: Open-Source Software for PD-PK-T Prediction. J Comput Chem. 2013;34(7):604–610. doi: 10.1002/jcc.23173. [DOI] [PubMed] [Google Scholar]
- 103.Liew CY, Lim YC, Yap CW. Mixed Learning Algorithms and Features Ensemble in Hepatotoxicity Prediction. J Comput-Aided Mol Des. 2011;25(9):855–871. doi: 10.1007/s10822-011-9468-3. [DOI] [PubMed] [Google Scholar]
- 104.Marxer M, Ingram K, Keiser J. Development of an in Vitro Drug Screening Assay Using Schistosoma Haematobium Schistosomula. Parasites Vectors. 2012;5(1):165. doi: 10.1186/1756-3305-5-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mansour NR, Bickle QD. Comparison of Microscopy and Alamar Blue Reduction in a Larval Based Assay for Schistosome Drug Screening. PLoS Neglected Trop Dis. 2010;4(8):e795. doi: 10.1371/journal.pntd.0000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved Structure, Function and Compatibility for CellProfiler: Modular High-Throughput Image Analysis Software. Bioinformatics. 2011;27(8):1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.