

Abstract
Excitotoxic Zn2+ and Ca2+ accumulation contributes to neuronal injury after ischemia or prolonged seizures. Synaptically released Zn2+ can enter postsynaptic neurons via routes including voltage sensitive Ca2+ channels (VSCC), and, more rapidly, through Ca2+ permeable AMPA channels. There are also intracellular Zn2+ binding proteins which can either buffer neuronal Zn2+ influx or release bound Zn2+ into the cytosol during pathologic conditions. Studies in culture highlight mitochondria as possible targets of Zn2+; cytosolic Zn2+ can enter mitochondria and induce effects including loss of mitochondrial membrane potential (ΔΨm), mitochondrial swelling, and reactive oxygen species (ROS) generation. While brief (5 min) neuronal depolarization (to activate VSCC) in the presence of 300 μM Zn2+ causes substantial delayed neurodegeneration, it only mildly impacts acute mitochondrial function, raising questions as to contributions of Zn2+-induced mitochondrial dysfunction to neuronal injury.
Using brief high (90 mM) K+/Zn2+ exposures to mimic neuronal depolarization and extracellular Zn2+ accumulation as may accompany ischemia in vivo, we examined effects of disrupted cytosolic Zn2+ buffering and/or the presence of Ca2+, and made several observations: 1. Mild disruption of cytosolic Zn2+ buffering—while having little effects alone—markedly enhanced mitochondrial Zn2+ accumulation and dysfunction (including loss of ΔΨm, ROS generation, swelling and respiratory inhibition) caused by relatively low (10 – 50 μM) Zn2+ with high K+. 2. The presence of Ca2+ during the Zn2+ exposure decreased cytosolic and mitochondrial Zn2+ accumulation, but markedly exacerbated the consequent dysfunction. 3. Paralleling effects on mitochondria, disruption of buffering and presence of Ca2+ enhanced Zn2+-induced neurodegeneration. 4. Zn2+ chelation after the high K+/Zn2+ exposure attenuated both ROS production and neurodegeneration, supporting the potential utility of delayed interventions. Taken together, these data lend credence to the idea that in pathologic states that impair cytosolic Zn2+ buffering, slow uptake of Zn2+ along with Ca2+ into neurons via VSCC can disrupt the mitochondria and induce neurodegeneration.
Keywords: zinc, calcium, excitotoxicity, mitochondria, ischemia, neuronal cultures, VSCC, Ca2+ channel, metallothionein, reactive oxygen species
Introduction
Ischemic stroke is a leading cause of morbidity and mortality to the aging population, but no neuroprotective therapy exists, partly reflecting limited understanding of relevant injury mechanisms. Excitotoxicity, caused by excessive glutamate release, is considered to be a major contributor to neurodegeneration. Prior studies of excitotoxic injury have largely focused on rapid Ca2+ entry through N-methyl-D-aspartate (NMDA) receptors, and have suggested mitochondria to be critical targets of the cellular Ca2+ loads (Choi et al., 1988; Nicholls and Budd, 2000; Rothman and Olney, 1986), but NMDA receptor targeted therapies have shown limited clinical efficacy (Hoyte et al., 2004; Ikonomidou and Turski, 2002).
Additional studies have implicated another highly prevalent divalent cation, Zn2+, which accumulates in neurons after ischemia and prolonged seizures, and contributes to neurodegeneration (Frederickson et al., 1989; Koh et al., 1996; Tonder et al., 1990). Zn2+ is stored in presynaptic vesicles, can be released upon activation, and enters postsynaptic neurons (“Zn2+ translocation”) through routes including voltage sensitive Ca2+ channels (VSCC) (Weiss et al., 1993), and with greater rapidity, through the subset of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid channels that are both Ca2+ and Zn2+ permeable (Ca-AMPA) (Jia et al., 2002; Sensi et al., 1999a; Yin and Weiss, 1995). Zn2+ has potent effects on isolated mitochondria (Brown et al., 2000; Dineley et al., 2005; Gazaryan et al., 2007; Jiang et al., 2001; Link and von Jagow, 1995; Sensi et al., 2003; Skulachev et al., 1967), and neuronal Zn2+ loading triggered by rapid entry through Ca-AMPA channels induces acute mitochondrial dysfunction, including reactive oxygen species (ROS) generation (Sensi et al., 1999a; Sensi et al., 2000), with greater potency than Ca2+, suggesting that mitochondria might be critical targets of Zn2+ effects. However, while slower Zn2+ entry through VSCC caused considerable delayed neurodegeneration, these exposures had relatively little impact on acute mitochondrial function (Sensi et al., 1999a; Weiss et al., 1993), raising doubt that Zn2+ translocation contributes importantly to mitochondrial dysfunction in pathological conditions (Pivovarova et al., 2014).
It is now apparent that in addition to direct entry of extracellular Zn2+, another critical determinant of cytosolic (and mitochondrial) Zn2+ accumulation is the presence of Zn2+ buffering proteins—like metallothioneins (MTs)—which normally buffer Zn2+ loads, but can also provide a source from which bound Zn2+ can be released by oxidative stress/acidosis, as can occur during pathological conditions (Malaiyandi et al., 2004; Maret, 2011; Maret and Vallee, 1998). Indeed, strong disruption of these intracellular Zn2+ pools causes acute cytosolic and mitochondrial Zn2+ accumulation even without Zn2+ translocation (Sensi et al., 2003), and can trigger slow Zn2+ dependent neuronal injury (Aizenman et al., 2000).
However, little is known about the respective contributions of each of these sources to mitochondrial dysfunction; indeed, only few studies to date have begun to explore the idea that the integrity of cytosolic buffering may critically modulate the effects of exogenous Zn2+ entry on mitochondrial function in cultured neurons (Clausen et al., 2013; Sensi et al., 2003). Furthermore, as most early studies were carried out in Ca2+ free media to ensure observation of Zn2+ specific effects, there is debate about the respective contributions of Ca2+ and Zn2+ to mitochondrial dysfunction observed in vivo, with some proposing synergy between these ions (Gazaryan et al., 2007; Jiang et al., 2001; Sensi et al., 2000) while others see little evidence for Zn2+ contributions (Devinney et al., 2009; Pivovarova et al., 2014).
The present study seeks to model early Zn2+ dependent events in ischemic neuronal injury to quantitatively examine how disrupted cytosolic Zn2+ buffering and the presence of Ca2+ modulate the consequences of moderate exogenous Zn2+ loads on mitochondrial function and cell death. To this aim, we use brief high K+/Zn2+ exposures (to mimic neuronal depolarization and extracellular Zn2+ accumulation as may accompany ischemia in vivo), and find that both disrupted buffering and the presence of Ca2+ strongly increase the impact of low Zn2+ exposures on mitochondrial function and cell death, with greater synergistic effects when combined. These findings support the hypothesis that slow Zn2+ entry into depolarized neurons could well contribute to mitochondrial dysfunction and neurodegeneration in vivo. Furthermore, Zn2+ chelation after the Zn2+ load diminishes both mitochondrial ROS generation and cell death, supporting the idea that delayed interventions targeting mitochondrial Zn2+ could provide therapeutic benefits.
Material and methods
Ethics statement
This study was carried out in accordance with the recommendations from the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Cortical cultures
Primary mixed cortical cultures were prepared as described previously (Yin et al., 1994). Briefly, cell suspensions from neocortical regions of CD-1 mouse embryos (gender not determined) from 15–16 gestational day timed-pregnant mice (ordered from Charles River Crl:CD1[ICR]) were extracted and plated on astrocytic monolayers in glass-bottomed dishes, on culture treated 24 well microplates, or on Seahorse XF24 cell culture microplates. Cells were maintained in media consisting of Minimum Essential Medium (MEM) supplemented with 10% heat-inactivated horse serum, 10% fetal bovine serum, 2 mM glutamine, and 25 mM glucose, and kept in 37°C/5% CO2 incubator. 2–3 days after dissection, the cultures were switched to an identical maintenance medium lacking fetal bovine serum and non-neuronal cell division halted by adding 10 μM cytosine arabinoside for 24 hrs. To prepare glial cultures (used to establish astrocytic monolayers described above), the same protocol was used, except the following: 1) tissues were obtained from 1–3 days old postnatal mice (gender not determined), 2) plating media was supplemented with epidermal growth factor (10 ng/ml), and 3) suspensions were directly plated on poly-D-lysine and laminin-coated coverslips and/or tissue culture treated plates.
Reagents and indicators
Hydroethidine (HEt) was purchased from Assay Biotech (Sunnyvale, CA). Newport Green, FluoZin-3 AM, MitoTracker Green, Pluronic F-127, MEM, fetal bovine serum, glutamine, and horse serum were purchased from Life Technologies (Grand Island, NY). N-methyl-D-aspartate (NMDA), 2,2′-dithiodipyridine (DTDP), Rhodamine 123 (Rhod123), and N,N,N,N-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN) were purchased from Sigma-Aldrich (St. Louis, MO). Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) was purchased from Tocris Bioscience (Ellisville, MO), apocynin obtained from Acros Organics (Morris Plains, NJ), and XF Base Medium (minimal Dulbecco’s Modified Eagle’s Medium) from Agilent Technologies (Santa Clara, CA). All other chemicals and reagents were purchased from common commercial sources.
Zn2+/Ca2+ loading
Prior to all experiments, cultured neurons were removed from the incubator and placed in HEPES-buffered media (consisting of [in mM] 120 NaCl, 5.4 KCl, 0.8 MgCl2, 20 Hepes, 15 glucose, 10 NaOH, in pH 7.4) with either 1.8 mM CaCl2 (1.8 Ca2+ HSS) or 0 mM CaCl2 (0 Ca2+ HSS) at room temperature. Cultures were maintained in HSS (± Ca2+) for 10 min, followed where indicated by addition of “pre-treatment” (with DTDP and/or TPEN) for 10 min prior to induction of Zn2+ and/or Ca2+ loading. To do so, neurons were exposed to indicated levels of Zn2+ (0 – 300 μM) and/or 1.8 mM Ca2+ in 90 mM K+ HSS (“high K+”; HSS modified with 90 mM K+ and Na+ adjusted to 35 mM to maintain osmolarity) for 5 min to depolarize neurons, inducing ion entry through VSCC. When Ca2+ was present during exposure (with or without Zn2+), the NMDA antagonist MK-801 (10 μM) was added to inhibit Ca2+ entry through NMDA receptors. After the 5 min exposure in high K+, neurons were washed 3 times into HSS (± Ca2+, DTDP and TPEN as present before the exposure) for durations indicated. In addition, as we have found highly Ca2+ or Zn2+ permeable Ca-AMPA channels to be expressed in a small subset (~ 13%) of cultured cortical neurons (Yin et al., 1994), we carried out additional controls using the AMPA channel antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX; 10 μM) during the exposures (Sensi et al., 1999a), and found no differences.
