

Abstract
There is the tendency to assume that endangered species have been both genetically and demographically healthier in the past, so that any genetic erosion observed today was caused by their recent decline. The Iberian lynx (Lynx pardinus) suffered a dramatic and continuous decline during the 20th century, and now shows extremely low genome- and species-wide genetic diversity among other signs of genomic erosion. We analyze ancient (N = 10), historical (N = 245), and contemporary (N = 172) samples with microsatellite and mitogenome data to reconstruct the species' demography and investigate patterns of genetic variation across space and time. Iberian lynx populations transitioned from low but significantly higher genetic diversity than today and shallow geographical differentiation millennia ago, through a structured metapopulation with varying levels of diversity during the last centuries, to two extremely genetically depauperate and differentiated remnant populations by 2002. The historical subpopulations show varying extents of genetic drift in relation to their recent size and time in isolation, but these do not predict whether the populations persisted or went finally extinct. In conclusion, current genetic patterns were mainly shaped by genetic drift, supporting the current admixture of the two genetic pools and calling for a comprehensive genetic management of the ongoing conservation program. This study illustrates how a retrospective analysis of demographic and genetic patterns of endangered species can shed light onto their evolutionary history and this, in turn, can inform conservation actions.
Keywords: Iberian lynx, ancient DNA, paleogenetics, genetic erosion, endangered species
Introduction
Species are becoming extinct at rates unprecedented in recent history as a consequence of human activity (Pimm et al. 2014; Ceballos et al. 2015). Populations of vertebrate species have decreased in size by an average of 52% in the last 40 years (McLellan 2014), and 15–40% of living species may have gone extinct by 2050 (Thomas and Williamson 2012). Habitat modification, fragmentation and destruction, pollution, climate change, overexploitation, and the spread of invasive species have been identified as the main drivers of the current biodiversity crisis. However, a hidden secondary threat in the form of genetic erosion often builds up as populations become small and isolated. Population genetics theory predicts that small and isolated populations progressively lose genetic diversity and accumulate genetic load as a consequence of genetic drift, and this may be exposed through inbreeding, resulting in inbreeding depression (Hedrick 2001). This makes them less able to adapt to environmental change and diminish their reproduction and survival. At the same time, population declines are often accompanied by fragmentation resulting in progressively reduced gene flow. Then, the above-mentioned processes operate independently in the resulting patches leading to increased genetic differentiation, unrelated to local adaptation (Frankham et al. 2002; Allendorf and Luikart 2007).
These processes have typically been assessed by comparing current genetic patterns among populations or species with different recent demographic history. However, current patterns of low genetic diversity and high differentiation can be the consequence of an alternative demographic and evolutionary history which entails contrasting risks of extinction: an equilibrium scenario derived from a long history of small population and restricted gene flow. This latter scenario entails a lower risk of inbreeding depression, since selection may have purged most deleterious variation. Besides, a genetic differentiation due to a stable migration-genetic drift equilibrium could be associated to local adaptation. The assessment of the relative weight of these two evolutionary histories is therefore of critical importance to evaluate the relative risks (inbreeding vs. outbreeding depression) and benefits of alternative management strategies (i.e., to mix or not to mix).
Fortunately, recent developments in the field of ancient DNA have allowed the characterization of genetic variation in past periods spanning decades—from museum specimens—to millennia—from archaeological and palaeontological samples. Historical and ancient genetic data can provide inferences of demographic history from the deep past throughout recent declines, and inform species conservation by providing landmarks of genetic diversity and differentiation statistics (Leonard 2008; Hofman et al. 2015). Furthermore, given a proper sampling of time periods and genetic markers, diachronic genetic studies can illuminate the dynamics of these processes, their dependence on demographic and geographical factors and their contribution to extinction.
