

Abstract
Inference of how evolutionary forces have shaped extant genetic diversity is a cornerstone of modern comparative sequence analysis. Advances in sequence generation and increased statistical sophistication of relevant methods now allow researchers to extract ever more evolutionary signal from the data, albeit at an increased computational cost. Here, we announce the release of Datamonkey 2.0, a completely re-engineered version of the Datamonkey web-server for analyzing evolutionary signatures in sequence data. For this endeavor, we leveraged recent developments in open-source libraries that facilitate interactive, robust, and scalable web application development. Datamonkey 2.0 provides a carefully curated collection of methods for interrogating coding-sequence alignments for imprints of natural selection, packaged as a responsive (i.e. can be viewed on tablet and mobile devices), fully interactive, and API-enabled web application. To complement Datamonkey 2.0, we additionally release HyPhy Vision, an accompanying JavaScript application for visualizing analysis results. HyPhy Vision can also be used separately from Datamonkey 2.0 to visualize locally executed HyPhy analyses. Together, Datamonkey 2.0 and HyPhy Vision showcase how scientific software development can benefit from general-purpose open-source frameworks. Datamonkey 2.0 is freely and publicly available at http://www.datamonkey.org, and the underlying codebase is available from https://github.com/veg/datamonkey-js.
Keywords: natural selection, statistical methods, web application, recombination, evolutionary inference
Since its introduction in 2005, Datamonkey (Kosakovsky Pond and Frost 2005a) has been a leading resource for a broad research community of evolutionary biologists and biomedical researchers. Datamonkey’s primary role is to provide a free and open web platform for researchers to conduct comparative analyses of sequence alignments using statistical models developed in HyPhy (Kosakovsky Pond et al. 2005). These analyses can take anywhere from minutes to hours to complete depending on specific analysis being conducted and the size of the data set.
Using a dedicated computing cluster, Datamonkey has processed nearly 1,000,000 jobs and contributed approximately a millennium of cluster CPU time to molecular evolutionary research. The original Datamonkey was written using the then-prevailing paradigm of web application development: Custom scripts in Perl and other languages, all processing done on the server, site styling assuming a desktop client, and many third-party dependencies for visualization. Dramatic advances in modern web application development have rendered this design obsolete and, as a consequence, not maintainable.
Sophisticated modern web platforms have conditioned users to expect certain features that are frequently lacking from scientific software: Professional interface design, snappy interactivity and full functionality within a browser, seamless performance across desktop, tablet, and mobile devices, and applications without page reloads. In parallel, sequence analysis methodology has continued its relentless march forward, with improved methods for studying natural selection (Smith et al. 2015; Murrell et al. 2013; Wertheim et al. 2015) often superseding older methods (Kosakovsky Pond and Frost 2005b; Kosakovsky Pond et al. 2010).
Here, we describe the complete re-engineering process leading to Datamonkey 2.0, available at http://www.datamonkey.org, which adopts modern web application design patterns and provides the most recent analytical tools for studying the evolution of coding sequences. We have exploited the explosive growth of JavaScript (following the V8 open source JavaScript engine release in 2008) to offer a much improved user experience in Datamonkey 2.0. The Datamonkey 2.0 codebase therefore eases module development by recognizing and standardizing design patterns common to web applications, and better accommodates long-running job processes and increasingly large data sets. By incorporating modern software features, we have made Datamonkey more intuitive to users, who will in turn be able to make more efficient use of the underlying data analysis methods.
New Approaches
Software Engineering
Datamonkey 2.0 is written almost entirely in JavaScript (JS, specifically the node.js framework), which has become the world’s most widely used programming language when counting the number of projects using it as the primary language (O’Grady 2017). The JS development ecosystem has rapidly matured (Wittern et al. 2016), and computational benchmark performance gains have been similarly dramatic (Gouy 2017). Key benefits to developing in JS are that the exact same code can run both on the server side and the client side, that JS is natively supported on virtually all popular devices and operating system platforms, and that sophisticated user interfaces are incredibly easy to develop. We conduct dependencies management via webpack, ensuring that installation and updates can be carried quickly and efficiently.