Quantitative imaging studies
10–16 days in vitro (DIV) cultures were mounted on the stage of a Nikon Diaphot inverted microscope equipped with a 75 W xenon lamp, a computer-controlled filter wheel, a 40x (1.3 numerical aperture) epifluorescence oil-immersion objective along with a green FITC fluorescence cube (Ex: 480 nm, dichroic: 505 nm, Em: 535 nm) and a red TRITC fluorescence cube (Ex: 540 nm, dichroic: 565 nm, Em: 605 nm). Emitted signals were acquired once every min with a Sensys Photometrics intensified charge-coupled device camera and digitized through the MetaFluor Version 7.0 software (Molecular Devices LLC, Sunnyvale, CA). Camera gain and exposure were adjusted to give baseline maximal fluorescence levels of 200–300 arbitrary units of a maximal 12-bit signal output of 4,096 for all fluorophores. While the precise experiment schematic is described below, generally, baseline was measured for 10 min, followed by 10 min pre-treatment with DTDP and/or TPEN (if indicated), then by 5 min Zn2+ and/or Ca2+ loading (with high K+), and finally by wash into HSS, with addition of FCCP if indicated. For imaging, fields with ≥ 15 healthy looking cells (defined as non-clustered neurons with robust processes), were selected. Background fluorescence (defined as the lowest fluorescence value in a neuron free region of the field) was subtracted from images, and fluorescence measurements for each cell (Fx) were normalized to the average fluorescence intensity of the 10 min baseline recording (F0) to track normalized fluorescence (Fx/F0) over time. Fx/F0 of each cell from the field was averaged to produce one value, constituting one repetition of an experiment.
To assess cytosolic Zn2+ levels, cultures were first loaded in the dark with 5 μM of either the low affinity Zn2+ indicator, Newport Green diacetate (Kd ~ 1 μM, Ex: 490 nm, Em: 530 nm), or the high affinity Zn2+ indicator FluoZin-3 AM (Kd ~ 15 nM, Ex: 494 nm, Em: 516 nm) in 0 Ca2+ HSS containing 0.2 % Pluronic F-127 and 1.5 % dimethlyl sulfoxide for 30 min at room temperature, then washed into 1.8 Ca2+ HSS and kept in the dark for additional 30 min at room temperature to de-esterify indicators prior to imaging (using a green FITC fluorescence cube). Mitochondrial membrane potential (ΔΨm) was assessed by examining fluorescence changes in Rhod123 (Ex: 507 nm, Em: 529 nm), a positively charged dye that accumulates in active mitochondria, where its fluorescence intensity is quenched. Upon loss of ΔΨm, Rhod123 is released into the cytosol, resulting in increased fluorescence(Duchen et al., 2003). Neurons were incubated in the dark with 2 μM of Rhod123 in 1.8 Ca2+ HSS for 30 min in room temperature prior to imaging (using a green FITC fluorescence cube). At the end of each experiment, the mitochondrial protonophore FCCP was added (1 μM, 5 min) to induce maximal loss of ΔΨm (and record the corresponding maximal Rhod123 ΔF). ROS generation was assessed using the superoxide sensitive dye HEt (Ex: 510–560 nm; Em: > 590 nm; using a red TRITC fluorescence cube). Cultures were loaded in the dark at room temperature with 5 μM HEt in either 0 or 1.8 Ca2+ HSS for 30 min prior to imaging; ROS production is assessed as the rate of fluorescence increase (HEt ΔF) as the HEt is oxidized into highly fluorescent ethidium.
Confocal imaging of mitochondrial morphology
Confocal microscopy of mitochondrial morphology was performed using an inverted stage Nikon Eclipse Ti chassis microscope with a Yokogawa CSUX spinning disk head. Images were collected with a 1000x (1.49 numerical aperture) oil-immersion objective using a Hamamatsu electromultiplying CCD camera, scanned sequentially with excitation (488 nm) via a Coherent sapphire laser source synchronized with the camera, and emission monitored with a 525 (50) nm filter, and acquired using MicroManager Image Acquisition software (version 1.4.16). Briefly, cells were loaded with 200 nM MitoTracker Green (Ex: 490 nm, Em: 516 nm) in 1.8 Ca2+ HSS for 30 min at room temperature in the dark, and then switched to either 0 or 1.8 Ca2+ HSS for the imaging experiment. Imaging rig was maintained at 37°C via heat fan, and camera gain, laser power, and exposure times were adjusted to give baseline fluorescence intensity of MitoTracker Green approximately 1.5x that of the background fluorescence value. Healthy looking neuron(s) were selected for imaging and exposed to treatments as described. Images were taken at baseline, 10 min after DTDP (if used), 5 min after Zn2+ and/or Ca2+ exposure and 10 min after wash (as detailed in Fig. 6). Acquired images were blinded to both treatment groups and time points, then imported into ImageJ software, which was used to make adjustment to whole image to provide optimal discrimination of mitochondrial morphology. Lengths and width of distinct mitochondria were measured manually, the values of which were then used to calculate the length/width (L/W) ratio of the mitochondria. To assess relative changes in morphology over time, the L/W ratio was normalized to baseline values. The L/W ratios of the mitochondria found in the selected field were averaged to produce one average L/W ratio, representing one repetition of an experiment.
Figure 6. Effects of Ca2+, Zn2+, and disruption of cytosolic Zn2+ buffering on mitochondrial morphology.
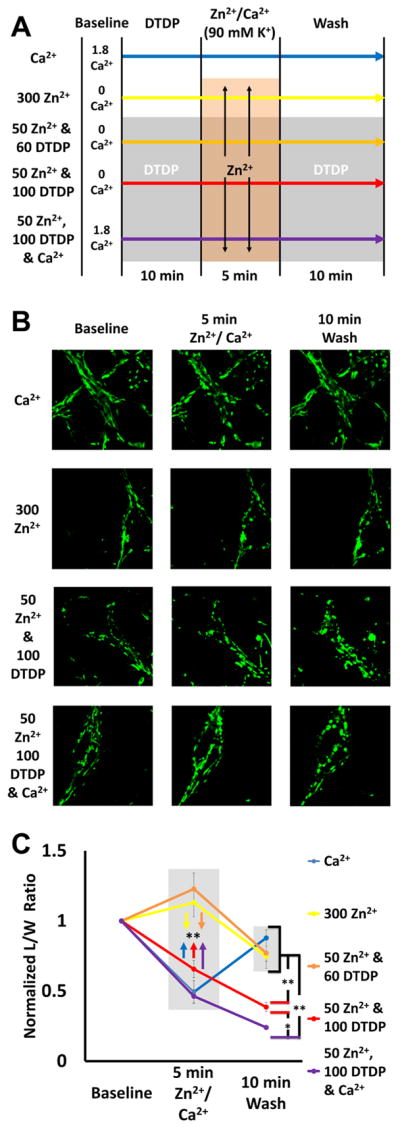
A). Experiment schematic: Neurons loaded with the mitochondrial dye MitoTracker Green (200 nM) were placed in 0 or 1.8 Ca2+ HSS, then exposed to DTDP (60 or 100 μM; where indicated), high K+/MK-801 with Zn2+ (0, 50, or 300 μM) and/or 1.8 mM Ca2+, followed by wash into HSS ± DTDP, as described.
B). Representative images: Confocal images (1000x) were taken at baseline, 5 min after Zn2+ and/or Ca2+ exposure, and 10 min after wash.
C). Zn2+ and Ca2+ induce different patterns of morphology change: The length and width of individual mitochondria were measured blindly, and length/width (L/W) ratios calculated and normalized to baseline. Values for baseline, 5 min after exposure, and after 10 min wash are displayed. Traces show mean ± SEM normalized L/W ratio for each time point, each representing ≥ 5 experiments consisting of ≥ 50 mitochondria (* indicates p < 0.05, ** indicates p < 0.01, by one-way ANOVA with Tukey post hoc). Note that while the Ca2+ induces a rapid but transient morphologic change, Zn2+ triggers more progressive changes (that increase with the degree of Zn2+ loading).
Mitochondrial respiration assay
Mitochondrial respiration was assessed by measuring changes in O2 consumption rate (OCR) using the Seahorse XF24 Extracellular Flux Analyzer, as described previously with adjustments (Yao et al., 2009). Briefly, neurons plated on top of astrocytic monolayers on Seahorse XF24 Cell Culture Microplates were exposed to DTDP, Zn2+ and/or Ca2+ in either 0 or 1.8 Ca2+ HSS (as detailed in Fig. 7), washed into XF Base Medium (supplemented with 2 mM glutamine, 15 mM glucose, and 1 mM sodium pyruvate), and maintained at 37°C for 1 hr. After the 1 hr incubation, the cultures were placed in the Seahorse XF24 instrument, which measures the OCR of cells during baseline and after the sequential exposure to 1 μM oligomycin, 2 μM FCCP, and antimycin A/rotenone (both at 1 μM). The concentrations of oligomycin, FCCP, and antimycin A/rotenone were based on those from the literature (Yao et al., 2009) as well as empirical titration, with FCCP specifically adjusted to induce at least 1.5x the baseline OCR. Prior to each experiment, Seahorse XF24 instrument was calibrated following the manufacturer’s protocols, and all culture wells were visually inspected (to ensure equal distribution of cells) and randomly assigned to treatment groups. Seahorse XF24 program was used to run the assay and Seahorse Wave Software used to analyze data. As neurons were plated on pre-established astrocyte monolayers (using the same culturing methods as described above), we carried out validation studies comparing responses of neuron-astrocyte co-culture to those of astrocyte-only cultures, confirming that astrocytes made minimal (≤ 5%) contributions to observed basal OCR and OCR changes (data not shown).
Figure 7. Zn2+-induced inhibition of mitochondrial respiration: synergy with Ca2+ and effects of disrupted buffering.

A). Schematic of experiment: Neurons were exposed to a series of treatments (detailed in B and C, left), incubated for 1 hr, then placed in the Seahorse assay, which measures O2 consumption rate (OCR) at baseline and after sequential application of oligomycin (Oligo; 1 μM), FCCP (2 μM), and antimycin A & rotenone (AA/Rot; both 1 μM) to characterize various respiratory parameters. Traces (B and C, right) show time course of OCR and represent mean ± SEM of 3 separate experiments, each consisting of 3 – 4 wells of cultured neurons, with arrows indicating time point at which mitochondrial inhibitors were added. Grey bars indicate time points of comparison (* indicates p < 0.05, ** indicates p < 0.01, by one-way ANOVA with Tukey post hoc).
B). Ca2+ and Zn2+ synergistically inhibit mitochondrial respiration: Neurons were placed in 0 or 1.8 Ca2+ HSS, exposed to high K+/MK-801 with 300 μM Zn2+, 1.8 mM Ca2+ or both Zn2+ and Ca2+ as described (left). After 1 hr incubation OCR was measured (right). Note that simultaneous exposure to Zn2+ and Ca2+ induced significant inhibition of mitochondrial respiration, despite the ions having minimal effects individually.
C). Disrupted Zn2+ buffering significantly exacerbates Zn2+ effects on mitochondrial respiration: Neurons were placed in 0 Ca2+ HSS, exposed to DTDP (100 μM; where indicated), high K+/MK-801 with Zn2+ (300, 10 or 0 μM, as indicated; left). After 1 hr incubation OCR was measured (right). Note the near complete inhibition of mitochondrial respiration by 10 μM Zn2+ exposure with DTDP.
Assessment of cell death
Cell death was assessed in neurons plated on culture treated 24 well microplates. Briefly, 14–16 DIV neurons were switched into 0 or 1.8 Ca2+ HSS, and exposed to a combination of DTDP, TPEN, Ca2+ and/or Zn2+ (concentration and durations detailed below). Cultures were then washed into MEM supplemented with 25 mM glucose and kept at 37°C/5% CO2 for 24 hrs (unless otherwise specified), prior to assessment of neuronal injury via direct examination (under phase-contrast microscopy) and by lactate dehydrogenase (LDH) efflux assay as described (Koh and Choi, 1987). The small amount of background LDH present in the media of cultures carried though a sham-wash protocol was subtracted from values obtained in treated cultures. In each experiment, LDH values were scaled to the mean value obtained by a standard exposure to 300 μM NMDA for 24 hr (an exposure that reliably destroys most of the neuronal population without glial damage).
