

Abstract
Background
Severe cardiac hypertrophy can lead to cardiac remodeling and even heart failure in the end, which is a leading cause of cardiovascular disease–related mortality worldwide. A disintegrin and metalloprotease‐22 (ADAM22), a member of the transmembrane and secreted metalloendopeptidase family, participates in many biological processes, including those in the cardiovascular system. However, there is no explicit information on whether ADAM22 can regulate the process of cardiac hypertrophy; the effects that ADAM22 exerts in cardiac hypertrophy remain elusive.
Methods and Results
We observed significantly increased ADAM22 expression in failing hearts from patients with dilated cardiomyopathy and hypertrophic cardiomyopathy; the same trend was observed in mice induced by transaortic constriction and in neonatal rat cardiomyocytes treated by angiotensin II. Therefore, we constructed both cardiac‐specific ADAM22 overexpression and knockout mice. At 4 weeks after transaortic constriction, cardiac‐specific ADAM22 knockout, by the CRISPR/Cas9 (clustered regularly interspaced palindromic repeat (CRISPR)–Cas9) system, deteriorated the severity of cardiac hypertrophy in mice, whereas cardiac‐specific ADAM22 overexpression mitigated the degrees of cardiac hypertrophy in mice. Similarly, altered ADAM22 expression modulated the angiotensin II–mediated cardiomyocyte hypertrophy in neonatal rat cardiomyocytes. After screening several signaling pathways, we found ADAM22 played a role in inhibition of protein kinase B (AKT) activation. Under the cardiac‐specific ADAM22 knockout background, AKT activation was enhanced in transaortic constriction–induced mice and angiotensin II–stimulated neonatal rat cardiomyocytes, with a severe degree of cardiac hypertrophy. Treatment of a specific AKT inhibitor attenuated the transaortic constriction–enhanced AKT activation and cardiac hypertrophy in mice.
Conclusions
The findings demonstrated that ADAM22 negatively regulates the AKT activation and the process of cardiac hypertrophy and may provide new insights into the pathobiological features of cardiac hypertrophy.
Keywords: a disintegrin and metalloprotease‐22, AKT, cardiac hypertrophy, CRISPR/Cas9 system, transaortic constriction
Subject Categories: Hypertrophy
Clinical Perspective
What Is New?
A disintegrin and metalloprotease‐22 lacks the metalloprotease activity and can regulate many biological processes; however, there is no information on whether a disintegrin and metalloprotease‐22 can regulate the process of cardiac hypertrophy.
In our research, we, for the first time, found altered a disintegrin and metalloprotease‐22 expression on the process of cardiac hypertrophy was associated with modulating the protein kinase B signaling in vivo and in vitro.
What Are the Clinical Implications?
These findings provide new insights into the pathogenesis of cardiac hypertrophy, and we believe that these findings should be interesting to cardiologists and other clinical physicians.
Introduction
Cardiac hypertrophy, characterized by cardiomyocyte hypertrophy, interstitial cell proliferation, and myocardial remodeling, is an adaptive manner in response to hemodynamic overload and cardiomyocyte injury.1, 2 Severe cardiac hypertrophy can lead to cardiac remodeling and heart failure.3 Although the mechanical tension is an initial factor for cardiac hypertrophy, the subsequent physiological changes, regulated by metabolic enzymes, activation of related gene transcription, and the signaling pathway, are the key contributors.4, 5, 6 Previous studies have indicated numerous signaling pathways are involved in mechanical stress‐induced hypertrophic responses in cardiomyocytes and the pathological process of cardiomyocyte hypertrophy.5, 7, 8, 9, 10 However, no precise mechanism underlying the pathogenic process of cardiomyocyte hypertrophy had been reported. Currently, metalloproteinase and integrin in extracellular matrix are considered to be the major factors related with cardiac remodeling.11 Integrins usually act as mediator and mechanical pressure transmitters between cells and extracellular matrix, activate the integrin signaling pathway, and then affect various cellular functions.12, 13
A disintegrin and metalloprotease‐22 (ADAM22) is a member of the ADAM family, which is transmembrane and secreted metalloendopeptidases that can regulate cell adhesion, inflammation, and cancer development.14, 15 It is characterized by their structural features, including a prodomain, a disintegrin domain, a metalloprotease domain, a cysteine‐rich domain, an epidermal growth factor like and a transmembrane domain, and a C‐terminal cytoplasmic tail. The ADAMs are the only cell surface proteins that contain disintegrin domain and can effectively digest integrins.16
In this study, we used human heart tissues, a mouse model, and primarily cultured rat cardiomyocytes by modulating ADAM22 expression to determine the potential role of ADAM22 in the development of cardiac hypertrophy.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Human Heart Samples
The left ventricular (LV) tissue samples were collected from 4 patients with dilated cardiomyopathy (DCM) and 4 patients with hypertrophic cardiomyopathy (HCM). A total of 4 non‐DCM LV tissues were obtained from patients with brain death or those who died from an accident and whose hearts were unsuitable for heart transplantation because of noncardiac reasons.17 Written informed consents were obtained from individual patients or their legal family members. This study was conducted according to the Declaration of Helsinki and approved by the Ethics Committees of the Central Hospital of Wuhan.
Animal Procedures and Models
Generation of cardiac‐specific ADAM22‐transgenic mice
ADAM22‐transgenic C57BL/6J mice were established for cardiac‐specific conditional overexpression of ADAM22 using a standard protocol.18 Briefly, the full length of cDNA for mouse ADAM22 was obtained by polymerase chain reaction (PCR). After being sequenced, the cDNA sequence was inserted into pCAG‐loxP‐CAT‐loxp‐lacZ by replacing the LacZ gene to generate the plasmid of pCAG‐CAT‐mADAM22, which contains the CAG promoter and the LoxP‐flanked CAT gene. After the linearization, the vector DNA was used for pronuclear microinjection of mouse embryos, which were transferred into recipient mothers. The genotypes of offspring mice were characterized by PCR on their tail DNA samples to identify potential transgenic founders. The generated CAG‐CAT‐mADAM22 mice were bred with α‐myosin heavy chain (α‐MHC)–MerCreMer (MCM) C57BL/6 mice to generate CAG‐CAT‐mADAM22/α‐MHC–MCM double‐transgenic mice. The CAG‐CAT‐mADAM22/α‐MHC–MCM mice, at 6 weeks of age, were injected intraperitoneally with 80 mg/kg tamoxifen (T‐5648; Sigma‐Aldrich) or vehicle alone daily for 5 consecutive days to generate ADAM22‐transgenic mice or use as the controls, respectively. Transgenic 1, with the highest expression, was used.
Generation of cardiac‐specific ADAM22 knockout mice
The cardiac‐specific conditional ADAM22 knockout mice were established using the CRISPR/Cas9 system. The exon 3 coding sequence region of mouse ADAM22 gene was cloned and flanked by 2 mouse Lox P (mLoxP) sequences. The recombinant DNA fragment was further cloned into a vector containing 2 homology arms of 957 and 1376 bp, to generate a circular donor vector, which was used as template for repairing the double‐stranded breaks by homologous recombination. Furthermore, 2 single‐guided RNAs targeting 2 locations in the coding sequence region were designed using available online tools (http://crispr.mit.edu/) and synthesized, and their specificity and function were validated in vitro. Subsequently, the Cas9 mRNA, single‐guided RNAs, and donor vector were injected into C57BL/6J mouse zygotes, which were transplanted into surrogate mother mice to generate 2 founder mice with the floxed coding sequence regions on the same allele. To confirm whether the floxed allele functioned as expected, the genomic DNA was isolated from resistant ES cells and tested for in vitro Cre/loxP‐mediated recombination using 2 pairs of primers (F1/R1 and F2/R2) to detect the deletion products and the circle product, respectively. All PCR products were confirmed by sequencing. The founder mice 5 to 9 were mated with C57BL/6J female mice to obtain ADAM22‐floxed mice, which were crossed with α‐MHC–MCM transgenic mice (MEM‐Cre; MEM‐Cre‐Tg [Myh6‐cre/Esr1, 005650]; Jackson Laboratory, Bar Harbor, ME) to produce ADAM22‐floxed/α‐MHC–MCM mice. Furthermore, the ADAM22‐floxed/α‐MHC–MCM mice, at 6 weeks of age, were injected intraperitoneally with tamoxifen (80 mg/kg) daily for 5 consecutive days to generate ADAM22 knockout mice. The α‐MHC–MCM mice and ADAM22‐floxed mice were treated with vehicle alone and served as the controls. The sequences of primers were as follows: F1/R1, 5′‐GATGTGGCAACTTCTTTGAAA‐3′ (forward) and 5′‐ATTGGCTCACTGGCAGACTT‐3′ (reverse); and F2/R2, 5′‐CAAGGTGTCCATGCTGCTAA‐3′ (forward) and 5′‐TCACGAAACACTGCGTATCC‐3′ (reverse). The mice were phenotyped 7 days after the tamoxifen injections.