To accommodate hundreds of long-running jobs daily, the Datamonkey 2.0 web application is divided into two independent components (fig. 1). The web application component performs model-view-controller (Leff and Rayfield 2001) duties and is hosted on a standard webserver. Key duties include accepting and managing job submissions from users, displaying progress and result pages, and maintaining a persistent database of jobs and results. The second component is the High Performance Computing (HPC) Job Provisioner, which is tasked with scheduling, running, and monitoring jobs (e.g. via job managers such as Torque), and reporting status updates to the web application. This component is hosted on an HPC cluster, which is generally distinct from the device running the web application. Importantly, by maintaining these components separately, Datamonkey 2.0 can distribute computationally intensive jobs across multiple or alternative HPC resources with minimal effort. Our current implementation can easily scale to thousands of jobs (typical Datamonkey 2.0 queue size is on the order of tens to hundreds jobs with average wait times of only a few minutes for the job to start running), and is limited by the available hardware and the backend task schedulers. The reason for developing a custom job provisioning module, instead of adopting an existing framework (such as Galaxy, described in Goecks et al. 2010), was the requirement of providing complex continuous user feedback on long running jobs, and maintaining an all-JS framework.
Fig. 1.
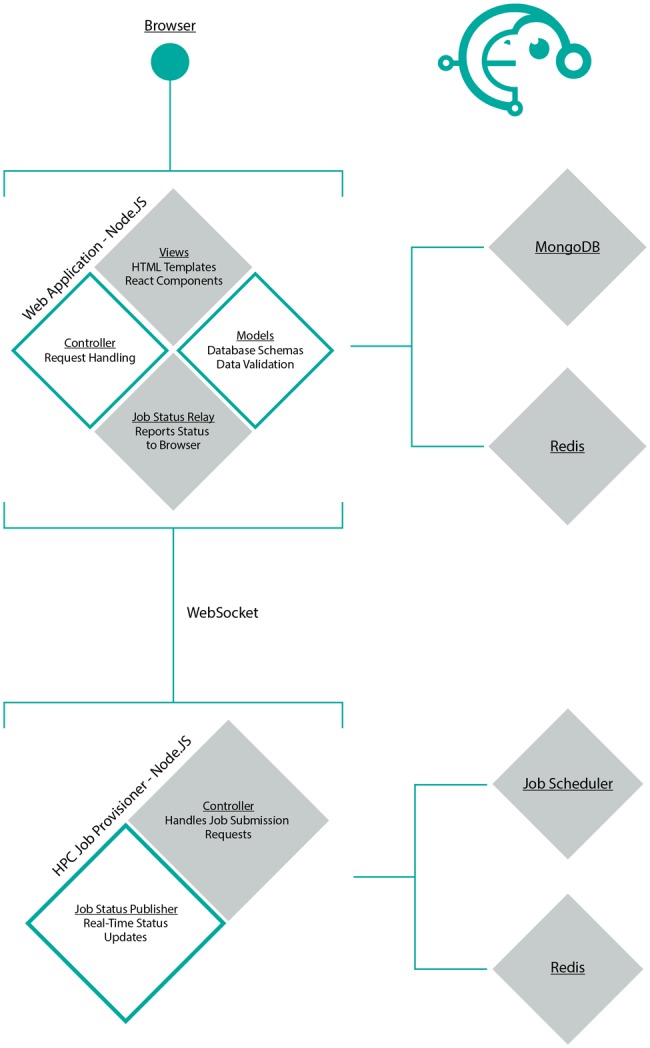
Component architecture of Datamonkey 2.0.
The two components communicate via a publish-subscribe pattern, implemented using Websockets and Redis. Through this setup, the web application subscribes to events that are published by the job provisioner, for example, “job progress update available.” The publish-subscribe pattern is computationally lightweight: The subscriber receives a notification only after the event has been published (as opposed to the polling model, where the web application would be periodically pinging the job provisioner for updates), and is robust to network communication issues.
Persistent unique analysis identifiers allow users to access long-running or past jobs. Documented web routes, for example, www.datamonkey.org/method/job-id enable programmatic scripting of analyses. Because all job requests and analysis results are represented in JSON (JavaScript Object Notation) format, users can prepare requests and process results in nearly every common programming language (e.g. Python, JavaScript, R). Importantly, our use of monolithic JSON files fosters reproducibility by storing a complete record of a given analysis in a single file. For example, a single analysis output file would contain information including test P-values, model parameter estimates, site-specific reports, and trees with estimated selective regimes.