Statistical analysis and repetition
To assess significance, either two-tailed t-test or one-way ANOVA with Tukey post-hoc analysis (indicated in each figure legend) was used, depending on the number of groups of comparison. All values are displayed as mean ± standard error of the mean (SEM). All experiments were repeated at least 3 times. Note that because of some differences in precise behavior of batches of cells reflecting biologic variables—including age and health—all comparisons reflect matched sets of cells, using the same sets of culture dissections with interleaved experiments.
Results
Slow Zn2+ translocation through VSCC induces only mild mitochondrial dysfunction
As discussed above, neuronal Zn2+ accumulation contributes to delayed neurodegeneration in pathological conditions like ischemia. One likely important source of this Zn2+ is co-release (with glutamate) from presynaptic terminals, and its entry into postsynaptic neurons (“translocation”) via various routes (including VSCC and Ca-AMPA channels). Inside neurons, Zn2+ can enter mitochondria and disrupt their function (Clausen et al., 2013; Jiang et al., 2001; Sensi et al., 2003; Sensi et al., 2002; Sensi et al., 2000). While mitochondrial Ca2+ accumulation has long been considered the major contributor to their dysfunction in ischemia (Halestrap, 2006; Nicholls and Budd, 2000), clues have emerged that Zn2+ may also be an important contributor (Bonanni et al., 2006; Calderone et al., 2004; Medvedeva et al., 2017; Medvedeva et al., 2009; Medvedeva and Weiss, 2014), particularly after rapid influx through highly Zn2+ permeable Ca-AMPA channels (Jia et al., 2002; Sensi et al., 1999a; Yin and Weiss, 1995). However, although brief exposure of cultured neurons to Zn2+ under depolarizing conditions (triggering entry largely through VSCC) led to considerable delayed degeneration (Weiss et al., 1993), this induced only mild acute effects on mitochondria (Pivovarova et al., 2014; Sensi et al., 1999a), raising uncertainty as to contributions of slower Zn2+ translocation to mitochondrial dysfunction in disease conditions.
Present studies seek to further clarify whether and under what circumstances low to moderate extracellular Zn2+ accumulation and its slower entry into neurons can impact the mitochondria and promote neuronal injury. We chose to model Zn2+ translocation by inducing entry through the VSCC, as may occur in ischemia with extracellular Zn2+ accumulation around depolarized neurons, because VSCC are ubiquitously expressed (unlike Ca-AMPA channels, which are only present on some neurons) and permit slower entry, resulting in more uniform and moderate Zn2+ loads. Our first aim was to extend prior studies by quantitatively examining the relationship between extracellular Zn2+ exposure, cytosolic and mitochondrial Zn2+ accumulation, and the consequent acute mitochondrial dysfunction.
Neurons were loaded with the low affinity cytosolic Zn2+ indicator Newport Green (Kd ~ 1 μM) (Sensi et al., 1999a) in 0 Ca2+ HSS (to ensure observation of Zn2+-specific effects). After measuring baseline fluorescence for 10 min, cultures were exposed to 25, 75, or 300 μM Zn2+ with high (90 mM) K+ for 5 min, washed into 0 Ca2+ HSS for 5 min, then treated to the mitochondrial protonophore, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 μM) for 10 min. FCCP dissipates the proton gradient across the inner mitochondrial membrane, resulting in rapid loss of mitochondrial membrane potential (ΔΨm) and release of mitochondrially sequestered Zn2+ (Clausen et al., 2013; Medvedeva et al., 2017; Sensi et al., 2003; Sensi et al., 2002). Thus, the rise in cytosolic Zn2+ after FCCP (assessed as Newport Green fluorescence change; Newport Green ΔF) reflects the amount of Zn2+ that had been taken up into mitochondria. Our findings indicate a direct relationship between the magnitude of the exogenous Zn2+ load with both the degree of cytosolic Zn2+ rise and the consequent Zn2+ uptake into mitochondria (Fig. 1A), with high K+/300 μM Zn2+ exposure resulting in far greater cytosolic and mitochondrial Zn2+ accumulation than the 25 or 75 μM exposures.
Figure 1. High K+/Zn2+ exposures induce dose-dependent mitochondrial Zn2+ uptake but only mild acute dysfunction.

A. High K+/Zn2+ exposures cause dose-dependent mitochondrial Zn2+ accumulation. Cultures were loaded with the low affinity cytosolic Zn2+ indicator Newport Green (Kd ~ 1 μM), exposed to 90 mM K+ (high K+) with Zn2+ (25, 75, 300 μM) for 5 min in 0 Ca2+ HSS, followed by wash into 0 Ca2+ HSS for additional 5 min prior to application of FCCP (1 μM). Left: Representative images: Brightfield image (i) shows appearance of neurons at baseline, and pseudocolor images show Newport Green fluorescence from the same field at baseline (ii), 5 min after high K+/300 μM Zn2+ exposure (iii), and 5 min after FCCP (iv). (Arrows highlight the same neurons in these images.) Right: Traces show time course of Newport Green ΔF (background subtracted and normalized to baseline [Fx/F0]; arrows indicate the time points illustrated in the images), and represent means ± standard error of the mean (SEM) of 6 experiments, ≥ 120 neurons. Grey bar indicates time points of comparison (** indicates p < 0.01 by one-way ANOVA with Tukey post hoc). Note the Zn2+ exposure concentration-dependent mitochondrial Zn2+ accumulation, indicated by the increase in ΔF after FCCP
B). These exposures only induce mild mitochondrial dysfunction: Cultures were loaded with the ΔΨmito sensitive indicator, Rhod123, or the superoxide preferring ROS indicator, HEt, and exposed to high K+/Zn2+ for 5 min, followed by wash into 0 Ca2+ HSS, as above. After 20 min, FCCP (1 μM) was applied to Rhod123-loaded cultures to induce full loss of ΔΨm. Traces show time course of Rhod123 ΔF (left) or HEt ΔF (right), normalized to baseline values (after background subtraction, as above; Fx/F0), and represent mean ± SEM 6 experiments, ≥ 120 neurons. Grey bars indicate time points of comparison (* indicates p < 0.05, ** indicates p < 0.01, by one-way ANOVA with Tukey post hoc). Note that only 300 μM Zn2+ exposure induced discernable effects.
We next sought to examine the acute consequences of these mitochondrial Zn2+ loads on ΔΨm and reactive oxygen species (ROS) generation. To assess changes in ΔΨm, we used the cationic indicator Rhodamine 123 (Rhod123), which accumulates in mitochondria in proportion to their ΔΨm (where its fluorescence is quenched); upon loss of ΔΨm, Rhod123 is released into the cytosol and the fluorescence increases (Rhod123 ΔF) (Duchen et al., 2003). ROS generation was assessed using the superoxide preferring oxidant sensitive indicator, hydroethidine (HEt), which is oxidized by superoxide radicals into the highly fluorescent compound, ethidium. The fluorescence of ethidium is amplified upon its binding to DNA, providing high sensitivity and resulting in predominant visualization of the signal in the nucleus (Bindokas et al., 1996). Because the oxidized ethidium accumulates, the rate of fluorescence increase indicates ROS production rate, and the increase in HEt fluorescence (HEt ΔF) over baseline reflects total ROS production. Rhod123 and HEt loaded cultures were exposed to Zn2+ in high K+ and washed into 0 Ca2+ HSS, similarly as above. After the 300 μM Zn2+ exposures, we detected modest loss of Δψm (as indicated by Rhod123 ΔF prior to FCCP-induced maximal depolarization) and ROS production (as indicated by HEt ΔF); the 25 and 75 μM Zn2+ exposures had little effects (Fig. 1B). Of note, although Zn2+ exposure has been found to cause delayed activation of the superoxide generating enzyme NADPH oxidase (NOX) (Noh and Koh, 2000), our prior studies indicated that rapid Zn2+ triggered ROS production is almost entirely of mitochondrial origin (Clausen et al., 2013; Sensi et al., 1999a). Thus, above data suggest that while neuronal Zn2+ entry through VSCC (modeling slow Zn2+ translocation) induces dose dependent mitochondrial Zn2+ accumulation, acute mitochondrial dysfunction was only detected with the strongest (300 μM) Zn2+ exposures.
Critical role of cytosolic buffering in Zn2+-triggered mitochondrial dysfunction
We next sought to examine the degree to which endogenous Zn2+ buffering, likely in large part via MTs, impacts Zn2+ dependent modulation of mitochondrial function. Indeed, whereas Zn2+ mobilization from cytosolic buffers appears able to impact mitochondrial function and trigger slow Zn2+ dependent injury even in the absence of exogenous Zn2+ entry (Aizenman et al., 2000; Sensi et al., 2003), there has been little quantitative assessment of these effects. To disrupt endogenous Zn2+ buffering, we used the disulfide compound 2,2-dithiodipyridine (DTDP) which oxidizes the sulfhydryls linking Zn2+ to cysteines, thus releasing bound Zn2+ from buffering proteins (like MTs) and preventing the released Zn2+ from being bound again (Aizenman et al., 2000; Maret and Vallee, 1998; Sensi et al., 2003).
To assess Zn2+ release from the buffers and its ability to enter mitochondria, we loaded cells (in 0 Ca2+ HSS) with the higher affinity Zn2+ indicator, FluoZin-3 (Kd ~ 15 nM) (Gee et al., 2002; Sensi et al., 2003), and monitored changes in fluorescence before and after application of 100 μM DTDP and subsequent addition of FCCP. In agreement with our prior observations (Sensi et al., 2003), this fairly high DTDP exposure caused a prompt but relatively modest Zn2+ rise, followed by a further sharp rise in cytosolic Zn2+ upon FCCP exposure, indicating robust mitochondrial Zn2+ accumulation (likely due to strong disruption of cytosolic buffering). With lower (60 μM) DTDP, we still observed an acute cytosolic Zn2+ rise and mitochondrial Zn2+ uptake, but these were markedly diminished, consistent with impaired—but not fully disrupted—Zn2+ buffering (Fig. 2A).
Figure 2. Disruption of cytosolic Zn2+ buffering leads to Zn2+-dependent mitochondrial dysfunction.

Cultures were loaded with the high affinity Zn2+ indicator FluoZin-3 (Kd ~ 15 nM), Rhod123 or HEt in 0 Ca2+ HSS, then exposed to DTDP (60 or 100 μM; to disrupt cytosolic Zn2+ buffering), with FCCP (1 μM) or TPEN (20 μM) applied as indicated. Traces represent mean ± SEM Fx/F0 values for each indicator and represents ≥ 5 experiments consisting of ≥ 100 neurons. Grey bars indicate time points of comparison (* indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, by two-tailed t-test).
A). DTDP induces dose-dependent cytosolic Zn2+ release and mitochondrial Zn2+ accumulation: FluoZin-3 loaded neurons were exposed to DTDP followed by addition of FCCP, as indicated. Note the dose dependent effects of DTDP, with 100 μM causing both greater cytosolic Zn2+ rise and mitochondrial Zn2+ loading (as indicated by the FCCP-induced ΔF) than 60 μM.
B). Disruption of buffering via DTDP can induce mitochondrial dysfunction: Rhod123- (left) or HEt- (right) loaded neurons were subjected to the indicated DTDP and FCCP exposures. Note that the 100 μM DTDP exposure resulted in substantial loss of ΔΨmito within 25 min (as indicated by the minimal response to FCCP) and significant ROS production, while 60 μM DTDP had far smaller effects.