A mouse model of pressure overload‐induced cardiac hypertrophy was established by transaortic constriction (TAC) via an aortic‐banding surgery. Adult male mice (8–10 weeks old, with body weights [BWs] of 24–27 g) were subjected to TAC or a sham operation 1 week later, after finishing the tamoxifen injection, as described previously.18 We identified the heart function of mice and performed other experiments at 4 weeks after TAC. Some mice from wild‐type or ADAM22 knockout were injected intraperitoneally with vehicle or 50 mg/kg LY294002 (a specific protein kinase B [AKT] inhibitor, L9908; Sigma) daily for 4 weeks.
All animal experiments were conducted according to the guidelines of the US National Institutes of Health and approved by the Animal Research and Care Committee of the Central Hospital of Wuhan.
Physical exercise
The swimming physical exercise was performed to induce cardiac hypertrophy and remodeling, according to the previous protocol.19 The details are as follows: 8‐ to 10‐week‐old mice were forced to swim for 10 minutes twice per day on day 1, and each round increased 10 minutes per day until the amount reached 90 minutes on day 9. All the mice kept on swimming in the next 14 days, with two rounds of 90 minutes. After then, the mice were killed for research purposes.
Echocardiographic measurements
Four weeks after a sham or TAC operation, individual mice were anesthetized using 1.5% to 2% inhaled isoflurane. The internal diameter and wall thickness of the LV of individual mice were measured by echocardiography using a MyLab30CV (Esaote) ultrasound system with a 15‐MHz transducer. The LV end diastolic diameter (LVEDD), LV end systolic diameter, and LV posterior wall thickness during diastole were measured by the M‐mode trace derived from the short axis of the LV at the level of papillary muscles. The percentage of LV fractional shortening was calculated using the following formula: [(LVEDD−LV end systolic diameter)/LVEDD]×100%.
The mice were euthanized, and their heart and tibial tissues were collected to calculate the ratios of heart weight (HW)/BW (mg/g), HW/tibial length (mg/mm), and lung weight/BW.
Histological analyses
The heart tissues were fixed in 10% buffered formalin, dehydrated, paraffin embedded, and cut into sections for histological analysis. The cross sections (5 μm thick) were midventricular and stained with hematoxylin and eosin for evaluation of its architecture. Picrosirius red staining was used for evaluating cardiac collagen deposition/interstitial fibrosis, and wheat germ agglutinin was used for assessment of cell area of ventricles. The images were captured under microscopy and assessed by Image‐Pro Plus, version 6.0 (Media Cybernetics).
Immunofluorescence staining
The cross sections of heart tissues (5 μm thick) underwent immunofluorescence staining to investigate the expression and localization of ADAM22. Rabbit anti‐mouse ADAM22 antibody (catalog No. ARP46413‐p050) and a mouse–cardiac troponin T–specific monoclonal antibody (ab8295; Abcam) were used as primary antibodies, and then the sections were incubated with goat anti‐rabbit IgG (A‐11011) and anti‐mouse IgG (A‐11001) as secondary antibodies, respectively (Thermo Fisher Scientific, Waltham, MA). We also performed immunofluorescence staining of ADAM22 in cardiomyocytes isolated from neonatal rat, using rabbit anti‐mouse ADAM22 antibody (catalog No. ARP46413‐p050) and α‐actinin antibody (catalog No. A7811; Sigma‐Aldrich) as primary antibodies; goat anti‐rabbit IgG (A‐11011) and anti‐mouse IgG (A‐11001) were used as secondary antibodies, respectively. We obtained all the immunofluorescence images via fluorescence microscope (Olympus, Tokyo, Japan) and DP2‐BSW software.
Western blot analysis
Total proteins were extracted from the LV tissues and primarily cultured cardiomyocytes. The concentrations of proteins in individual samples were measured by bicinchoninic acid using a Pierce BCA Protein Assay Kit, according to the manufacturer's instructions (Pierce). Culture supernatant was harvested and centrifuged to detect ADAM22 in the cell culture medium. Immunoprecipitation was performed using Protein A/G Mag Sepharose (GE Healthcare). Rabbit antibodies against ADAM22 (catalog No. ARP46413‐p050) was bound to magnetic beads for 2 hours at 4°C. Then, the antibody‐bound magnetic beads were added to the supernatant and incubated overnight at 4°C. Individual protein samples (30 μg/lane) were subjected to SDS‐PAGE and transferred onto polyvinylidene difluoride membranes (Millipore). After being blocked with 5% nonfat dry milk in Tris‐buffered saline, the membranes were incubated overnight at 4°C with rabbit antibodies against P‐mitogen‐activated protein (MAP)/extracellular signal–regulated kinase (ERK) (1:1000; catalog No. 9154), T‐MAP/ERK (1:1000; catalog No. 9122), P‐ERK1/2 (1:2000; catalog No. 4370), T‐ERK1/2 (1:1000; catalog No. 4695), P‐c‐Jun N‐terminal kinase 1/2 (1:1000; catalog No. 4668), T‐c‐Jun N‐terminal kinase 1/2 (1:1000; catalog No. 9252), P‐P38 (1:1000; catalog No. 4511), T‐P38 (1:1000; catalog No. 9212), P‐AKT (1:2000; catalog No. 4060), T‐AKT (1:1000; catalog No. 4691), P‐glycogen synthetase kinase 3β (1:1000; catalog No. 9322), T‐glycogen synthetase kinase 3β (1:1000; catalog No. 9315), P‐mammalian target of rapamycin (1:1000; catalog No. 2971), T‐mammalian target of rapamycin (1:1000; catalog No. 2983), P‐P70S6K (1:1000; catalog No. 9208), and T‐P70S6K (1:1000; catalog No. 2708; all from Cell Signaling Technology). Rabbit antibodies against ADAM22 (1:200; catalog No. ARP46413‐p050), atrial natriuretic peptide (ANP; 1:200; catalog No. Sc20158), and mouse antibodies against β‐MHC (1:200; catalog No. Sc53090; all from Santa Cruz Biotechnology) were used for Western blot analysis. The bound antibodies were detected with peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch Laboratories; 1:10 000 dilution). The protein signals were visualized and analyzed using the ChemiDocTM XRS+ system (Bio‐Rad, Hercules, CA).
Quantitative real‐time PCR
Total RNA was extracted from the LV samples and cultured cells using the TRIZol reagent (Invitrogen) and reversed transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche), following the manufacturer's protocol. Quantitative real‐time–PCR was performed using the SYBR Green PCR Master Mix (Roche) and specific primers. The sequences of primers were as follows: ANP, 5′‐ACCTGCTAGACCACCTGGAG‐3′ (forward) and 5′‐CCTTGGCTGTTATCTTCGGTACCG G‐3′ (reverse); β‐MHC, 5′‐CCGAGTCCCAGGTCAACAA‐3′ (forward) and 5′‐CTTCACGGGCACCCTTGGA‐3′ (reverse); GAPDH, 5′‐ACTTGAAGGGTGGAGCCAAA‐3′ (forward) and 5′‐GACTGTGGTCATGAGCCCTT‐3′ (reverse); B‐type natriuretic peptide (BNP; mouse), 5′‐GAGGTCACTCCTATCCTCTGG‐3′ (forward) and 5′‐GCCATTTCCTCCGACTTTTCTC‐3′ (reverse); collagen I (mouse), 5′‐AGGCTTCAGTGGTTTGGATG‐3′ (forward) and 5′‐CACCAACAGCACCATCGTTA‐3′ (reverse); collagen III (mouse), 5′‐CCCAACCCAGAGATCCCATT‐3′ (forward) and 5′‐GAAGCACAGGAGCAGGTGTAGA‐3′ (reverse); acetyl‐Coenzyme A acetyltransferase 1 (ACAT1) (mouse), 5′‐GCTGTGTTCCCATCCATCGT‐3′ (forward) and 5′‐GCAGGCACGTTGAAGGTCTC‐3′ (reverse); ADAM22 (mouse), 5′‐CAACTGAGACCCTGTCACCT‐3′ (forward) and 5′‐GAGGCATAGGGTACGGGTATTT‐3′ (reverse); and ADAM22 (human), 5′‐GGTAACCTGGGAGGCAACAA‐3′ (forward) and 5′‐TCCCATAGCCTGGCACTTTG‐3′ (reverse). The relative levels of targeted gene mRNA to control GAPDH transcripts were analyzed by 2−ΔΔCt.