The Iberian lynx (Lynx pardinus), because of its currently low genetic diversity and high differentiation, coupled with a fairly well-documented recent history of demographic decline and fragmentation, makes an outstanding model to characterize the genetic processes acting upon a species on its way to extinction. The species suffered a dramatic and continuous decline during the second half of the 20th century (Rodríguez and Delibes 2002; Ferreras et al. 2010; Palomares et al. 2011). With only two extant isolated populations—one located in the Doñana coastal plains and the other in the Eastern Sierra Morena mountains, ca. 240 km apart—and scarcely 100 individuals by 2002, the species was classified as critically endangered by IUCN (2002, 2006 and 2008 red lists; Rodríguez and Calzada 2015), and is generally recognized as the most endangered felid in the world (Nowell et al. 1996). Conservation actions implemented since then, including habitat protection, prey management, captive breeding, translocations and reintroductions have increased lynx numbers to more than 400 by 2015 (http://www.iberlince.eu/images/docs/3_InformesLIFE/Informe_Censo_2015.pdf; last accessed August 25, 2017), leading to its reclassification as “Endangered” in 2015 (Rodríguez and Calzada 2015).
Current Iberian lynx genetic diversity is low, especially in Doñana, and the two populations are highly differentiated (Johnson et al. 2004; Casas-Marce et al. 2013). Moreover, the Iberian lynx features one of the lowest genome- and species-wide diversity reported to date, as well as other signatures of genomic erosion, including high rates of potentially deleterious segregating and fixed mutations (Abascal et al. 2016). These patterns have been interpreted as the direct consequence of the species’ recent decline (Johnson et al. 2004; Casas-Marce et al. 2013; Abascal et al. 2016). However, the complete lack of diversity in a short region of the mitochondrial genome in both historical and ancient samples has suggested that low-genetic diversity is an intrinsic feature of the species (Rodríguez et al. 2011). It remains thus unclear how much of the genetic impoverishment currently observed has been caused by recent versus long-term demography.
Here, we use the Iberian lynx as a model to characterize the micro-evolutionary processes operating on a species on the route to extinction. We compare current, historical, and ancient patterns of diversity and differentiation using nuclear microsatellite markers and mitogenomic sequences. We reconstruct past demography, quantify the relative contribution of the recent decline to current genomic erosion, assess the degree of accumulated genetic erosion in different populations and evaluate its relationship to past demography and to the final persistence or extinction of populations. Finally, we discuss the implications for the ongoing management and conservation programs.
Results
We extracted 230 modern tissue samples, and 296 historical samples from museum and private collection specimens (Casas-Marce et al. 2012) (fig. 1). We obtained good quality genotypes at 20 microsatellite markers for all the fresh samples and for 155 out of the 296 historical samples (52.4%). Genotyping success of historical samples varied among types of tissue in the same fashion as previously reported (Casas-Marce et al. 2010). Average allelic dropout rates were 1.7% (range 0–7.9%) and 2.0% (range 0–11.8%) across loci and samples, respectively, when calculated by comparing the replicates with the consensus genotypes, and 5% (range 0–25%) and 3.1% (range 0–30%) when comparing the 18 consensus genotypes obtained from historical samples with their respective genotypes from fresh samples. False alleles and Type 1 errors were 0.5% among loci and samples; other kinds of errors were not observed.
Fig. 1.
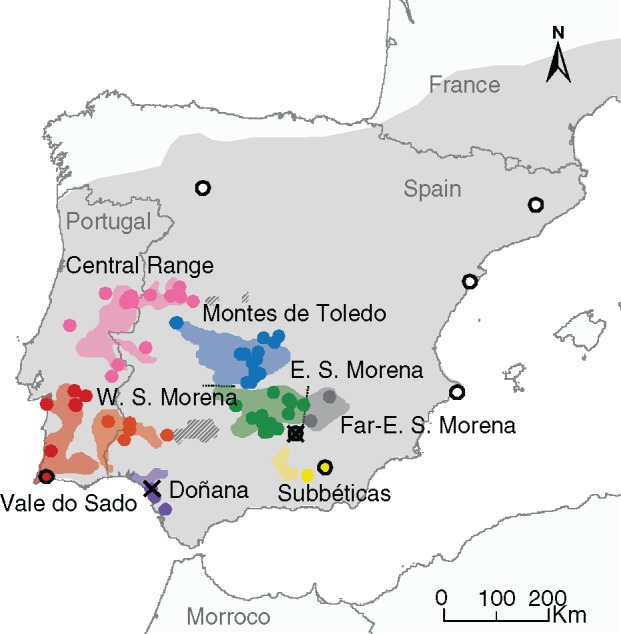
Distribution of sampling across ancient and historical Iberian lynx ranges. Ancient range in light grey taken from Rodríguez and Delibes (2002). In Colour historical distribution according to country-wide surveys in the 1980s in Spain and 1989–1994 in Portugal, with populations delimited as in Rodríguez and Delibes (1992), except that we subdivided the largest Eastern Sierra Morena-Montes de Toledo population as suggested by genetic structure analyses. Points represent sampled localities, with outlined points corresponding to ancient samples and crosses respresenting contemporary samples; note that each point may represent several samples. Unsampled populations are shown in striped fill.