We implement result visualization (described below) as a JS application using react.js as a high-level framework for integrating various reusable encapsulated page components in a single cohesive document. We use d3.js for data visualization (e.g. charts, trees), and bootstrap for styling and standard user interface elements (e.g. buttons, menus).
User Experience
Datamonkey 2.0 offers a streamlined interface that aims to relieve users of “option glut” and to guide them to the most appropriate statistical analysis based on the biological question they are seeking to answer (fig. 2). Once the user uploads a valid multiple sequence alignment and a corresponding phylogeny, and selects any analysis-specific options, the job is submitted to the computing cluster. The user can either directly watch analysis in a browser window or return at any later point to the stable job URL where the current status is shown. Once a job has completed, the computing cluster returns a single JSON file containing a complete description of all relevant results to the web application component of Datamonkey 2.0.
Fig. 2.
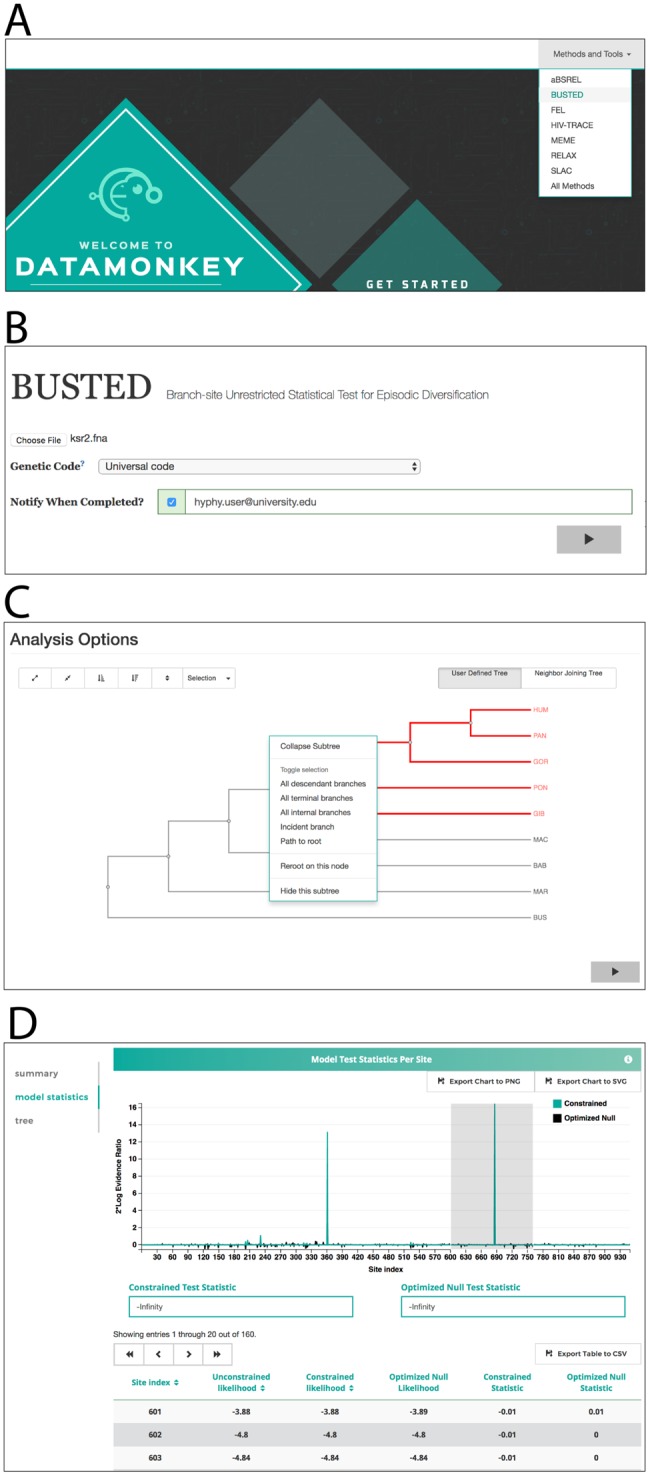
Key stages of the Datamonkey 2.0 user interface, using BUSTED as an example. (A) The landing page with analysis guidance tools, or direct selection of a method. (B) Data upload and preliminary configuration page (data-independent). (C) Continued analysis configuration which is data-dependent, for example, which tree branches to test for selection. (D) The final results page, which is a fully interactive JS application, showing in this case a plot of sites that indicate support for
At this stage, HyPhy Vision, maintained in the hyphy-vision (https://github.com/veg/hyphy-vision) repository, parses and reveals results in an analysis-specific manner (fig. 2D). Users can take advantage of HyPhy Vision directly to view JSON results from any compatible HyPhy analysis, whether executed locally or in Datamonkey 2.0, at http://vision.hyphy.org. The relationship between Datamonkey 2.0 and HyPhy Vision highlights the ease with which JS components can be included in other applications or used on their own.