Primary cardiomyocyte culture, recombinant adenoviral infection, and immunofluorescence
Neonatal rat cardiomyocytes (NRCMs) were prepared from the hearts of Sprague‐Dawley rat neonates of <24 hours. Briefly, the whole hearts from newborn rats were excised, cut into small pieces, and digested with trypsin under constant stirring. The primary cells were collected by passing through a 40‐μm cell strainer and removing fibroblasts using different attachment techniques. The suspending NRCMs were cultured in DMEM/F12 medium containing 20% fetal bovine serum, bromodeoxyuridine (to inhibit fibroblast proliferation), and penicillin/streptomycin.
To knock down ADAM22 expression, rat shADAM22 fragments (KR69986G; SABiosciences) were cloned into vector to generate specific adenovirus. Nontargeting AdshRNA was used as the control. To induce ADAM22 overexpression, the entire rat ADAM22 cDNA fragment was cloned into pcDNA3 and subcloned into a replication‐defective adenoviral vector to produce adenovirus AdADAM22. A similar adenoviral vector encoding the green fluorescent protein gene was used as a control.
After being cultured for 48 hours, NRCMs were infected with AdshADAM22, AdshRNA, AdADAM22, or adenoviral vector encoding the green fluorescent protein gene at a multiplicity of infection of 100 for 24 hours. The cells were serum starved and cultured in DMEM/F12 medium containing 1% fetal bovine serum (serum‐free DMEM/F12 medium) for an additional 12 hours. Subsequently, the NRCMs were stimulated with angiotensin II (AngII; 1 μmol/L; Sigma‐Aldrich) or vehicle PBS for 48 hours. The different groups of NRCMs were harvested for quantitative reverse transcription–PCR and Western blot assays. Some cells from each group were fixed with 100% methanol for 20 minutes at room temperature to quench the green fluorescent protein signal, washed three times, permeabilized with 0.1% Triton X‐100, and stained with fluorescein isothiocyanate–conjugated anti–α‐actinin (1:100 dilution; catalog No. A7811; Sigma‐Aldrich), followed by photoimaging under a fluorescent microscope. The cells sizes were measured using ImageJ software.
Statistical Analysis
For data sets with normal distribution, the differences among the groups were analyzed using 1‐way ANOVA, followed by a post hoc Bonferroni or a Tamhane T2 test using SPSS software, version 16.0. The data are shown as mean±SD. For data sets (human samples) with skewed distribution, nonparametric statistical analyses were performed using the Mann‐Whitney test. The results are presented as median and range. P<0.05 was considered statistically significant.
Results
ADAM22 Expression Is Upregulated in Human Failing Hearts and Murine Hypertrophic Hearts and Cardiomyocytes
To explore the potential role of ADAM22 in the process of cardiac hypertrophy, ADAM22 mRNA level and protein level were characterized in 4 LV tissues from patients with DCM and patients with HCM and 4 non‐DCM LV tissues by reverse transcription–PCR and Western blot. The characteristics of human samples are in Table S1. The relative mRNA levels and protein levels of ADAM22 expression in the LV tissues from patients with DCM and HCM were significantly higher than those in the non‐DCM LV tissues (P<0.05; Figure 1A, 1C, and 1D). The same patterns of ANP and β‐myosin heavy chain (β‐MHC) expression were detected in these 2 groups of the heart samples (Figure 1C and 1D). Similarly, the relative mRNA level and expression levels of ADAM22, ANP, and β‐MHC expression in the heart tissues increased in the mice with TAC, compared with those in the sham controls (Figure 1B and 1E). Furthermore, treatment with AngII simulated higher levels of ADAM22, ANP, and β‐MHC expression in NRCMs in a time‐dependent manner (Figure 1F). These independent lines of evidence indicated that ADAM22 expression was upregulated in the heart tissues during the process of cardiac hypertrophy. We also performed immunofluorescence to locate the expression of ADAM22 on cardiomyocytes. The data showed ADAM22 expressed on cardiomyocyte membrane in both tissue sections and isolated cardiomyocytes (Figure 1G, Figure S1). ADAM22 also expresses in fibroblasts and endotheliocytes, but there is no change in expression level with treatment of AngII (data not shown).
Figure 1.
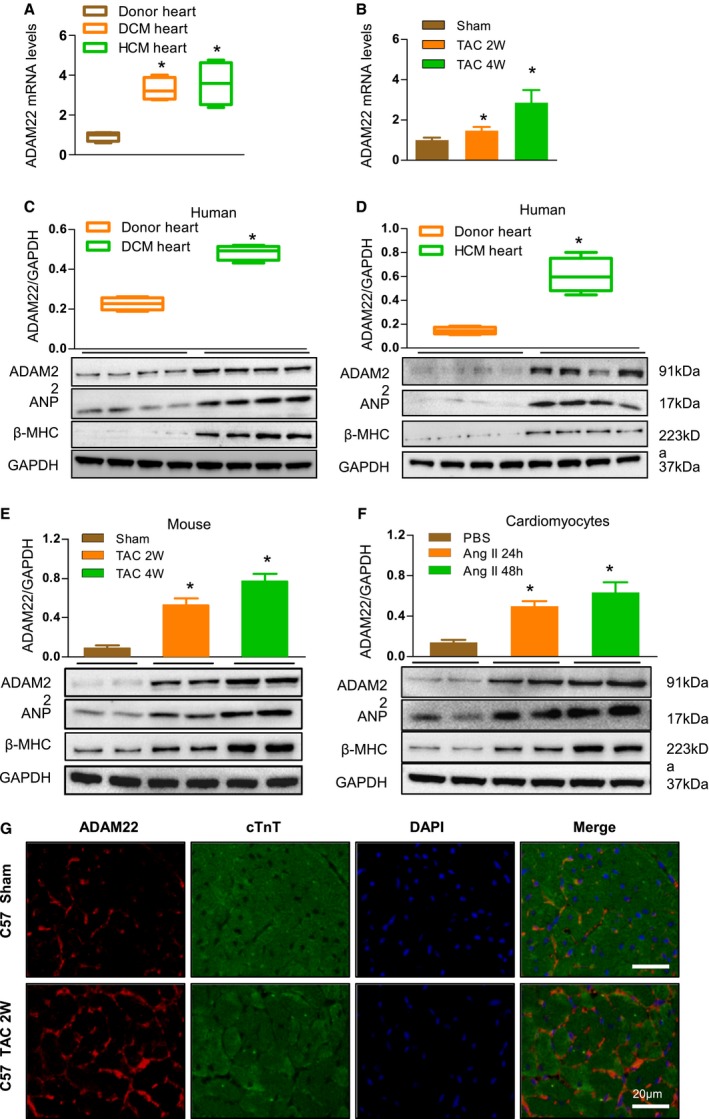
Upregulated a disintegrin and metalloprotease‐22 (ADAM22) expression in human dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) hearts, mouse hypertrophic hearts, and cardiomyocytes. A, Quantitative analysis of ADAM22 mRNA levels in the left ventricles (LVs) from non‐DCM (donor heart), patients with DCM, and patients with HCM (n=4 samples per group). B, Quantification of ADAM22 mRNA levels in the hearts of mice at 2 and 4 weeks after the sham or transverse aortic constriction (TAC) surgery (n=6 mice per group). C, Western blot analysis and quantification of ADAM22, atrial natriuretic peptide (ANP), and β‐myosin heavy chain (β‐MHC) expression in the LVs from non‐DCM (donor heart) and patients with DCM (n=4 samples per group). D, Western blot analysis and quantification of ADAM22, ANP, and β‐MHC expression in the LVs from non‐DCM (donor heart) and patients with HCM (n=4 samples per group). E, Western blot analysis and quantification of ADAM22 mRNA levels in the hearts of mice at 2 and 4 weeks after the sham or TAC surgery (n=6 mice per group). F, Western blot analysis and quantification of ADAM22, ANP, and β‐MHC expression in primarily cultured neonatal rat cardiomyocytes that had been treated with PBS or angiotensin II (AngII) for 24 or 48 hours (n=6 samples per group). G, Immunofluorescence of cross sections (5 μm thick) from TAC or sham operation C57 mice heart tissues (bar=20 μm; n=6 mice in TAC group, and n=5 mice in sham group). Data are representative images or expressed as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. *P<0.05 vs the donor hearts, † P<0.05 vs the sham group, ‡ P<0.05 vs the PBS‐treated group.