We also reconstructed whole mitochondrial genomes (16,449 bp) for a total of 158 Iberian lynxes: 10 ancient (dated 2.5–>43.5 ka), 83 historical (dating to 1700–1990), and 65 contemporary (1991–2010) samples, with each period containing samples distributed across the corresponding species range (supplementary tables S1–S4, Supplementary Material online). Overall, we observed 23 different haplotypes (fig. 2; supplementary fig. S9 and table S5, Supplementary Material online) defined by 40 variable sites, of which 39 are transitions and 1 is a transversion. Thirty-five variable sites occur in coding regions, of which eight are nonsynonymous variants (supplementary table S6, Supplementary Material online).
Fig. 2.
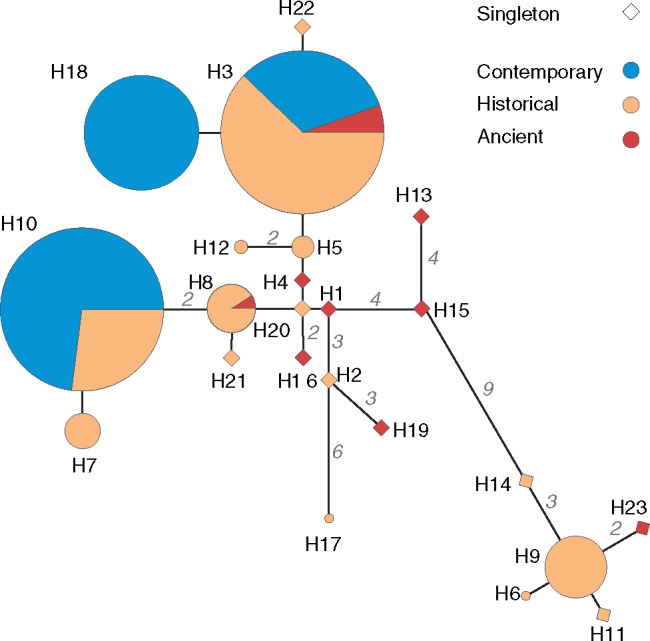
Median-joining networks of mitogenomic haplotypes. Observed haplotypes are represented by circles whose sizes are proportional to the number of observations in each period. Haplotypes are connected by lines of length proportional to the number of mutations separating them (also indicated by small numbers). Haplotypes observed only once are depicted as diamonds to improve visibility.
Demographic History
We used three different approaches to infer the demographic history of the species as a whole, and of each historical and contemporary population in different time periods. We first used a Bayesian Skyline Plot (BSP) (Drummond et al. 2012) based on whole mitochondrial genome data to explore long-term female effective population size. Then, an Approximate Bayesian computation (ABC) approach (Cornuet et al. 2014) was used with microsatellite data to gain insight into the more recent history spanning the last few centuries. Finally, we estimated census sizes from 1950 to 2015 and time of isolation for each historical population based on the available distribution and density data (Rodríguez and Delibes 2002).