C). DTDP effects on mitochondria are Zn2+-dependent: Rhod123- (left) or HEt- (right) loaded neurons were exposed to 100 μM DTDP ± Zn2+ chelator TPEN (applied 10 min before DTDP), followed by FCCP (only in Rhod123 loaded cultures), as indicated. Note that TPEN largely eliminated the DTDP induced loss of ΔΨmito (left) and markedly attenuated the ROS production (right).
To examine acute effects of these DTDP exposures on mitochondria, we used the ΔΨm indicator Rhod123 and the ROS indicator HEt, as above. With continuous exposure to 100 μM DTDP, we noted a gradual increase in Rhod123 ΔF that progressed to subtotal loss of Δψm within 40 min (as indicated by lack of response to FCCP), and a rise in HEt ΔF indicative of increased ROS production. In contrast, with 60 μM DTDP, there was little Rhod123 or HEt ΔF (Fig. 2B). Finally, to validate the Zn2+ dependence of these DTDP effects, we repeated strong (100 μM) DTDP exposures in the presence or absence of the membrane permeable Zn2+ chelator, N,N,N,N-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN; 20 μM, applied 10 min before and with DTDP). TPEN application provided near complete block of both the Rhod123 and HEt ΔF, indicating that the acute effects of the DTDP exposure on mitochondrial function are largely Zn2+ dependent, likely resulting from movement of Zn2+ from the endogenous buffers into the mitochondria (Fig. 2C). (As prior studies found DTDP to promote mild Ca2+ release from endoplasmic reticulum (McCord and Aizenman, 2013), we assessed effects of DTDP exposures on Ca2+, and confirmed very small rises that did not differ between 60 and 100 μM DTDP exposures; data not shown). Thus, these results confirm and extend prior studies (Sensi et al., 2003), and indicate dose dependent effects of protracted mobilization of endogenous Zn2+ stores on mitochondria, even in the absence of extracellular Zn2+ influx.
While above findings help clarify the degree to which the integrity of cytosolic Zn2+ buffering can impact mitochondria, in pathological conditions like ischemia, disruption of buffering (due to acidosis/oxidative stress) would most likely occur concurrently with extracellular Zn2+ accumulation (largely from vesicular release). Thus, we next sought to examine how relatively low extracellular Zn2+ accumulation (at levels that may occur in ischemia) can impact mitochondria when cytosolic Zn2+ buffering is impaired. To this aim, we combined two exposures that each caused little acute mitochondrial dysfunction when applied alone (modest Zn2+ entry triggered by 5 min high K+ exposure with 50 μM Zn2+, and partial disruption of buffering induced by 60 μM DTDP), and compared the consequences with those of a far greater exogenous Zn2+ exposure alone (high K+/300 μM Zn2+ in the absence of DTDP; see Fig. 1B, 2B). Using Newport Green, Rhod123, and HEt to assess mitochondrial Zn2+ loading, loss of Δψm and ROS generation respectively (as in Fig. 1), we found these exposures to induce similar cytosolic rises and levels of acute mitochondrial Zn2+ accumulation (as indicated by the similar Newport Green ΔF upon adding FCCP; Fig. 3A), but the combined exposure caused markedly greater loss of Δψm and ROS generation than the higher Zn2+ exposure alone (Fig. 3B, C). Furthermore, the loss of Δψm and ROS generation occurring after the high K+/50 μM Zn2+/DTDP exposure are not transient, but appeared to progress at a near constant rate (as indicated by the slope of the Rhod123 and HEt ΔF traces) for the duration of the recording period. Prior studies have estimated that peak cytosolic Zn2+ rises after brief exposure to high K+/300 μM Zn2+ are in the 100s of nM (a substantial increase from subnanomolar resting levels) (Canzoniero et al., 1999; Colvin et al., 2010; Frederickson et al., 2005; Maret, 2015; Sensi et al., 1999a). Thus, despite similar degrees of acute cytosolic and mitochondrial Zn2+ accumulation triggered by these exposures, the greater and longer-lasting mitochondrial dysfunction triggered by the lower exposure with DTDP likely reflects a greater persistence of the Zn2+ within the mitochondria, due to impaired ability to buffer the Zn2+ rises and recover Zn2+ homeostasis.
Figure 3. Impaired cytosolic Zn2+ buffering markedly enhances the acute impact of Zn2+ exposures on mitochondria.

Cultures were loaded with Newport Green, Rhod123, or HEt in 0 Ca2+ HSS, and exposed to high K+/300 μM Zn2+ alone (blue), or to high K+/50 μM Zn2+ with 60 μM DTDP (applied as indicated; red); FCCP (1 μM) was added as indicated. Traces represent mean ± SEM Fx/F0 values for each dye and represents 6 experiments consisting of ≥ 120 neurons. Grey bars indicate time points of comparison (NS indicates No Significance, * indicates p < 0.05, ** indicates p < 0.01, by two-tailed t-test).
A). Low exogenous Zn2+ exposure to neurons with impaired buffering results in similar degrees of mitochondrial uptake as much higher Zn2+ exposure with intact buffering: Note the similar magnitudes of mitochondrial Zn2+ loading caused by the high K+/300 μM Zn2+ (blue) and the high K+/50 μM Zn2+/DTDP exposures.
B, C). However, the lower Zn2+ exposure with impaired buffering results in greater mitochondrial dysfunction: Rhod123 (B) or HEt (C) loaded neurons were exposed as indicated. Note that the 50 μM Zn2+/DTDP exposure induced markedly greater loss of Δψm and ROS generation than 300 μM Zn2+ alone.
Finally, we examined the Zn2+ exposure dependence of the combined high K+/Zn2+/60 μM DTDP exposures by comparing effects obtained using 50 μM Zn2+ with those occurring with lower (10 μM) Zn2+ exposures. Not surprisingly, the effects were strongly dose dependent, with the higher exposure causing more mitochondrial Zn2+ accumulation (Fig. 4A) and greater acute effects on Δψm and ROS generation (Fig. 4B, C). Interestingly, the difference was greater for the ROS generation, with both exposures only causing partial loss of Δψm. In sum, above data indicate that even partial disruption of cytosolic Zn2+ buffering can significantly exacerbate the impact of neuronal Zn2+ entry on mitochondria, and thus is likely to be a critical determinant of the extent of Zn2+-triggered mitochondrial disruption after ischemia.
Figure 4. Zn2+ exposure dose-dependence of mitochondrial Zn2+ loading and acute dysfunction in neurons with impaired buffering.

Cultures were loaded with Newport Green, Rhod123 or HEt in 0 Ca2+ HSS, and exposed to high K+ with 10 (blue) or 50 (red) μM Zn2+ in 60 μM DTDP. FCCP (1 μM) was added as indicated. Traces represent mean ± SEM Fx/F0 values for each dye and represents 6 experiments consisting of ≥ 120 neurons. Grey bars indicate time points of comparison (* indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, by two-tailed t-test).
A). Zn2+ exposure induces dose-dependent mitochondrial Zn2+ loading in neurons with disrupted buffering: Note the dose dependency of the cytosolic Zn2+ rise and mitochondrial Zn2+ uptake, with the 50 μM Zn2+ exposure causing far greater Zn2+ uptake than the 10 μM Zn2+.
B, C). Mitochondrial dysfunction reflects the extent of Zn2+ accumulation: Rhod123 (B) or HEt (C) loaded neurons were exposed as indicated. Note that the 50 μM Zn2+ exposure induced far greater loss of Δψm and ROS generation than 10 μM Zn2+. Further note that despite causing relatively strong ROS generation, 50 μM Zn2+ still only caused modest loss of Δψm.
Ca2+ reduces Zn2+ uptake but exacerbates consequent mitochondrial dysfunction
Above experiments (like many prior studies of Zn2+ effects) were carried out in Ca2+ free media, to ensure Zn2+-specificity of effects. However, as Ca2+ is always present in vivo, we felt it crucial to next examine effects of Zn2+ in the presence of physiologic (1.8 mM) levels of Ca2+. First, to assess effects of Ca2+ on cytosolic and mitochondrial Zn2+ accumulation, Newport Green loaded cultures were subjected to a range of Zn2+ loads (high K+/300 μM Zn2+ alone, or high K+ with 10 or 50 μM Zn2+/60 μM DTDP) for 5 min in 0 or 1.8 Ca2+ HSS. The NMDA antagonist, MK-801 (10 μM) was added during these exposures to prevent rapid Ca2+ influx through highly Ca2+ permeable NMDA channels. In each of these conditions, the presence of Ca2+ decreased both the cytosolic Zn2+ rises (likely reflecting competition for entry through VSCC) (Kerchner et al., 2000; Weiss et al., 1993) and the mitochondrial Zn2+ uptake (assessed as the ΔF upon application of FCCP; Fig. 5A).
Figure 5. Ca2+ attenuates mitochondrial Zn2+ accumulation despite exacerbating the consequent dysfunction.

Cultures were loaded with Newport Green, Rhod123 or HEt in 0 or 1.8 Ca2+ HSS, and exposed to high K+ with 300, 50,10 or 0 μM Zn2+ (as indicated, along with 10 μM MK-801, to inhibit Ca2+-entry via NMDA receptor activation); DTDP (60 μM), FCCP (1 μM) and/or apocynin (500 μM) were added as indicated. Traces represent mean ± SEM Fx/F0 values for each dye and represents ≥ 5 experiments consisting of ≥ 120 neurons. Grey bars indicate time points of comparison (NS indicates No Significance, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, by two-tailed t-test [A, C] or by one-way ANOVA with Tukey post hoc [B]).
A). Presence of Ca2+ decreases neuronal and mitochondrial Zn2+ uptake: Note that presence of Ca2+ attenuated both cytosolic Zn2+ rise during the exposure and FCCP-induced mitochondrial Zn2+ release.
B). Ca2+ and Zn2+ synergistically induce mitochondrial dysfunction: Neurons loaded with Rhod123 (left) or HEt (right) were exposed to high K+/DTDP/MK-801 with 10 μM Zn2+ (blue), 1.8 mM Ca2+ (red) or with both Zn2+ and Ca2+ (purple) for 5 min, then washed as indicated. Apocynin was added to HEt-loaded neurons (right) to inhibit contributions from Ca2+-dependent NOX activation. Note that despite relatively little effects from Ca2+ and Zn2+ individually, together they induced significant mitochondrial dysfunction.
C). Overwhelming mitochondrial Zn2+ loading induces rapid mitochondrial depolarization: Neurons loaded with Rhod123 (left) or HEt (right) in 1.8 Ca2+ HSS were exposed to high K+/DTDP/MK-801/Ca2+, with 50 (blue) or 300 μM (red) Zn2+ for 5 min, followed by wash as indicated. Note that 300 μM Zn2+ induced greater loss of ΔΨm than 50 μM Zn2+, despite both inducing similar levels of ROS generation.