The HyPhy Vision engine structures analysis-specific visualization as interactive reports with graphical components. All visualization apps include a precise, publication-ready summary statement of the primary analysis results. Analysis-specific graphical components include annotated tree renderings that can be manipulated and exported, filterable tables of site-level results, charts showing statistical evidence of selection at a specific site or branch, and rate distribution plots. We emphasize that all of the interactivity is implemented in JS, runs entirely in the browser, has responsive design (i.e. can be viewed on tablet and mobile devices), and is compatible with all modern browsers. Specific graphical components (charts, trees, etc.) can be exported as image files, and other data can be downloaded in machine- and human-readable formats such as CSV or JSON.
Methods Available
Datamonkey 2.0 includes statistical tests to characterize evolutionary forces that have acted on sequence alignment. We offer both methods designed for exploratory analyses (e.g. generate a list of individual sites subject to episodic diversifying selection) and targeted testing (e.g. test to see if an a priori designated lineage or set of lineages has experienced higher or lower selective pressures compared with a reference lineage). The analyses available with the initial release are described below, and we emphasize that more analyses will be continually added to Datamonkey 2.0 as we (and others) publish new or improved tools that address similar evolutionary questions. Whenever possible, our analyses account for the confounding effect of recombination via data partitioning (Scheffler et al. 2006), and allow synonymous substitution rates to vary across sites (Kosakovsky Pond and Muse 2005).
SLAC
Single Likelihood Ancestor Counting, or SLAC (Kosakovsky Pond and Frost 2005b) is a substitution counting-based method for identifying sites that may have experienced pervasive diversifying or purifying selection. It is able to handle larger data sets (e.g. >1,000 sequences), but generally has the lowest statistical power of all site-specific methods. For example, Banke et al. (2009) used SLAC to screen a large number of viral sequence isolates for selection and found that positive selection was driving secondary mutations in the gag gene of HIV-1 in the presence of drug resistance mutations in the protease gene.
FEL
Fixed Effects Likelihood, or FEL (Kosakovsky Pond and Frost 2005b) is a maximum likelihood method used for identifying sites that may have experienced pervasive diversifying or purifying selection by individually testing whether or not at each site in the alignment. As an example, Brault et al. (2007) ran FEL to screen the genome of the West Nile virus for evidence of positive selection, and then used functional assays to confirm that the single site identified by FEL conferred increased virogenesis in American crows.
FUBAR
Fast Unconstrained Bayesian AppRoximation, or FUBAR (Murrell et al. 2013) is a method designed to identify sites that may have experienced pervasive diversifying or purifying selection. It uses a principled statistical approximation to limit the number of expensive likelihood evaluations and is suitable for large data sets (e.g. >1,000 sequences). For example, Ladner et al. (2015) used FUBAR to analyze a data set of 920 complete Ebola Virus genomes, identifying 11 sites subject to pervasive positive selection.
MEME
Mixed Effects Model of Evolution, or MEME (Murrell et al. 2012) includes a likelihood ratio test for detecting individual sites subject to episodic diversifying selection. Unlike SLAC, FEL, and FUBAR, MEME is able to identify sites where only some of the branches have experienced selective pressure. With a sufficiently large data set, MEME provides the most power in Datamonkey 2.0 for detecting site-level selection. For small numbers of sequences (e.g. <30) FEL may instead be more powerful. One example of an analysis using MEME is from Neverov et al. (2014), who employed MEME to detect sites subject to episodic selection in Influenza A virus (H3N2) and understand how reassortment events modulate adaptive evolution of the virus.
aBSREL
The adaptive Branch-Site Random Effects Likelihood method, or aBSREL (Smith et al. 2015) assesses evidence of positive selection affecting individual branches, which can be specified a priori or examined exhaustively. The method also determines which branches support more complex evolutionary models, and can therefore be used for more accurate estimation of branch lengths in heterotachous coding-sequences for molecular clock dating (Wertheim and Kosakovsky Pond 2011). As an example, Zhou et al. (2015) applied aBSREL to examine patterns of lineage-specific selection in hymenoptera and found that increased episodes of positive selection correlate with the transition to a eusocial lifestyle.