BSP shows a nearly stable population of around 4,000 females throughout most of the covered lynx history, followed by a decline that started around 5,000 years ago and accelerated suddenly 400–450 years ago, when the population dramatically dropped from 2,000 to 20 females (supplementary fig. S1, Supplementary Material online). Next, we used ABC to adjust a model in which remnant populations diverged from an ancestral panmictic population formed by all other historical samples (supplementary fig. S2, Supplementary Material online). The divergence between Doñana and the rest of the historical population was estimated at the beginning of the 19th century (39.3 [31.3, 60.3] generations ago, which corresponds to an estimated decade of 1800s [1700s, 1840s]; mode [95% CI]), and its effective population size as 20 [14, 39] individuals (mode [95% CI]; considering a constant size over time; supplementary fig. S3B, Supplementary Material online). In the case of extant Eastern Sierra Morena, the estimated date is around 1950 [1880s, 1950s]) (11.1 [9.31, 23.3] generations ago), with a size of 29 [19, 56] individuals (supplementary fig. S3B, Supplementary Material online). These estimated isolation dates, especially for Doñana population, are much older than what had been inferred by range reconstructions. Note that if we had used a model where divergence of populations was accompanied by gene flow or had allowed for larger population size in the past, the estimated divergence times would be even older, although the known demography and the results from other analyses (e.g., genetic structure) indicate that the current estimates are reasonable. On the other hand, the scarcity of samples from some areas and periods of historical populations impeded the consideration of further substructure for the ABC analysis. Although this could have led to an overestimation of the divergence times, the estimated dates are not far from those when Doñana and contemporary Eastern Sierra Morena start forming clusters separated from the historical population in STRUCTURE analyses (around 1900 or earlier for Doñana; between 1970 and 1990 for Eastern Sierra Morena; see Genetic structure section). Because the genetic differentiation must have postdated isolation by a few generations, the ABC estimations of population divergence dates do not seem highly overestimated if at all.
Finally, census sizes estimated for the historical populations over time based on records from 1950 to 2015 revealed that populations varied widely in size and trend. Two populations (Montes de Toledo or Eastern Sierra Morena) were relatively large by 1950 (above 750 individuals) but declined quite abruptly, while others (Far-E. Sierra Morena and Doñana) remained rather small (around 100 individuals) during the entire period (supplementary fig. S4, Supplementary Material online). All populations except Eastern Sierra Morena and Doñana were extinct by 2000, although the lack of data for the period 1985–2000 impeded estimation of the date of extirpation for most populations. The remnant Eastern Sierra Morena and Doñana reached 60 and 42 individuals, respectively, in 2000. Both populations increased their census sizes later following the adoption of conservation measures. In addition, the two larger populations remained connected until the mid 1980s, whereas most of the others became isolated at least 40 years earlier (figs. 3 and 4; supplementary fig. S4, Supplementary Material online). These two larger and connected populations (Montes de Toledo and Eastern Sierra Morena) are thus predicted to be the ones least affected by recent genetic drift and the best representation of the genetic variation of the species previous to the 20th century decline. Therefore, they are hereon considered central to the rest of the metapopulation.
Fig. 3.