Despite above findings of reduced mitochondrial Zn2+ loading in the presence of Ca2+, some prior studies have suggested that these ions may have synergistic effects on mitochondrial function (Gazaryan et al., 2007; Jiang et al., 2001; Sensi et al., 2000). Thus, we next used Rhod123 and HEt loaded cultures to examine how the presence of Ca2+ influences Zn2+ effects on Δψm and ROS generation. To assess possible synergism, we first examined the low end of the Zn2+ exposure range (high K+ with 10 μM Zn2+/60 μM DTDP) that caused little loss of Δψm and ROS generation in 0 Ca2+ HSS (Fig. 4B). In a new set of experiments, cultures were exposed to high K+/60 μM DTDP (with 10 μM MK-801) in the presence of either 10 μM Zn2+, 1.8 mM Ca2+ or with both Zn2+ and Ca2+. In addition, as NOX activation has been reported to contribute, along with mitochondria, to acute Ca2+ triggered ROS generation (unlike acute Zn2+ triggered ROS, which, as discussed above, appears mostly to be of mitochondrial origin) (Brennan et al., 2009; Clausen et al., 2013), the NOX inhibitor apocynin (500 μM) was added in HEt loaded cultures to prevent ROS generation due to Ca2+ dependent activation of this enzyme. While the high K+ exposures with either the 10 μM Zn2+ or 1.8 mM Ca2+ alone had little acute impact on the mitochondria, the combined exposure resulted in substantially greater loss of Δψm and ROS generation (Fig. 5B), further substantiating the synergistic effects of Ca2+ and Zn2+ on mitochondria.
Finally, we examined the dose dependence of loss of Δψm and ROS generation at the high end of the exposure range, in light of our prior observations (Fig. 4B, C) that high K+/50 μM Zn2+/60 μM DTDP exposures caused substantial ROS generation but rather modest loss of Δψm. Rhod123 and HEt loaded cultures were exposed to high K+ with Zn2+ (50 or 300 μM) and 1.8 mM Ca2+ in 60 μM DTDP (with MK-801, and apocynin added in HEt loaded cultures as above). Whereas both of these exposures caused similar strong and persistent ROS generation, there was a clear dose dependency on the loss of Δψm, with the 50 μM Zn2+ exposure still causing only partial loss of Δψm, while the “maximal” (and likely supraphysiological) 300 μM Zn2+ exposure triggered near complete loss of Δψm (Fig. 5C). This highlights the ability of mitochondria to maintain at least partial Δψm despite quite strong Zn2+ loads that cause strong and persistent ROS generation.
Thus, despite the presence of Ca2+ in the extracellular fluid resulting in decreased cytosolic and mitochondrial Zn2+ accumulation, it markedly increases the consequent Zn2+ effects on the mitochondria, highlighting strong synergism of these 2 cations. Indeed, the degree of synergism is sufficient that even a quite brief and low extracellular Zn2+ exposure (5 min, 10 μM Zn2+) applied under pathophysiologically relevant conditions (with Ca2+ present in depolarized neurons with partially impaired Zn2+ buffering) triggered substantial acute effects on the mitochondria.
Mitochondrial Zn2+ accumulation induces rapid swelling and disruption of mitochondrial respiration
Above findings lend credence to the hypothesis that neuronal Zn2+ entry contributes to mitochondrial dysfunction in pathological conditions like ischemia. However, whereas the measures employed thus far (loss of Δψm and ROS production) are valuable indices of acute disruption, they are not indicative of long lasting mitochondrial dysfunction or loss of viability that may ultimately lead to neurodegeneration.
To address this, we first examined acute changes in mitochondrial morphology triggered by Zn2+ loads. While mitochondria are mostly rod shaped, in pathological conditions like ischemia, both Ca2+ and Zn2+ can trigger either transient or irreversible morphologic changes, including swelling (Brustovetsky et al., 2002; Halestrap, 2006; Jiang et al., 2001; Sugawara et al., 1999).
To assess mitochondrial morphology, cultures were loaded with the fluorescent mitochondrial marker, MitoTracker Green (200 nM), and neuronal mitochondria examined using confocal microscopy at 1000x (as described in Material and methods). This marker has the advantages that it covalently binds to mitochondrial proteins (and thus stays in neuronal mitochondria despite loss of Δψm) and maintains fluorescence in oxidative environments (Buckman et al., 2001; Jiang et al., 2001). Cultures were exposed as indicated to high K+/MK-801 with Zn2+ and/or Ca2+ (± DTDP) and images acquired at baseline, after the 5 min Zn2+/Ca2+ exposure, and, finally, 10 min after wash (into HSS ± DTDP) (as indicated in Fig. 6A, B). We examined effects of 60 μM as well as 100 μM DTDP, to assess possible consequences of Zn2+ loads with both partial and near maximal disruption of cytosolic buffering, as might occur during episodes of strong in vivo ischemia. To assess morphological changes, images were blinded to experimental condition and time point, and imported into Image J software, where the lengths and widths of distinct mitochondria were measured manually (see Material and methods); data are expressed as mean length/width (L/W) ratios normalized to baseline values (which ranged from 4–8; mean 5.9 ± 0.3).
There were distinct differences triggered by the different exposures (Fig. 6C): 1). Exposure to high K+/1.8 mM Ca2+ alone caused a significant swelling of mitochondria evident at the end of the 5 min exposure, that had largely recovered after 10 min wash. 2). High K+ with 300 μM Zn2+ alone, or with 50 μM Zn2+/60 μM DTDP (both in 0 Ca2+ HSS) did not cause swelling evident at the end of the exposure, but mild swelling was evident 10 min later in both treatments. 3). High K+/50 μM Zn2+/100 μM DTDP exposures in 0 Ca2+ HSS caused marked swelling at the end of the exposure that substantially progressed over the subsequent 10 min; with Ca2+ present, the swelling at both time points was even greater, with extreme rounding up of mitochondria that may be indicative of irrecoverable damage. Of note, we found little morphological changes after 10 min pre-treatment with DTDP (60 or 100 μM in 0 or 1.8 Ca2+ HSS) prior to Zn2+ exposure (data not shown), further supporting the need for contributions from both extracellular Zn2+ entry and intracellular Zn2+ mobilization to potently impact the mitochondria. In summary, these data suggest distinct effects of these ions, with Ca2+ being an effective trigger of acute transient—but largely recoverable—swelling, while Zn2+ induces swelling of slower onset, that, when “strengthened” (by DTDP and synergism with Ca2+) appears to be strongly progressive after termination of the exposure.
We next sought to examine delayed effects of the Zn2+ exposures on mitochondrial respiration. Indeed, as the main function of mitochondria is energy production, which is dependent upon the integrity of the electron transport chain, we felt it critical to assess such effects, which, in contrast to loss of Δψm and ROS production, provide a direct measure of the disruption of mitochondrial respiratory capacity after an insult. For these studies, we made use of a device (the Seahorse XF24 analyzer) which measures the O2 consumption rate (OCR) in cultures at baseline and in response to sequential application of the following drugs: (1) the ATP synthase inhibitor, oligomycin (1 μM), which prevents dissipation of the proton gradient across the inner membrane due to ATP synthesis, leading to membrane hyperpolarization and slowing of electron transport; the decrease in OCR upon its application provides an estimate of the portion of O2 consumption contributing to ATP production; (2) FCCP (2 μM), which dissipates the proton gradient, uncoupling the electron transport chain, yielding maximal oxidative capacity and OCR, and; (3) combined application the complex I blocker, rotenone, and the complex III blocker, antimycin A (both 1 μM), to fully inhibit the electron transport chain, and blocking all mitochondrial respiration (Fig. 7A) (Brand and Nicholls, 2011).
We carried out two sets of experiments. The first aimed to examine synergism between effects of Zn2+ and Ca2+ (via exposure to high K+/MK-801 with 300 μM Zn2+, 1.8 mM Ca2+, or both Zn2+ and Ca2+ Fig. 7B left), while the second aimed to examine the degree to which disruption of cytosolic Zn2+ buffering can exacerbate Zn2+-triggered respiratory inhibition (via exposure to high K+/MK-801 with 300 μM Zn2+, 100 μM DTDP alone, 10 μM Zn2+ + DTDP, all in 0 Ca2+ HSS; Fig 7C left). When present, DTDP was added 10 min prior to, during and for 20 min after the high K+ exposures, to ensure strong disruption of buffering at the time of and for a period after the Zn2+ loading. Our findings were generally consistent with those above, examining Zn2+ effects on Δψm, ROS generation, and mitochondrial morphology. Specifically, whereas OCR after high K+ exposures with either 300 μM Zn2+ or 1.8 mM Ca2+ alone was not different from that of control (wash into 1.8 Ca2+ HSS), with combined Zn2+ and Ca2+ exposure, both the basal and the maximal uncoupled OCR (upon FCCP exposure) were substantially decreased (by ~ 50%) (Fig. 7B right). Similarly, whereas OCR after exposure to either 100 μM DTDP or high K+/300 μM Zn2+ alone was little different from that of control (wash into 0 Ca2+ HSS), exposure to high K+/10 μM Zn2+/DTDP caused almost complete inhibition of respiration (Fig. 7C right). Of note, these effects on respiration were long-lasting, persisting even at the time of FCCP application (>2 h after the high K+/Zn2+ exposures).
In sum, these findings further support the hypothesis that mitochondrial Zn2+ accumulation (enhanced and prolonged by disrupted cytosolic Zn2+ buffering), can act synergistically with Ca2+ to disrupt mitochondrial function. The intense swelling and long lasting respiratory inhibition caused by the low Zn2+ exposures with 100 μM DTDP (but not by DTDP alone) lend further credence to the idea that Zn2+-triggered mitochondrial dysfunction may be irreversible and contribute to neuronal death in pathological conditions.
Mitochondrial Zn2+ accumulation contributes to dose-dependent cell death
While above studies showing pronounced mitochondrial swelling and long lasting respiratory inhibition might predict that these effects would contribute to subsequent neurodegeneration, they are not in themselves indicative of cell death. We thus felt it important to carry out neurotoxicity studies, to more directly address the cytotoxic consequences of the mitochondrial effects (Fig. 8).
Figure 8. Mitochondrial Zn2+ accumulation contributes to neuron death.
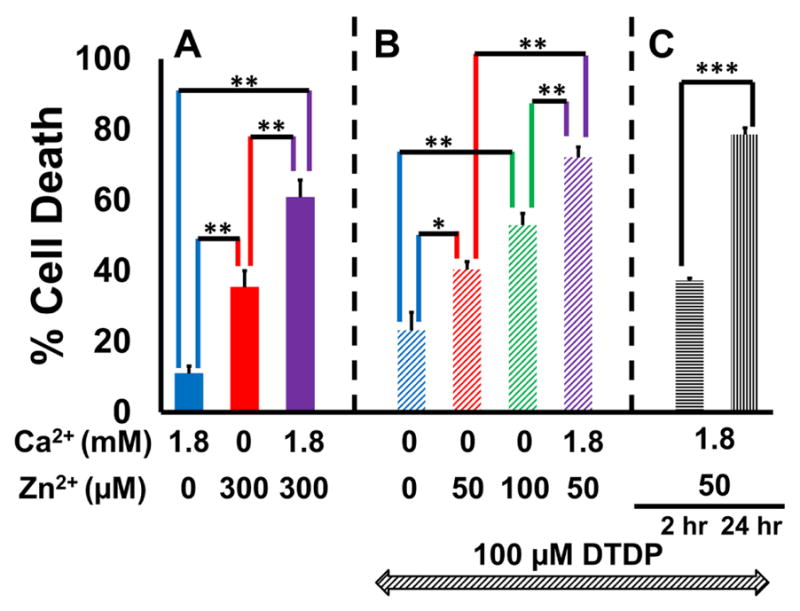
Neurons were exposed to a sequence of 10 min DTDP (100 μM; as indicated in B and C), 5 min high K+/MK-801 exposures with Zn2+ and/or Ca2+ (concentration as shown), washed for 10 min (with DTDP in B and C), transferred to MEM + 25 mM glucose and returned to the incubator for 24 hrs (or for only 2 hrs where indicated in C), prior to assessing cell death via LDH efflux assay. Bars show % cell death (see Material and methods), and represent mean ± SEM of 3 independent experiments, each consisting of 4 wells of cultured neurons (* indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001 by one-way ANOVA with Tukey post hoc [A and B] or by two-tailed t-test [C]).