RELAX
This is a specialized method that formally tests whether or not selection has been relaxed or intensified on a collection of a priori-specified branches in the tree relative to others (Wertheim et al. 2015). RELAX is also more generally useful for comparing selective regimes in different parts of the tree as illustrated by Macqueen and Gubry-Rangin (2016), who applied RELAX to identify convergent signatures of relaxed selection pressures in microbes during early stages of transition to an acidophilic lifestyle.
BUSTED
Branch-Site Unrestricted Statistical Test for Episodic Diversification, or BUSTED (Murrell et al. 2015) is a likelihood ratio test for evidence of diversifying selection affecting some (unspecified) sites in the alignment along some (unspecified) branches in the tree. It is best suited for screening relatively small alignments where site or branch-centric methods have little statistical power to detect local selective events. By aggregating signal over sites and branches, BUSTED can achieve increased power in small data sets. As an example of such an application, Enard et al. (2016) used BUSTED to comprehensively screen mammalian orthologs for gene-wide selection and discovered that viral-interacting proteins were enriched for positive selection.
GARD
Genetic Algorithm for Recombination Detection (Kosakovsky Pond et al. 2006) implements a genetic algorithm approach to screen alignments for evidence of phylogenetic incongruence, which is interpreted as a hallmark of recombination, gene conversion, or similar processes. The output of GARD is a partitioned alignment in NEXUS format, with partition-specific trees, representing putatively nonrecombinant fragments. This NEXUS output can be used directly as input to SLAC, FEL, FUBAR, MEME, or BUSTED for a recombination-aware selection analysis. For example, Khan et al. (2015) used such an approach to find evidence of positive selection on olfactory receptor (OR) ligand domains, and linked variation in the OR genes with ecological adaptation in Sauropsida.
Concluding Remarks
Robust and modern software tools are increasingly being recognized as integral to scientific rigor and reproducibility (Howison and Bullard 2016; List et al. 2017), and poor user experience coupled with scientific developers’ tendency to “reinvent the wheel” have been cited as barriers to scientific progress and tool adoption (Prlić and Procter 2012; Wilson et al. 2014). The popularity of Datamonkey and its range of applications attest to the strong demand for user-friendly tools that pave access to sophisticated and computationally intensive methods for sequence analyses. With Datamonkey 2.0, we have leveraged advances in rapid, complex, and robust web application development to bring a modern user experience to comparative sequence analysis, and by doing so we have eliminated learning-curve barriers and made it more likely that users will use this cutting-edge methodology. The “write-once, run everywhere” paradigm first popularized by Java in the 1990s is finally a reality with JavaScript-based web applications, and we hope that the Datamonkey 2.0 makes a convincing case for this approach to scientific tool development.
Further, the open-source components in Datamonkey 2.0 may facilitate future tool development by us and others by accelerating development cycles, thereby enabling a rapid expansion of the available method toolkit for the evolutionary and biomedical research community. For example, we have developed a process that reduced the time needed to add a HyPhy-based sequence analysis to Datamonkey 2.0 to only one or two days of additional development effort. This ease of implementation will make the barrier of developing user-friendly interfaces for novel methods easily surmountable. Finally, because Datamonkey 2.0 and HyPhy Vision are documented open-source projects, their components (e.g. manipulable tree-viewers) may be of use to others working on phylogenetic and molecular evolutionary analysis tools.
Acknowledgments
This work was supported in part by grants R01 GM093939 (NIH/NIGMS) and U01 GM110749 (NIH/NIGMS).
References
- Banke S, Lillemark MR, Gerstoft J, Obel N, Jørgensen LB.. 2009. Positive selection pressure introduces secondary mutations at gag cleavage sites in human immunodeficiency virus type 1 harboring major protease resistance mutations. J Virol. 83(17):8916–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault AC, Huang CY-H, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR.. 2007. A single positively selected west Nile viral mutation confers increased virogenesis in American crows. Nat Genet. 39(9):1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard D, Cai L, Gwennap C, Petrov DA.. 2016. Viruses are a dominant driver of protein adaptation in mammal. eLife 5:e12469.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, Galaxy Team T, and Galaxy Team 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11(8):R86.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy I. 2017. The computer language benchmarks game.