Induction of ADAM22 Overexpression Attenuates Pressure Overload–Induced Hypertrophy
To understand the role of ADAM22 in the process of cardiac hypertrophy, we generated ADAM22‐transgenic mice with cardiac‐specific ADAM22 overexpression using the CAG promoter driving ADAM22 expression by a Loxp/Cre system (Figure 2A). To evaluate the influence of ADAM22 on physiological cardiac hypertrophy, ADAM22‐transgenic and control mice were subjected to physical exercise. Whether under sedentary or exercise condition, the cardiac function of ADAM22‐transgenic and nontransgenic mice did not change, which was indicated by the ratio of HW/BW and the cardiac cross‐sectional area (Figure S2). We also identified the effect of tamoxifen on the phenotype of α‐MHC‐MCM before and after TAC. TAC operation did not affect the survival rate of transgenic mice (Figure S3), and tamoxifen injection did not affect the cardiac function of α‐MHC‐MCM (Figure S4). Western blot analysis revealed that ADAM22 expression increased in different strains of transgenic mice (Figure 2B). First, there was no significant difference in the architecture and function of the heart between the ADAM22‐transgenic and nontransgenic mice after the sham surgery (Figure 2). Four weeks after TAC, the hypertrophic degrees of ADAM22‐transgenic mice were significantly lower than those in the nontransgenic controls, evidenced by significantly decreased values of LV posterior wall thickness during diastole and LVEDD; however, the value of fractional shortening increased (P<0.05 for all; Figure 2C, 2D, 2E, Table S2), a hallmark of smaller LV dimensions and elevated systolic function. The ratios of HW/BW, HW/tibial length, and lung weight/BW significantly decreased, compared with those in the nontransgenic controls (Figure 2F through 2H). Histologically, the heart size, cardiomyocyte cross‐sectional areas, and the degrees of interstitial fibrosis in the ADAM22‐transgenic mice were significantly less than those in nontransgenic mice (Figure 2I through 2K). Similarly, the relative levels of ANP, BNP, β‐MHC, ACTA1, and collagen 1A2 and 3A1 mRNA transcripts in the hearts of ADAM22‐transgenic mice were significantly lower than those in the nontransgenic control (Figure 2L). Hence, induction of ADAM22 overexpression mitigated the TAC‐induced cardiac hypertrophy in mice.
Figure 2.
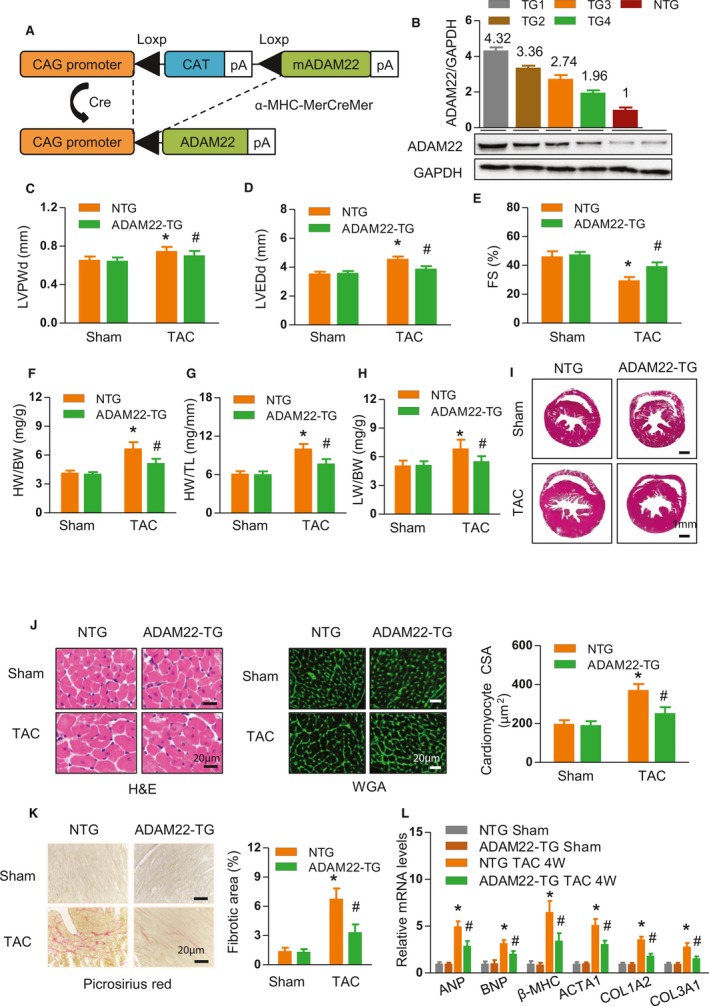
Cardiac a disintegrin and metalloprotease‐22 (ADAM22) overexpression attenuates the overload‐induced hypertrophic response. A, Schematic of the CAG promoter‐driven mouse cDNA of the ADAM22 transgenic construct. B, Immunoblot and quantification of ADAM22 expression in the hearts of 4 different transgenic founders: 1, 2, 3, and 4. C through E, Levels of left ventricular end diastolic posterior wall dimension (LVPWd), left ventricular end‐diastolic dimension (LVEDD), and fractional shortening (FS) in the indicated groups (n=7 mice in nontransgenic group, and n=12 mice in transgenic group) that received a sham or transverse aortic constriction (TAC) surgery. F through H, The heart weight (HW)/body weight (BW), HW/tibial length (TL), and lung weight (LW)/BW ratios (n=7 mice in nontransgenic group, and n=12 mice in transgenic group). I, Histological analyses of whole heart stained with hematoxylin and eosin (H&E) in the indicated groups (bar=1 mm). J, Histological analyses of the cardiomyocyte cross‐sectional areas (CSAs) after H&E and wheat germ agglutinin staining (bar=20 μm; n=5 mice in nontransgenic group, and n=6 mice in transgenic group). K, Characterization of cardiac interstitial fibrosis after picrosirius red staining (bar=20 μm; n=5 mice in nontransgenic group, and n=6 mice in transgenic group). L, Quantification of hypertrophic markers in the indicated groups (n=4 samples in nontransgenic group, and n=5 samples in transgenic group). Data are representative images or expressed as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. ACTA indicates acetyl–Coenzyme A acetyltransferase 1; ANP, atrial natriuretic peptide; BNP, B‐type natriuretic peptide; and MHC, myosin heavy chain. *P<0.05 vs nontransgenic/sham group, #P<0.05 vs ADAM22‐transgenic/sham group.