A). Ca2+ and Zn2+ synergistically induce cell death: Note that while 300 μM Zn2+ induced more cell death than 1.8 mM Ca2+, its impact was further exacerbated by the presence of Ca2+.
B). Dose-dependency of Zn2+-induced cell death under conditions of strongly disrupted buffering: Note the dose-dependent increase in cell death with increasing Zn2+ exposures, that was further enhanced by the presence of Ca2+.
C). Zn2+-induced cell death progresses gradually over hours: Note the significantly greater cell death at 24 hrs compared to 2 hrs.
Two sets of studies—generally paralleling those on mitochondrial respiration (Fig. 7)—were carried out to assess the importance of both Zn2+-Ca2+ synergy and integrity of cytosolic Zn2+ buffering to delayed neurodegeneration. In the first set, cultures were exposed to high K+/MK-801 with 300 μM Zn2+, 1.8 mM Ca2+, or both Zn2+ and Ca2+ for 5 min (Fig. 8A). In the second set, cultures were pre-exposed to 100 μM DTDP prior to 5 min high K+/MK-801 with 0, 50 or 100 μM Zn2+ (± Ca2+), followed by 10 min washout into DTDP (Fig. 8B). In both sets, neurons were transferred to MEM (supplemented with 25 mM glucose) and returned to the incubator for 24 hrs after which neuronal injury was assessed via direct morphological examination and by lactate dehydrogenase (LDH) efflux assay (as described in Material and methods). Our findings are again consistent with above studies on mitochondrial function, with Ca2+ strongly enhancing Zn2+ triggered cell death (in neurons with both intact and disrupted buffering), and with Zn2+ exposure inducing dose-dependent injury under conditions of strongly disrupted buffering (Fig. 8A, B). While these findings do not directly prove that Zn2+ induced mitochondrial dysfunction caused the cell death, the similarity of effects for both mitochondrial dysfunction and neurodegeneration strongly suggest a link between the two.
In addition, to examine the temporal relationship between mitochondrial disruption and cell death (as indicated by LDH release, reflecting loss of membrane integrity), cultures were pre-exposed to 100 μM DTDP prior to 5 min high K+/MK-801 with 50 μM Zn2+ + Ca2+ followed by 10 min washout into DTDP (as in Fig. 8B), but with LDH release measured after both 2 hrs (when we observed near complete inhibition of respiration, see Fig. 7C) and after 24 hrs. We found the LDH release at 2 hrs to be less than half that at 24 hrs, highlighting the progressive cellular disintegration occurring after severe mitochondrial disruption (Fig. 8C).
If mitochondrial Zn2+ accumulation does contribute to neurodegeneration in vivo (such as after transient ischemia), it would be valuable to determine whether targeted delayed interventions could abrogate its effects, yielding better outcomes. To begin to address this possibility, we used the membrane permeable Zn2+ chelator TPEN. Cultures were subjected to an exposure we had found to cause rapid mitochondrial swelling and extensive neurodegeneration (high K+/50 μM Zn2+/100 μM DTDP/MK-801 in 1.8 Ca2+ HSS, with apocynin added in HEt loaded cultures, as in Fig. 6, 8B). We first examined effects of this exposure on ROS generation, which can contribute to subsequent neuronal damage. This exposure caused a rapidly increasing HEt ΔF that persisted for at least 30 min after the 5 min Zn2+ exposure (much as in Fig. 5C). However, when TPEN (20 μM) was added immediately after washout of the high K+/Zn2+ exposure, the HEt ΔF was markedly attenuated (Fig. 9A). We then wondered whether this attenuation of ROS generation would be reflected by changes in subsequent neurodegeneration. To test this, cultures were exposed to high K+/50 μM Zn2+/100 μM DTDP/MK-801 in 1.8 Ca2+ HSS alone (as above; Fig. 8B) or with TPEN (10 μM) added either 10 min before, during and after the Zn2+ exposure (TPEN pre-treatment), or only after the Zn2+ exposure (TPEN post-treatment; Fig. 9B). While TPEN pre-treatment was markedly protective, validating the importance of Zn2+ to neuronal injury, it is notable that post-treatment was also modestly protective (Fig. 9B). As the effect of Zn2+ chelation on cell death parallels that on ROS generation (Fig. 9A), these findings further support the idea that mitochondrial dysfunction contributes to neurodegeneration and suggest the potential utility of delayed interventions. In sum, these findings not only strengthen the hypothesis that slow Zn2+ translocation via VSCC along with Ca2+, under conditions of disrupted cytosolic Zn2+ buffering (as likely occurs during pathologic conditions), can induce mitochondrial dysfunction and cell death, but also suggest the exciting possibility that delayed modulation of mitochondrial Zn2+ accumulation could provide neuroprotection.
Figure 9. Delayed Zn2+ chelation attenuates mitochondrial ROS generation and neuron death.

Neurons were exposed to high K+/50 μM Zn2+/MK-801, with DTDP (100 μM), and apocynin (500 μM, A only). TPEN was applied as indicated below. Traces (A) represent HEt Fx/F0 and bars (B) represent % cell death after 24 hr; all values are mean ± SEM and represents ≥ 3 independent experiments. Grey bar (in A) indicates time points of comparison (* indicates p < 0.05, ** indicates p < 0.01, by two-tailed t-test [A] or by one-way ANOVA with Tukey post hoc [B]).
A). Delayed Zn2+ chelation attenuates Zn2+ triggered ROS production: Note the rapid rise in HEt ΔF that was largely attenuated by TPEN (20 μM), added after the Zn2+ exposure.
B). Zn2+ chelation attenuates cell death even when delivered after the Zn2+ exposure: Cultured neurons were exposed as described, with TPEN (10 μM) present either for 10 min before, during and 10 min after high K+/Zn2+ exposure (TPEN pre), or only for 10 min after Zn2+ exposure (TPEN post). Cultures were then transferred to MEM + 25 mM glucose and returned to the incubator for 24 hrs, prior to assessing cell death via LDH efflux assay. Note that both the TPEN pre- and post-treatments attenuated neuron death.
Discussion
Summary of findings
In the present study, we sought to elucidate effects of slow Zn2+ uptake via VSCC on mitochondrial function and subsequent neuronal injury. We find that with disrupted buffering, as likely occurs during in vivo ischemia, brief exposure to low Zn2+ under depolarizing conditions (to elicit extracellular Zn2+ entry through the VSCC) induced acute mitochondrial dysfunction, including loss of ΔΨm, ROS generation, mitochondrial swelling, and inhibition of respiration, as well as delayed neurodegeneration. The presence of physiologic levels of Ca2+ exacerbated these deleterious Zn2+ effects despite attenuating both cytosolic and mitochondrial Zn2+ accumulation, suggesting strong synergism between these ions. While our findings do not prove that Zn2+ triggered mitochondrial dysfunction directly led to neurodegeneration, the strong correlation between the effects of Zn2+ exposures under varied conditions on mitochondria with the induction of cell death supports the hypothesis that mitochondrial Zn2+ accumulation—and the consequent dysfunction—is an important upstream contributor to neuronal injury. Finally, Zn2+ chelation after Zn2+ loading attenuated both mitochondrial ROS generation and neuron death, further supporting the hypothesis that Zn2+ triggered mitochondrial dysfunction is an early contributor to neuronal damage, and that delayed, targeted interventions can be protective.
Zn2+ triggered neurodegeneration: ongoing questions about sources and targets
In light of the debilitating consequences of ischemic stroke, there is a compelling need to develop a better understanding of neuronal injury mechanism in order to identify neuroprotective targets. While attention has long focused on Ca2+ as the critical ionic contributor to neuronal injury, emerging clues—including observations of cytosolic Zn2+ accumulation in neurons after ischemia and findings that selective Zn2+ chelation can be neuroprotective—have highlighted important contribution of Zn2+ (Calderone et al., 2004; Koh et al., 1996; Tonder et al., 1990). Indeed, most Ca2+ indicators also respond to Zn2+ with greater affinity than Ca2+, and it is likely that some effects previously attributed to Ca2+ are actually due to Zn2+ (Cheng and Reynolds, 1998; Stork and Li, 2006).
Our understanding of Zn2+ mechanisms in ischemia has been in flux. It was first assumed that the toxic Zn2+ accumulation seen after ischemia or prolonged seizures resulted from presynaptic release (Assaf and Chung, 1984; Howell et al., 1984) and its translocation into post-synaptic neurons (Koh et al., 1996; Tonder et al., 1990) through routes including VSCC and Ca-AMPA channels (Noh et al., 2005; Sensi et al., 2000; Yin et al., 2002). However, in studies using mice lacking presynaptic releasable Zn2+ (via knockout of the vesicular Zn2+ transporter, ZnT3) (Cole et al., 2000; Cole et al., 1999), prolonged seizures surprisingly still caused strong Zn2+ accumulation and Zn2+ dependent injury to CA1 pyramidal neurons, highlighting critical contributions from other sources (Lee et al., 2000). Further studies using ZnT3 knockouts as well as knockouts of the neuronal Zn2+ binding protein, metallothionein-III (MT-III) (Erickson et al., 1997), provided compelling evidence for distinct contributions of Zn2+ to neuronal injury between CA1 and CA3 pyramidal neurons, with Zn2+ translocation, likely in large part through Ca-AMPA channels, predominating in CA3, but mobilization from MT-III predominating in CA1 (Lee et al., 2000; Lee et al., 2003; Medvedeva et al., 2017). Thus, it is now apparent that synaptically released Zn2+ as well as Zn2+ mobilized from intracellular binding proteins (like MT-III) can contribute to neuronal accumulation and injury in pathological conditions, although the respective contributions from these sources likely differ between populations of neurons.
Another unsettled question concerns the target(s) through which Zn2+ mediates injury. While it is apparent that Zn2+ can activate multiple pathways that contribute to neurodegeneration (Shuttleworth and Weiss, 2011), several lines of evidence led us to consider that mitochondria may be an important early target. Zn2+ has potent and complicated effects on isolated mitochondria, entering them through the mitochondrial Ca2+ uniporter (MCU), inhibiting electron transport and other critical mitochondrial enzymes at submicromolar concentrations, and triggering swelling via activation of mitochondrial permeability transition pore (mPTP); highlighting the complexity of these effects, they are exposure dependent, with low levels increasing respiration and inhibiting ROS production while high levels induced opposite effects (Brown et al., 2000; Dineley et al., 2005; Gazaryan et al., 2007; Jiang et al., 2001; Link and von Jagow, 1995; Sensi et al., 2003; Skulachev et al., 1967; Wudarczyk et al., 1999). In cultured neurons, Zn2+ loading via rapid entry through Ca-AMPA channels resulted in acute mitochondrial dysfunction, including rapid loss of Δψm and ROS production (Sensi et al., 1999a; Sensi et al., 1999b, 2000), supporting the idea that mitochondria may be important Zn2+ targets in neuronal injury.