- Howison J, Bullard J.. 2016. Software in the scientific literature: problems with seeing, finding, and using software mentioned in the biology literature. J Assoc Inf Sci Technol. 67(9):2137–2155.http://dx.doi.org/10.1002/asi.23538 [Google Scholar]
- Khan I, Yang Z, Maldonado E, Li C, Zhang G, Gilbert MTP, Jarvis ED, O’Brien SJ, Johnson WE, Antunes A.. 2015. Olfactory receptor subgenomes linked with broad ecological adaptations in sauropsida. Mol Biol Evol. 32(11):2832–2843. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW.. 2005a. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21(10):2531–2533. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW.. 2005b. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 22(5):1208–1222. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV.. 2005. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 22(12):2375–2385.http://dx.doi.org/10.1093/molbev/msi232 [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW.. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 23(10):1891–1901. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Scheffler K, Gravenor MB, Poon AF, Frost SD.. 2010. Evolutionary fingerprinting of genes. Mol Biol Evol. 27(3):520–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner JT, Wiley MR, Mate S, Dudas G, Prieto K, Lovett S, Nagle ER, Beitzel B, Gilbert ML, Fakoli L, et al. 2015. Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell Host Microbe. 18(6):659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff A, Rayfield JT.. 2001. Web-application development using the Model/View/Controller design pattern. Proceedings Fifth IEEE International Enterprise Distributed Object Computing Conference, p. 118–127.
- List M, Ebert P, Albrecht F.. 2017. Ten simple rules for developing usable software in computational biology. PLoS Comput Biol. 13(1):e1005265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macqueen DJ, Gubry-Rangin C.. 2016. Molecular adaptation of ammonia monooxygenase during independent pH specialization in Thaumarchaeota. Mol Ecol. 25(9):1986–1999.http://dx.doi.org/10.1111/mec.13607 [DOI] [PubMed] [Google Scholar]
- Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL, Malik HS.. 2012. Detecting individual sites subject to episodic diversifying selection. PLOS Genet. 8(7):1–10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K.. 2013. Fubar: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 30(5):1196–1205.http://dx.doi.org/10.1093/molbev/mst030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, et al. 2015. Gene-wide identification of episodic selection. Mol Biol Evol. 32(5):1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverov AD, Lezhnina KV, Kondrashov AS, Bazykin GA, Malik HS.. 2014. Intrasubtype reassortments cause adaptive amino acid replacements in h3n2 influenza genes. PLOS Genet. 10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Grady S. 2017. The RedMonk Programming Language Rankings: June 2017.
- Pond SLK, Frost SDW, Muse SV.. 2005. Hyphy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679.http://dx.doi.org/10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- Prlić A, Procter JB.. 2012. Ten simple rules for the open development of scientific software. PLoS Comput Biol. 8(12):e1002802.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Martin DP, Seoighe C.. 2006. Robust inference of positive selection from recombining coding sequences. Bioinformatics 22(20):2493–2499.http://dx.doi.org/10.1093/bioinformatics/btl427 [DOI] [PubMed] [Google Scholar]
- Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL.. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 32(5):1342–1353.http://dx.doi.org/10.1093/molbev/msv022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim JO, Kosakovsky Pond SL.. 2011. Purifying selection can obscure the ancient age of viral lineages. Mol Biol Evol. 28(12):3355–3365.http://dx.doi.org/10.1093/molbev/msr170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K.. 2015. Relax: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol. 32(3):820–832.http://dx.doi.org/10.1093/molbev/msu400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson G, Aruliah DA, Brown CT, Chue Hong NP, Davis M, Guy RT, Haddock SHD, Huff KD, Mitchell IM, Plumbley MD, et al. 2014. Best practices for scientific computing. PLoS Biol. 12(1):e1001745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittern E, Suter P, Rajagopalan S. (2016). A look at the dynamics of the JavaScript package ecosystem. 2016 IEEE/ACM 13th Working Conference on Mining Software Repositories (MSR), p. 351–361.
- Zhou X, Rokas A, Berger SL, Liebig J, Ray A, Zwiebel LJ.. 2015. Chemoreceptor evolution in hymenoptera and its implications for the evolution of eusociality. Genome Biol Evol. 7(8):2407–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]