Cardiac‐Specific Conditional Knockout of ADAM22 Deteriorates the TAC‐Induced Hypertrophy
Next, we generated conditional ADAM22 knockout mice by crossing transgenic α‐MHC–MCM mice with ADAM22‐floxed mice (Figure S5) and demonstrated the conditional knockout founders by Western blot analysis (Figure 3A). Four weeks after the sham surgery, there was no significant difference in the value of any measure among the ADAM22 knockout and control (α‐MHC–MCM and ADAM22‐floxed) groups of mice (Figure 3). In contrast, there was a significant difference in the heart architecture and function between the ADAM22 knockout and control mice at 4 weeks after TAC. The survival rate of ADAM22 knockout mice decreased significantly (Figure S3). In comparison with the controls, ADAM22 knockout mice displayed significantly larger values of LV posterior wall thickness during diastole and LVEDd, but less fractional shortening (Figure 3B through 3D, Table S3), as well as significantly increased ratios of HW/BW, HW/tibial length, and lung weight/BW (Figure 3E through 3G). Histologically, ADAM22 knockout mice exhibited significantly increased levels of cardiac hypertrophy by increased heart size, cardiomyocyte cross‐sectional area, and degrees of interstitial fibrosis, accompanied by increased levels of ANP, BNP, β‐MHC, ACTA1, and collagen 1A2 and 3A1 mRNA transcripts (Figure 3H through 3M). Thus, ADAM22 deficiency enhanced the process of pressure overload‐induced cardiac hypertrophy.
Figure 3.
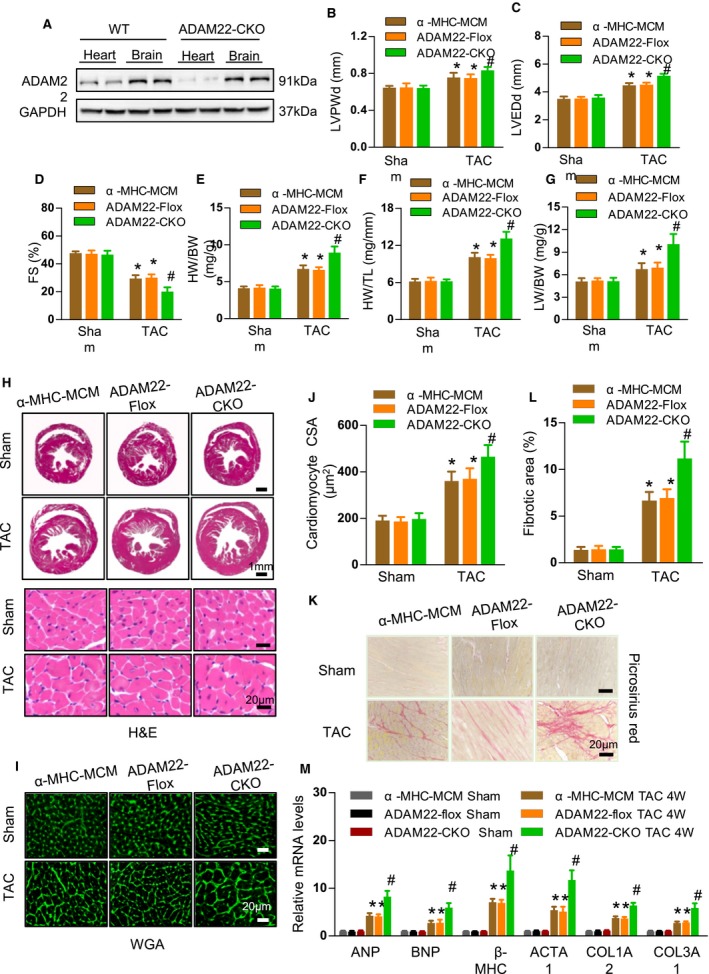
Cardiac a disintegrin and metalloprotease‐22 (ADAM22) knockout aggravates transverse aortic constriction (TAC)–induced hypertrophy. A, Western blot analysis of ADAM22 expression in different tissues of ADAM22 knockout and wild‐type (WT) mice (n=8 mice per group). B through D, The levels of left ventricular end diastolic posterior wall dimension (LVPWd), left ventricular end‐diastolic dimension (LVEDD), and fractional shortening (FS) in the indicated groups (n=8 mice per group; *P<0.05 vs α‐MHC‐MCM or ADAM22‐Flox group; # P<0.05 vs ADAM22 knockout group). E through G, The HW/BW, HW/TL, and LW/BW ratios (n=8 mice per group). H, Histological analyses of whole heart stained with hematoxylin and eosin (H&E) in the indicated groups (bar=1 mm). I, Histological analyses of cross‐sectional areas (CSAs) after wheat germ agglutinin (WGA) staining (bar=20 μm; n=5 mice in α‐myosin heavy chain–MerCreMer [α‐MHC–MCM] and ADAM22‐Flox group, and n=6 mice in ADAM22 knockout group). J through L, Characterization of cardiac interstitial fibrosis after picrosirius red staining (bar=20 μm; n=5 mice in α‐MHC–MCM and ADAM22‐Flox group, and n=6 mice in ADAM22 knockout group). M, Quantification of hypertrophic markers in the indicated groups (n=4 samples in α‐MHC–MCM and ADAM22‐Flox group, and n=5 samples in ADAM22 knockout group). Data are representative images or present as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. BW indicates body weight (g); HW, heart weight (mg); LW, lung weight; and TL, tibial length (mm). P<0.05 vs α‐MHC–MCM or ADAM22‐Flox group, † P<0.05 vs ADAM22 knockout group.
ADAM22 Rescues AngII‐Induced Cardiomyocyte Hypertrophy in Vitro
Because cardiac hypertrophy in mice is a complex process, we used the well‐established NRCM hypertrophy model in vitro to evaluate the specific role of ADAM22 in cardiomyocytes after modulation of ADAM22 expression by adenovirus‐mediated ADAM22 knockdown or overexpression. Infection with AdshADAM22 or AdADAM22 significantly decreased ADAM22 expression by 75% or increased by nearly 3‐fold in NRCMs, compared with values in the controls (Figure 4A). ADAM22 silencing or overexpression did not affect the morphological features of NRCMs in the absence of AngII stimulation. Treatment with AngII significantly increased cell size and the relative levels of ANP, BNP, β‐MHC, and ATCA1 mRNA transcripts, indicating cardiomyocyte hypertrophy in any group of cells (Figure 4B through 4E). In comparison with the control cells, induction of ADAM22 overexpression significantly mitigated the AngII‐enhanced cell size and the ANP, BNP, β‐MHC, and ATCA1 expression in NRCMs (Figure 4B and 4D). In contrast, ADAM22 silencing significantly deteriorated the severity of AngII‐mediated cardiomyocyte hypertrophy by further increasing cell size and the ANP and β‐MHC expression levels in NRCMs (Figure 4C and 4E). Therefore, ADAM22 protected against AngII‐mediated cardiomyocyte hypertrophy in vitro.
Figure 4.