However, not all studies support this idea. First, brief fairly high (300 μM) Zn2+ exposures to depolarized neurons (triggering slower Zn2+ translocation through VSCC) induced considerable delayed neurodegeneration but caused relatively little acute mitochondrial dysfunction (Pivovarova et al., 2014; Sensi et al., 1999a; Weiss et al., 1993). Furthermore, a recent study on isolated mitochondria in highly purified Ca2+ free buffer reported that in contrast to Ca2+, Zn2+ was a weak trigger of depolarization and did not trigger mPTP opening (Devinney et al., 2009). Thus, although Zn2+ appears to contribute to neurodegeneration after ischemia or prolonged seizures, there has been ongoing debate as to respective contributions of Zn2+ vs Ca2+ to mitochondrial dysfunction in these conditions.
Mitochondrial Zn2+ accumulation and its consequences
Despite evidence that both Zn2+ and Ca2+ contributed to neurodegeneration after ischemia or prolonged seizures, until recently there had been little attempt to discriminate their respective effects. To this aim, we carried out the first study seeking to track both ions simultaneously in single pyramidal neurons in hippocampal slices subjected to oxygen glucose deprivation (OGD) (Medvedeva et al., 2009), and found Zn2+ rises to precede and contribute to subsequent lethal Ca2+ overload; with Zn2+ chelation, the cell death was markedly delayed. Furthermore, the early Zn2+ effects appeared to depend upon its interaction with mitochondria (Medvedeva et al., 2009), with uptake of endogenous Zn2+ into mitochondria through the MCU contributing specifically to ROS production and neuronal cell death (Medvedeva and Weiss, 2014). Indeed, other studies have supported the idea that Zn2+ may contribute to mitochondrial dysfunction after in vivo ischemia, specifically promoting release of pro-apoptotic peptides from mitochondria and contributing to the activation of large channels in the mitochondrial outer membranes (Bonanni et al., 2006; Calderone et al., 2004).
As noted above, despite some studies suggesting that Zn2+ may only weakly impact mitochondrial function (Devinney et al., 2009; Pivovarova et al., 2014), there has been a growing body of evidence that Zn2+—as well as Ca2+—can enter mitochondria and impact their function. While the nature of interactions between these ions on mitochondria has been little explored, prior studies have provided early clues for possible synergism between these ions. Specifically, combined application of Ca2+ and Zn2+ caused greater swelling of isolated mitochondria than Ca2+ or Zn2+ alone (Jiang et al., 2001), and Zn2+ entry through Ca-AMPA channels yielded stronger and more persistent ROS generation when Ca2+ was also present (Sensi et al., 2000). Present studies extend these early clues via examination of effects of slower Zn2+ and Ca2+ entry through VSCC, and find potent synergism between Zn2+ and Ca2+ on multiple measures of mitochondrial dysfunction. These findings are particularly notable, as brief Ca2+ influx through VSCC does not generally cause significant injury, and the presence of Ca2+ clearly resulted in a decreased amount of Zn2+ entering the neurons.
However, the nature of the interactions between Ca2+ and Zn2+ remains unclear and merits further study. One possibility is that the presence of Ca2+ may modify Zn2+ permeation through the MCU. While the MCU was previously considered to be quite selective for Ca2+, Zn2+ can also enter mitochondria through this route (Clausen et al., 2013; Gazaryan et al., 2007; Jiang et al., 2001; Malaiyandi et al., 2005; Saris and Niva, 1994), and Ca2+ was actually found to markedly facilitate Zn2+ entry through the MCU in isolated mitochondria (Saris and Niva, 1994). Studies of the recently identified MCU protein and associated regulatory peptides may yield further clues, with two peptides (termed MICU1 and MICU2) acting to regulate pore conductance as a function of the cytosolic Ca2+ concentration, inhibiting the channel when levels are low, and activating it when they rise (Kamer and Mootha, 2015; Marchi and Pinton, 2014; Murgia and Rizzuto, 2015). Thus, might some Ca2+ be needed for channel gating and Zn2+ entry, possibly accounting for the paucity of Zn2+ effects on isolated mitochondria carried out in purified Ca2+ free media (Devinney et al., 2009)? Indeed, a Ca2+ dependence for channel gating and Zn2+ entry through the MCU could contribute to the observed synergism between Ca2+ and Zn2+.
The other unresolved issue concerns whether the levels of Zn2+ readily achieved in neurons in pathological conditions are likely to induce acute disruption of mitochondrial function, in light of observations (discussed above) that brief strong Zn2+ exposures to depolarized neurons caused little acute disruption of mitochondrial function (Pivovarova et al., 2014; Sensi et al., 1999a). However, although such relatively slow Zn2+ entry through VSCC does not cause the acute ROS production and strong loss of ΔΨm seen with more rapid entry (through Ca-AMPA channels), these exposures do result in persistent (at least 2 hrs) Zn2+ accumulation in mitochondria, contributing to partial loss of ΔΨm and release of pro-apoptotic peptides (Jiang et al., 2001; Sensi et al., 2002). It is also notable that strong disruption of cytosolic Zn2+ buffering alone (in the absence of extracellular Zn2+) results in sufficient intracellular Zn2+ mobilization to cause milder disruption of mitochondrial function and trigger delayed neurodegeneration (Aizenman et al., 2000; Bossy-Wetzel et al., 2004; Sensi et al., 2003). Thus, perhaps it is not surprising that in combination, even relatively slow Zn2+ entry under conditions of impaired cytosolic Zn2+ buffering can acutely disrupt mitochondrial function.
In past studies we found that strong disruption of buffering enhanced ROS production caused by relatively low levels of Zn2+ entry (Clausen et al., 2013). Present studies markedly extend the understanding of conditions that determine effects of cellular Zn2+ loads on mitochondria. First, we find that even partial disruption of cytosolic Zn2+ buffering (that had little effect on its own), markedly increase mitochondrial dysfunction caused by brief Zn2+ exposures even at levels as low as 10 μM. Secondly, we find that the effects are markedly enhanced by the presence of physiological levels of Ca2+ entry (and may be artificially inhibited under the non-physiological conditions of limited or absent Ca2+ in which many prior studies of Zn2+ were carried out). We further find that the enhanced disruption of mitochondria extends beyond the usual measures of loss of ΔΨm and ROS production to include strong and progressive swelling and long lasting inhibition of respiration, effects that may be indicative of severe or irrecoverable disruption. Finally, we examine effects of the presence of Ca2+ and disrupted Zn2+ buffering on cell death. While we do not definitively demonstrate that the Zn2+ dependent mitochondrial disruption leads directly to cell death, we found the effects on mitochondrial function to be strongly correlated with those on cell death and that delayed Zn2+ chelation attenuated both the mitochondrial ROS generation and the subsequent neurodegeneration. Thus, our studies provide new support to the idea that Zn2+ triggered mitochondrial disruption is an important upstream event that contributes to delayed injury, the targeting of which could be protective.
Conclusions and possible therapeutic implications
We started with the comment that the available treatments for ischemia injury are inadequate, reflecting in part incomplete understanding of relevant targetable events. We wish to end by suggesting a new hypothesis: Early mitochondrial Zn2+ accumulation—and the consequent disruption of their function—is a critical and targetable event in the neurodegenerative sequence. The validity of this hypothesis depends upon a number of factors: 1. The occurrence of disrupted buffering. It is apparent that acidosis and oxidative stress occur in the context of ischemia, and can result in the disruption of cytosolic Zn2+ buffering and its toxic accumulation in neurons (Aizenman et al., 2000; Lee et al., 2000; Sensi et al., 2003; Shuttleworth and Weiss, 2011; Weiss et al., 2000). 2. The occurrence of Zn2+ accumulation in the extracellular space. It is difficult to accurately quantify Zn2+ accumulation in brain resulting from synaptic release; early estimates that peak levels reach 100–300 μM (Frederickson, 1989) are probably high. Widespread extracellular accumulation with ischemia or seizures certainly occurs, and recent studies suggest that levels in the 10–100 μM range may be achievable in areas of hippocampus and cortex where there is considerable synaptic Zn2+ (Frederickson et al., 2006; Ueno et al., 2002; Vogt et al., 2000). Although neurons expressing Ca-AMPA channels may get the strongest Zn2+ loads, present studies suggest that even brief presence of as little as 10 μM Zn2+ may result in sufficient entry into any depolarized neurons with impaired cytosolic Zn2+ buffering to trigger acute mitochondrial disruption. In light of observations that VSCC activity increases with aging and chronic hypoxia (Thibault and Landfield 1996, Campbell et al 1996, Webster et al 2006), these effects may be greater still in the aging populations most at risk of ischemic attack.
Present studies also provide a “proof of principle” for the potential utility of delayed delivery of Zn2+ targeting interventions – with Zn2+ chelation after the Zn2+ exposure decreasing both ROS generation and subsequent cell death (Fig. 9), consistent with other recent findings on OGD induced ROS production (Slepchenko et al., 2017). Optimal interventions may vary depending on when the intervention is delivered. In the early phases, interventions targeting Zn2+ entry into mitochondria might include Zn2+ chelators, MCU blockers, or antioxidants (to diminish oxidative disruption of buffering) while at later stages, mPTP blockers may decrease mitochondrial swelling and mediator release (Friberg and Wieloch, 2002). Of note, most drugs have multiple effects complicating the development of optimal interventions. For instance, whereas MCU blockers alone in early stages of ischemia might diminish mitochondrial Zn2+ accumulation, they could hasten injury by promoting cytosolic Ca2+ overload (Medvedeva and Weiss, 2014; Velasco and Tapia, 2000).
We further suggest that consequences of mitochondrial Zn2+ loading are variable; with very strong loads resulting in acute irreversible mitochondrial disruption and cell death (Medvedeva et al., 2009; Medvedeva and Weiss, 2014), while milder ischemia may result in alterations in mitochondrial function that may contribute to the activation of downstream delayed cell death pathways. Recent findings are compatible with this idea. CA1 pyramidal neurons undergo selective delayed degeneration after transient ischemia, which is associated with delayed mitochondrial swelling and cytochrome C release (Sugawara et al., 1999). Interestingly, in recent slice studies, we found persistent Zn2+ accumulation in CA1 mitochondria after transient sublethal OGD (Medvedeva et al., 2017). In light of the demonstrated potent ability of mitochondrial Zn2+ to trigger dysfunction including ROS generation, swelling, mPTP activation and cytochrome C release (Gazaryan et al., 2007; Jiang et al., 2001; Weiss et al., 2000) might the Zn2+ be a targetable trigger for the downstream death promoting events? One such mechanism may be the delayed insertion of Kv2.1 K+ channels that can cause a form of apoptotic neurodegeneration (Aizenman et al., 2000; McLaughlin et al., 2001). As mitochondrial ROS has been implicated in the activation of both p38 mitogen-activated protein kinase (MAPK) and apoptosis signal-regulating kinase 1 (ASK1) (Bossy-Wetzel et al., 2004; Soberanes et al., 2009), both of which are essential for insertion of the Kv2.1 K+ channels (Aras and Aizenman, 2005; McLaughlin et al., 2001), perhaps Zn2+ induced mitochondrial dysfunction is a critical upstream contributor to these events? In sum, we feel that considerable data—including findings from present study—highlights the likely importance of early interactions of Zn2+ with mitochondria as a trigger of acute and delayed injury after brain ischemia or prolonged seizures, and that efforts to target these early events could yield therapeutic benefits.
Highlights.