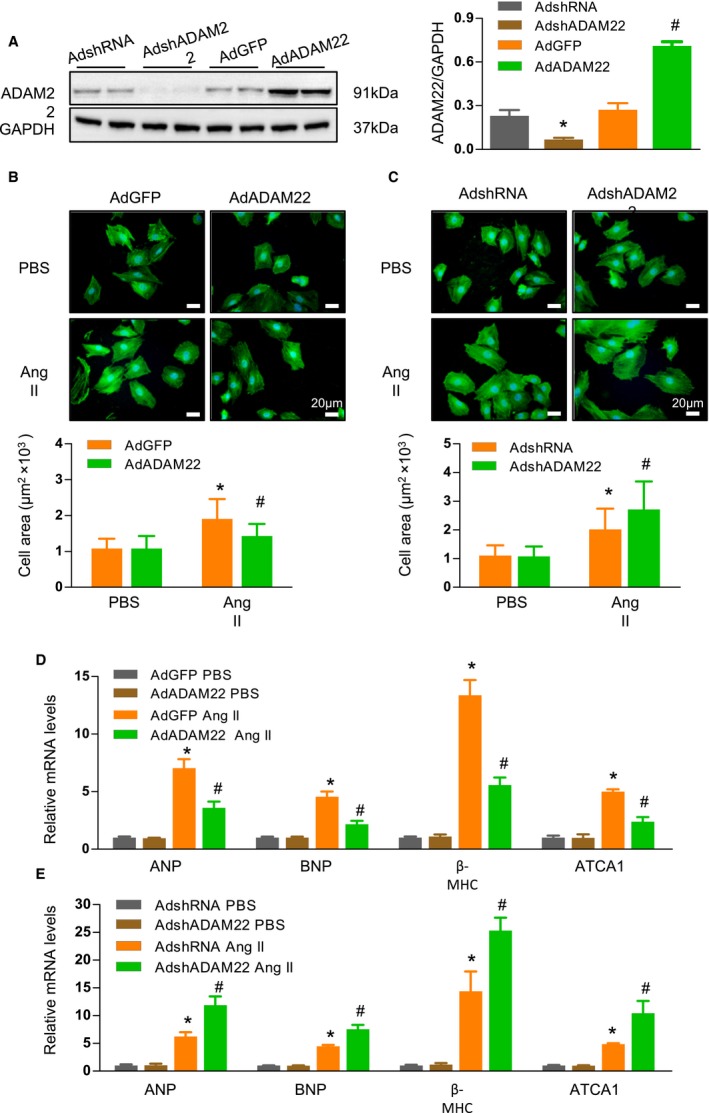
A disintegrin and metalloprotease‐22 (ADAM22) modulates angiotensin II (AngII)–induced cardiomyocyte hypertrophy in vitro. A, Western blot analysis of ADAM22 expression. B and C, Fluorescent analysis of neonatal rat cardiomyocytes (NRCMs) that had been infected with adenoviral vector encoding the green fluorescent protein gene (AdGFP) or overexpression ADAM22 (AdADAM22; B) or with nontargeting control (AdshRNA) or AdADAM22 (C), and treated with AngII (1 μmol/L) or PBS for 48 hours (blue, nuclear; green, α‐actinin; bar=20 μm). The cell surface areas in individual groups of cells were assessed (n>100 cells/group were examined). D and E, The relative levels of hypertrophic marker mRNA transcripts in NRCMs that had been infected with AdGFP or AdADAM22 (D) or with AdshRNA or knockdown ADAM22 expression (AdshADAM22; E) (n=7 samples per group). Data are representative images or present as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. ANP indicates atrial natriuretic peptide; ATCA, acetyl‐Coenzyme A acetyltransferase 1; BNP, B‐type natriuretic peptide; and MHC, myosin heavy chain. *P<0.05 vs AdshRNA or AdGFP/PBS group; † P<0.05 vs AdshRNA or AdGFP/AngII group.
ADAM22 Inhibits the Hypertrophic Stress‐Enhanced AKT Activation
Previous studies have shown that the AKT and MAP kinase (MAPK) pathways are crucial for the process of cardiomyocyte hypertrophy.20, 21 To understand molecular mechanisms underlying the action of ADAM22, we tested the effect of altered ADAM22 expression on the AKT and MAPK signaling. First, there was no significant difference in the relative levels of MAP/ERK, ERK1/2, c‐Jun N‐terminal kinase 1/2, and p38 expression and phosphorylation between the control and ADAM22 knockout or ADAM22‐transgenic mice, regardless of the sham and TAC (Figure 5A). These findings indicated that altered ADAM22 did not affect the MAPK signaling in the hearts of mice. Second, there was no significant difference in the levels of AKT signaling activation between the control and ADAM22 knockout or ADAM22‐transgenic mice after the sham surgery, but significantly increased levels of AKT activation were detected after TAC in all groups of mice (Figure 5B). In comparison with the controls, ADAM22 knockout mice displayed significantly higher levels of AKT, glycogen synthetase kinase 3β, mammalian target of rapamycin, and p70S6K phosphorylation in their heart tissues at 4 weeks after TAC (P<0.05 for all). In contrast, ADAM22‐transgenic mice exhibited significantly lower levels of AKT activation in their heart tissues at 4 weeks after TAC (P<0.05 for all). Similarly, although ADAM22 silencing significantly increased the AngII‐enhanced AKT activation, ADAM22 overexpression significantly decreased the AngII‐promoted AKT activation in NRCMs (P<0.05 for all; Figure 5C). Apparently, ADAM22 negatively regulated the AKT activation to protect against cardiomyocyte hypertrophy.
Figure 5.

A disintegrin and metalloprotease‐22 (ADAM22) inhibits the protein kinase B (AKT) activation on hypertrophic stresses. A, Western blot analysis of the mitogen‐activated protein kinase expression and phosphorylation in the indicated groups (n=6 mice per group). B, Western blot analysis of the AKT, glycogen synthetase kinase (GSK) 3β, mammalian target of rapamycin (mTOR), and p70S6K expression and phosphorylation in indicated groups (n=5 mice per group). C, Western blot analysis of the AKT, mTOR, GSK3β, and p70S6K expression and phosphorylation in the indicated groups (n=5 samples per group). Data are representative images or present as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. ERK indicates extracellular signal–regulated kinase; JNK, c‐Jun N‐terminal kinase; MEK, mitogen‐activated protein/ERK; NS, nonsignificant; and TAC, transverse aortic constriction. *P<0.05 vs ADAM22‐Flox or nontransgenic/sham group, † P<0.05 vs ADAM22‐Flox or nontransgenic/TAC 4‐week group, ‡ P<0.05 vs AdshRNA or AdGFP/PBS group, § P<0.05 vs AdshRNA or AdGFP/AngII group.
Inhibition of the AKT Activity Mitigates Pressure Overload–Induced Cardiac Hypertrophy
Given that ADAM22 knockout enhanced the AKT activation and deteriorated the process of cardiac hypertrophy, we tested whether inhibition of the AKT activity could mitigate the TAC‐induced cardiac hypertrophy in mice. ADAM22 knockout and ADAM22‐floxed mice were subjected to TAC surgery and treated with, or without, LY294002 for 4 weeks. Treatment with LY294002 significantly reduced the AKT activation in both ADAM22 knockout and control ADAM22‐Flox mice, particularly in the ADAM22 knockout mice (P<0.05 for all; Figure 6A). Furthermore, inhibition of the AKT activity significantly decreased the severity of cardiac hypertrophy by decreasing the values of LV posterior wall thickness during diastole, LVEDD, heart size, and cardiomyocyte cross‐sectional area, but increasing fractional shortening, and improving interstitial fibrosis in the hearts of both ADAM22 knockout and control ADAM22‐Flox mice (P<0.05 for all; Figure 6B through 6E). Therefore, inhibition of the AKT activity mitigated pressure overload–induced cardiac hypertrophy in mice.
Figure 6.
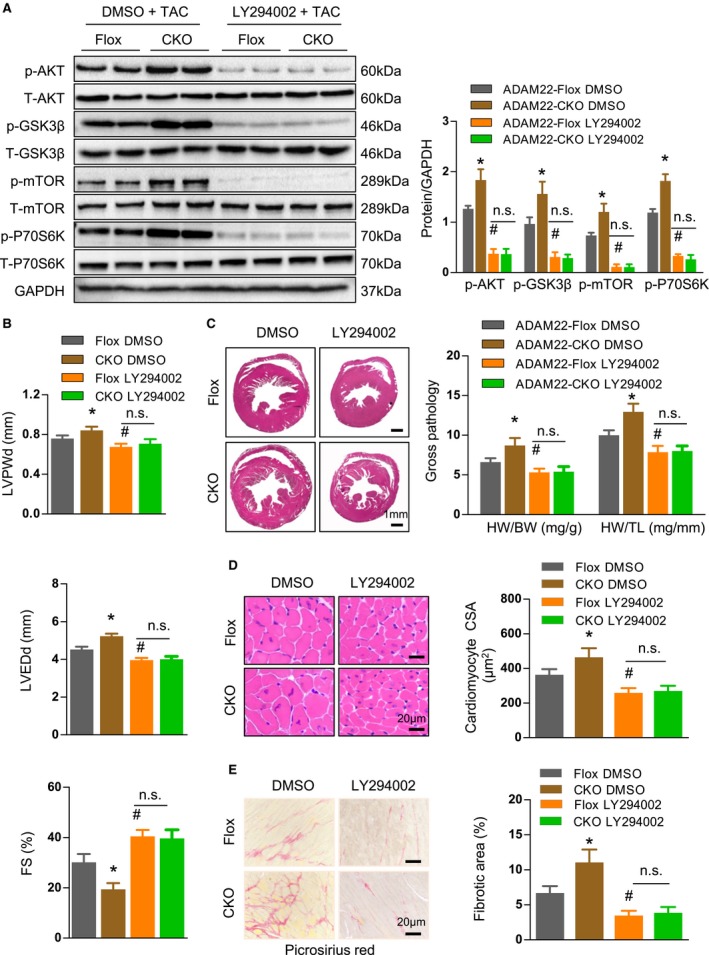
Inhibition of the protein kinase B (AKT) activity attenuates the transverse aortic constriction (TAC)–induced cardiac hypertrophy in vivo. A, Western blot analysis of the AKT, glycogen synthetase kinase (GSK) 3β, mammalian target of rapamycin (mTOR), and p70S6K expression and phosphorylation in the indicated groups after treatment with LY294002 or PBS for 4 weeks (n=6 mice per group). B, The levels of left ventricular end diastolic posterior wall dimension (LVPWd), left ventricular end‐diastolic dimension (LVEDD), and fractional shortening (FS) in indicated groups (n=7 mice in a disintegrin and metalloprotease‐22 [ADAM22]‐Flox group, and n=11 mice in ADAM22 knockout group). C, Histological analysis of whole heart and the heart weight (HW; mg)/body weight (BW; g) and HW/tibial length (TL; mm) ratios (n=11 mice in ADAM22‐Flox group, and n=12 mice in ADAM22 knockout group). D, Staining of the heart cross‐sectional areas (bar=20 μm; n=6 mice per group were measured). E, Characterization of cardiac interstitial fibrosis after picrosirius red staining (bar=20 μm; n=6 mice per group). Data are representative images or present as the mean±SD of each group from at least 3 independent experiments. Statistical analysis was performed by 1‐way analysis of variance and post hoc tests. DMSO indicates dimethyl sulfoxide; and NS, nonsignificant. *P<0.05 vs ADAM22‐Flox /DMSO group, #P <0.05 vs ADAM22–CKO /DMSO group.
Discussion
Cardiac hypertrophy is associated with heart failure, which is a leading cause of cardiovascular disease–related mortality worldwide. The pathogenesis and regulation of cardiac hypertrophy are poorly understood. In this study, we provided novel evidence to demonstrate that ADAM22 acted as a protective regulator of cardiac hypertrophy by attenuating the cardiac hypertrophy–related AKT activation in cardiomyocytes.
ADAM22 is a transmembrane and secreted protein and can interact with many other proteins, including LGI1, Kv 1.2, SRC‐1, PSD‐95, and PSD‐93. The main functions of ADAM22 in previous studies were mainly in the brain and endocrine resistance in breast cancer.22, 23, 24 In this study, we, for the first time, detected significantly upregulated expression of ADAM22 in failing hearts from patients with DCM and HCM. DCM represents the end stage of heart failure, whereas cardiac hypertrophy is the early stage of pathological change in cardiomyocytes. The upregulated ADAM22 expression in the failing hearts from patients with DCM did not accurately reflect its role in regulating the early process of myocardial hypertrophy. Given that the HCM shares similar mechanisms with cardiac hypertrophy, we investigated the relative levels of ADAM22 expression in the myomectomy samples from 4 patients with HCM by reverse transcription–PCR and Western blot assays. Our findings indicated that ADAM played an important role in cardiac hypertrophy. Expression of ADAM22 was also upregulated in mice with TAC‐induced pressure overload hypertrophy and AngII‐stimulated NRCMs (Figure 1). However, cardiac‐specific ADAM22 knockout deteriorated the severity of cardiac hypertrophy, whereas induction of cardiac‐specific ADAM22 overexpression mitigated the TAC‐induced cardiac hypertrophy, in mice. Similarly, altered ADAM22 expression modulated AngII‐induced cardiomyocyte hypertrophy in NRCMs. It is possible that upregulated ADAM22 expression in cardiomyocytes may reflect an inducible regulation to inhibit the process of cardiac hypertrophy. Hence, ADAM22 may act as a protector during the process of cardiac hypertrophy. We are interested in further investigating the relationship between the cardiac hypertrophy and increased ADAM22 expression.
Previous studies have shown that many signaling pathways, such as AKT and MAPK signaling, participate in the pathological features of cardiac hypertrophy.25, 26, 27, 28 Actually, the AKT activation can be induced by the process of cardiac hypertrophy in a time‐dependent manner, and it enhances the severity of cardiac hypertrophy in animals.29, 30, 31 In this study, we found that TAC not only significantly upregulated the MAP/ERK, ERK, c‐Jun N‐terminal kinase, and p38 MAPK signaling, but also enhanced the AKT/mammalian target of rapamycin signaling, in the hearts of mice. However, altered cardiac‐specific ADAM22 expression only modulated the AKT, but not MAPK, signaling in mice. Evidentially, ADAM22 knockout significantly enhanced the TAC‐promoted AKT signaling, but not MAPK activation, in mice. ADAM22 overexpression attenuated the TAC‐enhanced AKT activation in mice, accompanied by modulation of the severity of cardiac hypertrophy. Similar patterns of the effect of altered ADAM22 expression on AngII‐induced cardiomyocyte hypertrophy and AKT activation were observed in NRCMs. More important, inhibition of the AKT activity also attenuated the TAC‐induced cardiac hypertrophy in mice. These data indicated that ADAM22 inhibited cardiac hypertrophy by attenuating the AKT activation in cardiomyocytes, independent of the MAPK signaling.
ADAM22 lacks a zinc‐binding motif in metalloprotease domain32 and functions as an integrin ligand independent of the metalloprotease domain.33 Given that integrins play a critical role in the pathogenic progress of cardiac fibrosis and failure,13, 34, 35 we hypothesize that the involvement of ADAM22 in the pathogenic process of cardiac hypertrophy is mainly through the disintegrin activity. Until now, several integrins, including integrin αvβ3, integrin dimers containing α6 or α9, integrin β1, and integrin β3, were identified to interact with ADAM2236; in addition, integrin‐linked kinase (ILK) is also reported as a target of ADAM22. Overexpression of ILK could rescue growth inhibition mediated by ADAM22, suggesting a negative mediation relationship existed between ILK and ADAM22.36 ILK is a serine/threonine kinase and actively phosphorylates AKT in a phosphatidylinositol‐3 kinase–dependent manner.37 Previous studies revealed that ILK could induce hypertrophy in transgenic mice and stimulate phosphorylation of p70S6K by dependent activity.38, 39 Combined with our data, we speculated the potentially critical role of ADAM22 in inhibiting pressure overload–induced myocardial hypertrophy might relate to ILK that inactivates the AKT signaling.
This study still has some limitations. First, our findings were produced by one animal model, which has its own limitation. Second, there is evidence that ILK interacts with ADAM22 and affects the AKT signaling during the process of cardiac hypertrophy, which needs an in‐depth study in our future research.
In summary, the findings from this study, to the best of our knowledge, provide novel evidence to demonstrate that ADAM22 negatively regulates both TAC‐ and agonist‐induced cardiac hypertrophy in mice and NRCMs by attenuating the AKT activation. These findings may provide new insights into molecular regulation on the process of cardiac hypertrophy.
Author Contributions
Ren, Yang, S. Chen, and C. Wu contributed equally to this work. Ren, M. Chen, and Xia participated in the research design. Yang, S. Chen, C. Wu, Zhang, Huang, Wang, Deng, and Ding participated in the performance of the research. S. Chen, M. Chen, and Xia participated in the writing of the article. Ye, J. Wu, M. Chen, and Xia participated in the data analysis.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (grant 81470482) and, in part, by the Nature Science Foundation of Hubei Province (2013CFA009, 2014BHE0022, 2014CFB154, 2017CFB357).
Disclosures
None.
Supporting information
Table S1. Characteristics of Donor, DCM and HCM Subject
Table S2. Echo Data of ADAM22‐TG and ADAM22‐NTG Mice With TAC or Sham Operation
Table S3. Echo Data of ADAM22‐CKO and ADAM22‐Flox Mice With TAC or Sham Operation
Figure S1. The expression of ADAM22 in cardiomyocytes.