Excitotoxic Zn2+ and/or Ca2+ accumulation can trigger neuronal injury in vitro
Slow neuronal Zn2+ entry causes mitochondrial uptake and mild functional disruption
These effects are markedly increased upon disruption of cytosolic Zn2+ buffering
Ca2+ impedes neuronal Zn2+ entry but markedly enhances its impact on mitochondria
Mitochondrial Zn2+ accumulation may be a viable target for neuroprotection
Acknowledgments
Supported by NIH grants NS065219 and NS096987 (JHW), and a grant from the American Heart Association (JHW). The authors declare no competing financial interests.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Ca-AMPA
Ca2+ permeable AMPA channels
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- DTDP
2,2′-dithiodipyridine
- HEt
hydroethidine
- MT
metallothionein
- NMDA
N-methyl-D-aspartate
- TPEN
N,N,N,N-tetrakis(2-pyridylmethyl)ethane-1,2-diamine
- OGD
oxygen glucose deprivation
- ROS
reactive oxygen species
- Rhod123
rhodamine 123
- VSCC
voltage sensitive Ca2+ channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Aras MA, Aizenman E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett. 2005;387:136–140. doi: 10.1016/j.neulet.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H, Ofengeim D, Flannery RJ, Miyawaki T, Cho CH, Polster BM, Pypaert M, Hardwick JM, Sensi SL, Zukin RS, Jonas EA. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci. 2006;26:6851–6862. doi: 10.1523/JNEUROSCI.5444-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between Nitric Oxide and Zinc Pathways to Neuronal Cell Death Involving Mitochondrial Dysfunction and p38-Activated K(+) Channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nature neuroscience. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Kristal BS, Effron MS, Shestopalov AI, Ullucci PA, Sheu KF, Blass JP, Cooper AJ. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2000;275:13441–13447. doi: 10.1074/jbc.275.18.13441. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- Buckman JF, Hernandez H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. Journal of neuroscience methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LW, Hao SY, Thibault O, Blalock EM, Landfield PW. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J Neurosci. 1996;16:6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzoniero LM, Turetsky DM, Choi DW. Measurement of intracellular free zinc concentrations accompanying zinc-induced neuronal death. J Neurosci. 1999;19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Reynolds IJ. Calcium-sensitive fluorescent dyes can report increases in intracellular free zinc concentration in cultured forebrain neurons. J Neurochem. 1998;71:2401–2410. doi: 10.1046/j.1471-4159.1998.71062401.x. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen A, McClanahan T, Ji SG, Weiss JH. Mechanisms of Rapid Reactive Oxygen Species Generation in response to Cytosolic Ca2+ or Zn2+ Loads in Cortical Neurons. Plos One. 2013;8:e83347. doi: 10.1371/journal.pone.0083347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000;39:153–169. doi: 10.1016/s0920-1211(99)00121-7. [DOI] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics: integrated biometal science. 2010;2:306–317. doi: 10.1039/b926662c. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, Malaiyandi LM, Vergun O, DeFranco DB, Hastings TG, Dineley KE. A comparison of Zn2+- and Ca2+-triggered depolarization of liver mitochondria reveals no evidence of Zn2+-induced permeability transition. Cell calcium. 2009;45:447–455. doi: 10.1016/j.ceca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KE, Richards LL, Votyakova TV, Reynolds IJ. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion. 2005;5:55–65. doi: 10.1016/j.mito.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Surin A, Jacobson J. Imaging mitochondrial function in intact cells. Methods in enzymology. 2003;361:353–389. doi: 10.1016/s0076-6879(03)61019-0. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Hollopeter G, Thomas SA, Froelick GJ, Palmiter RD. Disruption of the metallothionein-III gene in mice: analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J Neurosci. 1997;17:1271–1281. doi: 10.1523/JNEUROSCI.17-04-01271.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, Krezel A, McAdoo DJ, Mueller RN, Zeng Y, Balaji RV, Masalha R, Thompson RB, Fierke CA, Sarvey JM, de Valdenebro M, Prough DS, Zornow MH. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol. 2006;198:285–293. doi: 10.1016/j.expneurol.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Hernandez MD, McGinty JF. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989;480:317–321. doi: 10.1016/0006-8993(89)90199-6. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nature reviews Neuroscience. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Gazaryan IG, Krasinskaya IP, Kristal BS, Brown AM. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem. 2007;282:24373–24380. doi: 10.1074/jbc.M611376200. [DOI] [PubMed] [Google Scholar]
- Gee KR, Zhou ZL, Ton-That D, Sensi SL, Weiss JH. Measuring zinc in living cells. A new generation of sensitive and selective fluorescent probes. Cell calcium. 2002;31:245–251. doi: 10.1016/S0143-4160(02)00053-2. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochemical Society transactions. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Current molecular medicine. 2004;4:131–136. doi: 10.2174/1566524043479248. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? The Lancet. Neurology. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Jia Y, Jeng JM, Sensi SL, Weiss JH. Zn2+ currents are mediated by calcium-permeable AMPA/kainate channels in cultured murine hippocampal neurones. J Physiol. 2002;543:35–48. doi: 10.1113/jphysiol.2002.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH. Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem. 2001;276:47524–47529. doi: 10.1074/jbc.M108834200. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nature reviews Molecular cell biology. 2015;16:545–553. doi: 10.1038/nrm4039. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Canzoniero LM, Yu SP, Ling C, Choi DW. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J Physiol. 2000;528(Pt 1):39–52. doi: 10.1111/j.1469-7793.2000.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. Journal of neuroscience methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci. 2000;20:RC79. doi: 10.1523/JNEUROSCI.20-11-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim JH, Palmiter RD, Koh JY. Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol. 2003;184:337–347. doi: 10.1016/s0014-4886(03)00382-0. [DOI] [PubMed] [Google Scholar]
- Link TA, von Jagow G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J Biol Chem. 1995;270:25001–25006. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- Malaiyandi LM, Dineley KE, Reynolds IJ. Divergent consequences arise from metallothionein overexpression in astrocytes: zinc buffering and oxidant-induced zinc release. Glia. 2004;45:346–353. doi: 10.1002/glia.10332. [DOI] [PubMed] [Google Scholar]
- Malaiyandi LM, Vergun O, Dineley KE, Reynolds IJ. Direct visualization of mitochondrial zinc accumulation reveals uniporter-dependent and -independent transport mechanisms. J Neurochem. 2005;93:1242–1250. doi: 10.1111/j.1471-4159.2005.03116.x. [DOI] [PubMed] [Google Scholar]
- Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. 2014;592:829–839. doi: 10.1113/jphysiol.2013.268235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W. Redox biochemistry of mammalian metallothioneins. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2011;16:1079–1086. doi: 10.1007/s00775-011-0800-0. [DOI] [PubMed] [Google Scholar]
- Maret W. Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics: integrated biometal science. 2015;7:202–211. doi: 10.1039/c4mt00230j. [DOI] [PubMed] [Google Scholar]
- Maret W, Vallee BL. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc Natl Acad Sci U S A. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord MC, Aizenman E. Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2.1 K+ currents. Proc Natl Acad Sci U S A. 2013;110:13988–13993. doi: 10.1073/pnas.1306238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Ji SG, Yin HZ, Weiss JH. Differential Vulnerability of CA1 versus CA3 Pyramidal Neurons After Ischemia: Possible Relationship to Sources of Zn2+ Accumulation and Its Entry into and Prolonged Effects on Mitochondria. J Neurosci. 2017;37:726–737. doi: 10.1523/JNEUROSCI.3270-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci. 2009;29:1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Weiss JH. Intramitochondrial Zn(2+) accumulation via the Ca(2+) uniporter contributes to acute ischemic neurodegeneration. Neurobiology of disease. 2014;68:137–144. doi: 10.1016/j.nbd.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia M, Rizzuto R. Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell calcium. 2015;58:11–17. doi: 10.1016/j.ceca.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci. 2000;20:RC111. doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci U S A. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Stanika RI, Kazanina G, Villanueva I, Andrews SB. The interactive roles of zinc and calcium in mitochondrial dysfunction and neurodegeneration. J Neurochem. 2014;128:592–602. doi: 10.1111/jnc.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic--ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- Saris NE, Niva K. Is Zn2+ transported by the mitochondrial calcium uniporter? FEBS Lett. 1994;356:195–198. doi: 10.1016/0014-5793(94)01256-3. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Weiss JH. Mitochondrial sequestration and Ca(2+)-dependent release of cytosolic Zn(2+) loads in cortical neurons. Neurobiology of disease. 2002;10:100–108. doi: 10.1006/nbdi.2002.0493. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A. 1999a;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Weiss JH. Glutamate triggers preferential Zn2+ flux through Ca2+ permeable AMPA channels and consequent ROS production. Neuroreport. 1999b;10:1723–1727. doi: 10.1097/00001756-199906030-00018. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Weiss JH. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. The European journal of neuroscience. 2000;12:3813–3818. doi: 10.1046/j.1460-9568.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Weiss JH. Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol Sci. 2011;32:480–486. doi: 10.1016/j.tips.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP, Chistyakov VV, Jasaitis AA, Smirnova EG. Inhibition of the respiratory chain by zinc ions. Biochemical and biophysical research communications. 1967;26:1–6. doi: 10.1016/0006-291x(67)90242-2. [DOI] [PubMed] [Google Scholar]
- Slepchenko KG, Lu Q, Li YV. Cross talk between increased intracellular zinc (Zn2+) and accumulation of reactive oxygen species in chemical ischemia. American journal of physiology Cell physiology. 2017;313:C448–C459. doi: 10.1152/ajpcell.00048.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberanes S, Urich D, Baker CM, Burgess Z, Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM, Kamp DW, Chandel NS, Schumacker PT, Mutlu GM, Budinger GR. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284:2176–2186. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CJ, Li YV. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: a question on calcium overload. J Neurosci. 2006;26:10430–10437. doi: 10.1523/JNEUROSCI.1588-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Tonder N, Johansen FF, Frederickson CJ, Zimmer J, Diemer NH. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett. 1990;109:247–252. doi: 10.1016/0304-3940(90)90002-q. [DOI] [PubMed] [Google Scholar]
- Ueno S, Tsukamoto M, Hirano T, Kikuchi K, Yamada MK, Nishiyama N, Nagano T, Matsuki N, Ikegaya Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. The Journal of cell biology. 2002;158:215–220. doi: 10.1083/jcb.200204066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco I, Tapia R. Alterations of intracellular calcium homeostasis and mitochondrial function are involved in ruthenium red neurotoxicity in primary cortical cultures. Journal of neuroscience research. 2000;60:543–551. doi: 10.1002/(SICI)1097-4547(20000515)60:4<543::AID-JNR13>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C. Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging. 2006;27:439–445. doi: 10.1016/j.neurobiolaging.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Hartley DM, Koh JY, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Wudarczyk J, Debska G, Lenartowicz E. Zinc as an inducer of the membrane permeability transition in rat liver mitochondria. Archives of biochemistry and biophysics. 1999;363:1–8. doi: 10.1006/abbi.1998.1058. [DOI] [PubMed] [Google Scholar]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Turetsky D, Choi DW, Weiss JH. Cortical neurones with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiology of disease. 1994;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Sensi SL, Ogoshi F, Weiss JH. Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J Neurosci. 2002;22:1273–1279. doi: 10.1523/JNEUROSCI.22-04-01273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HZ, Weiss JH. Zn(2+) permeates Ca(2+) permeable AMPA/kainate channels and triggers selective neural injury. Neuroreport. 1995;6:2553–2556. doi: 10.1097/00001756-199512150-00025. [DOI] [PubMed] [Google Scholar]