Figure S2. The influence of ADAM22 on physiological cardiac hypertrophy.
Figure S3. Survival data in mice upon TAC.
Figure S4. The effect of tamoxifen injection on cardiac function of αMHCMerCreMer.
Figure S5. Generation of cardiac‐specific conditional ADAM22 knockout mice and identification.
Acknowledgments
We thank Dr Hongliang Li (Wuhan University, Wuhan, China) for generously providing the ADAM22 conditional knockout and transgenic mice and for other experimental and technological assistance.
(J Am Heart Assoc. 2018;7:e005696 DOI: 10.1161/JAHA.117.005696.)29358191
Contributor Information
Manhua Chen, Email: cmh_centre@163.com.
Jiahong Xia, Email: jiahong.xia@hust.edu.cn.
References
- 1. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. [DOI] [PubMed] [Google Scholar]
- 2. O'Mahony C, Tome‐Esteban M, Lambiase PD, Pantazis A, Dickie S, McKenna WJ, Elliott PM. A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/American Heart Association risk stratification and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart. 2013;99:534–541. [DOI] [PubMed] [Google Scholar]
- 3. Moore‐Morris T, Cattaneo P, Puceat M, Evans SM. Origins of cardiac fibroblasts. J Mol Cell Cardiol. 2016;91:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage‐gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akazawa H. Mechanisms of cardiovascular homeostasis and pathophysiology: from gene expression, signal transduction to cellular communication. Circ J. 2015;79:2529–2536. [DOI] [PubMed] [Google Scholar]
- 6. Wang D, Oparil S, Feng JA, Li P, Perry G, Chen LB, Dai M, John SW, Chen YF. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide‐null mouse. Hypertension. 2003;42:88–95. [DOI] [PubMed] [Google Scholar]
- 7. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito K, Akazawa H, Tamagawa M, Furukawa K, Ogawa W, Yasuda N, Kudo Y, Liao CH, Yamamoto R, Sato T, Molkentin JD, Kasuga M, Noda T, Nakaya H, Komuro I. PDK1 coordinates survival pathways and beta‐adrenergic response in the heart. Proc Natl Acad Sci U S A. 2009;106:8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. [DOI] [PubMed] [Google Scholar]
- 10. Akazawa H, Yasuda N, Komuro I. Mechanisms and functions of agonist‐independent activation in the angiotensin II type 1 receptor. Mol Cell Endocrinol. 2009;302:140–147. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Guo L, Shen D, Xu X, Wang J, Han S, He W. The role of c‐SKI in regulation of TGFbeta‐induced human cardiac fibroblast proliferation and ECM protein expression. J Cell Biochem. 2017;118:1911–1920. [DOI] [PubMed] [Google Scholar]
- 12. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. [DOI] [PubMed] [Google Scholar]
- 13. Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug‐of‐war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. [DOI] [PubMed] [Google Scholar]
- 14. Cai Z, Zhang A, Choksi S, Li W, Li T, Zhang XM, Liu ZG. Activation of cell‐surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26:886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. [DOI] [PubMed] [Google Scholar]
- 16. Blobel CP, White JM. Structure, function and evolutionary relationship of proteins containing a disintegrin domain. Curr Opin Cell Biol. 1992;4:760–765. [DOI] [PubMed] [Google Scholar]
- 17. Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1‐dependent signalling. Nat Commun. 2016;7:11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, Zhao Y, Gao L, Yang Q, Zhu XH, Wei X, Pu J, Li H. Suppressor of IKKε is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac‐specific deletion of gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. [DOI] [PubMed] [Google Scholar]
- 20. Kramann N, Hasenfuss G, Seidler T. B‐RAF and its novel negative regulator reticulocalbin 1 (RCN1) modulates cardiomyocyte hypertrophy. Cardiovasc Res. 2014;102:88–96. [DOI] [PubMed] [Google Scholar]
- 21. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lovero KL, Fukata Y, Granger AJ, Fukata M, Nicoll RA. The LGI1‐ADAM22 protein complex directs synapse maturation through regulation of PSD‐95 function. Proc Natl Acad Sci U S A. 2015;112:4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muona M, Fukata Y, Anttonen AK, Laari A, Palotie A, Pihko H, Lonnqvist T, Valanne L, Somer M, Fukata M, Lehesjoki AE. Dysfunctional ADAM22 implicated in progressive encephalopathy with cortical atrophy and epilepsy. Neurol Genet. 2016;2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yokoi N, Fukata Y, Kase D, Miyazaki T, Jaegle M, Ohkawa T, Takahashi N, Iwanari H, Mochizuki Y, Hamakubo T, Imoto K, Meijer D, Watanabe M, Fukata M. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med. 2015;21:19–26. [DOI] [PubMed] [Google Scholar]
- 25. Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. [DOI] [PubMed] [Google Scholar]
- 26. Streicher JM, Ren S, Herschman H, Wang Y. MAPK‐activated protein kinase‐2 in cardiac hypertrophy and cyclooxygenase‐2 regulation in heart. Circ Res. 2010;106:1434–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D, Abe J, Neyses L, Weston AH, Cartwright EJ, Wang X. Targeted deletion of the extracellular signal‐regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload‐induced apoptosis in the heart. Circ Res. 2010;106:961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yao H, Han X, Han X. The cardioprotection of the insulin‐mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drugs. 2014;14:433–442. [DOI] [PubMed] [Google Scholar]
- 29. Horikawa YT, Panneerselvam M, Kawaraguchi Y, Tsutsumi YM, Ali SS, Balijepalli RC, Murray F, Head BP, Niesman IR, Rieg T, Vallon V, Insel PA, Patel HH, Roth DM. Cardiac‐specific overexpression of caveolin‐3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling. J Am Coll Cardiol. 2011;57:2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MV, Napoli C, Sadoshima J, Croce CM, Ross J Jr. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sagane K, Yamazaki K, Mizui Y, Tanaka I. Cloning and chromosomal mapping of mouse ADAM11, ADAM22 and ADAM23. Gene. 1999;236:79–86. [DOI] [PubMed] [Google Scholar]
- 33. Zhu P, Sang Y, Xu H, Zhao J, Xu R, Sun Y, Xu T, Wang X, Chen L, Feng H, Li C, Zhao S. ADAM22 plays an important role in cell adhesion and spreading with the assistance of 14‐3‐3. Biochem Biophys Res Commun. 2005;331:938–946. [DOI] [PubMed] [Google Scholar]
- 34. Chen C, Li R, Ross RS, Manso AM. Integrins and integrin‐related proteins in cardiac fibrosis. J Mol Cell Cardiol. 2016;93:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte‐specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. [DOI] [PubMed] [Google Scholar]
- 36. D'Abaco GM, Ng K, Paradiso L, Godde NJ, Kaye A, Novak U. ADAM22, expressed in normal brain but not in high‐grade gliomas, inhibits cellular proliferation via the disintegrin domain. Neurosurgery. 2006;58:179–186. [DOI] [PubMed] [Google Scholar]
- 37. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide‐3‐OH kinase‐dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin‐linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu H, Fedak PW, Dai X, Du C, Zhou YQ, Henkelman M, Mongroo PS, Lau A, Yamabi H, Hinek A, Husain M, Hannigan G, Coles JG. Integrin‐linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114:2271–2279. [DOI] [PubMed] [Google Scholar]
- 39. Hannigan GE, Coles JG, Dedhar S. Integrin‐linked kinase at the heart of cardiac contractility, repair, and disease. Circ Res. 2007;100:1408–1414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of Donor, DCM and HCM Subject
Table S2. Echo Data of ADAM22‐TG and ADAM22‐NTG Mice With TAC or Sham Operation
Table S3. Echo Data of ADAM22‐CKO and ADAM22‐Flox Mice With TAC or Sham Operation
Figure S1. The expression of ADAM22 in cardiomyocytes.
Figure S2. The influence of ADAM22 on physiological cardiac hypertrophy.
Figure S3. Survival data in mice upon TAC.
Figure S4. The effect of tamoxifen injection on cardiac function of αMHCMerCreMer.
Figure S5. Generation of cardiac‐specific conditional ADAM22 knockout mice and identification.