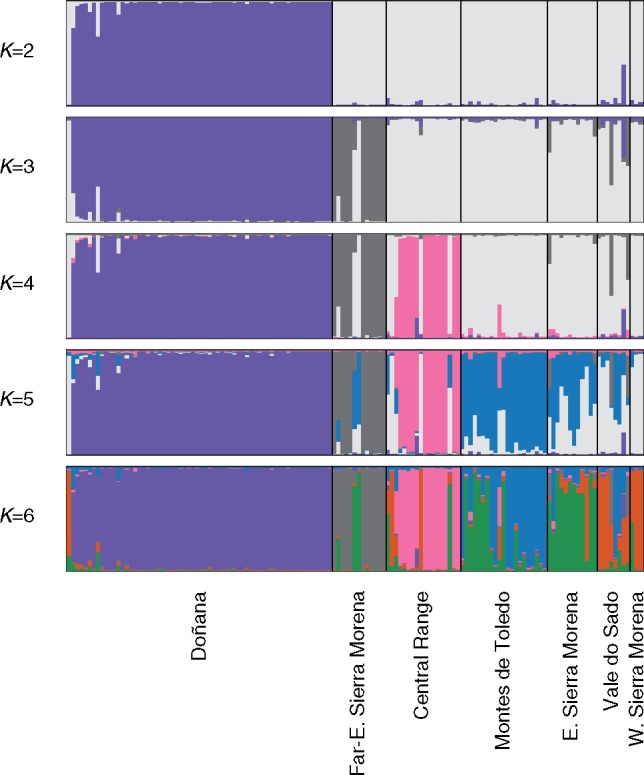
Results of STRUCTURE analyses of historical microsatellite variation. Samples were first subdivided into two clusters, separating almost all Doñana samples from the rest (K = 2). A few older samples were partially assigned to the second historical cluster, with the oldest sample (dated in 1856) completely assigned to it. As K increases, other lynx populations are assigned to the new clusters, becoming differentiated from the rest: Eastern Sierra Morena (K = 3), Central Range (K = 4), Montes de Toledo-Eastern Sierra Morena (K = 5), and Western Sierra Morena-Vale do Sado (K = 6). Older samples from Eastern Sierra Morena and Montes de Toledo—two populations that have remained large and interconnected until the second half of the 20th century—are assigned to the same cluster (green), indicating that they were part of a single panmictic population that only recently became genetically differentiated. This genetic pool is probably the closest representation of the ancestral genetic variation of the species. See also supplementary figure S7, Supplementary Material online.
Fig. 4.

Dynamics of population isolation and contraction, and genetic variation from ancient to contemporary times. The Iberian lynx population is represented by a cylinder projected on the distribution map, that becomes progressively fragmented into subpopulations which contract, become genetically differentiated and eventually go extinct. Maps represent the distribution of microsatellite (left) and mitogenomic variation (right) among ancient (top), historical (middle), and contemporary populations (bottom). Microsatellite pies represent the average coefficient of assignment to each of the clusters identified by STRUCTURE from historical (K = 6) and contemporary (K = 2) data sets (fig. 3 and supplementary fig. S7, Supplementary Material online). Mitogenome pies represent the distribution of mitochondrial genome haplotypes. Haplotypes observed only once are represented in shades of gray. Numbers within pies refer to sample size. See figure 2 and supplementary figure S9, Supplementary Material online, for networks depicting the relationship among haplotypes.
Genetic Structure
We first analyzed different temporal and geographical partitions of microsatellite data with Factorial Correspondence Analyses (FCA) and with clustering algorithms implemented in STRUCTURE (Pritchard et al. 2000; Falush et al. 2007). The 3-D FCA plot shows a central cloud formed by the oldest samples from which more modern samples of the populations of extant Doñana, Eastern Sierra Morena and extinct Central Range and Far-E. Sierra Morena progressively separate through independent routes (supplementary fig. S5, Supplementary Material online).
In the STRUCTURE analysis of all samples together (contemporary and historical), the modern samples of remnant populations are neatly separated from each other and from now extinct historical populations at K = 3, but older samples tend to cluster with the latter (supplementary fig. S6, Supplementary Material online). All individuals from Doñana are consistently grouped together in a separate Doñana cluster, but older samples are partially (Q = 0.14–0.27) and the oldest (1856) is completely assigned to the historical cluster (Q > 0.95). Similarly, most modern individuals (1970–2010) from Eastern Sierra Morena form a separate cluster, whereas older samples (1942–1973) show shared ancestry with the rest of historical samples from now extinct populations (supplementary fig. S6, Supplementary Material online). These results suggest that remnant populations became genetically differentiated from other historical populations between 1850 and 1900 in Doñana and between 1970 and 1990 in Eastern Sierra Morena, which is compatible with their respective dates of isolation estimated with ABC (supplementary figs. S2 and S3, Supplementary Material online).
The analyses of only historical samples (before 1990) of all populations confirmed the early and deep differentiation of Doñana (K = 2), and also revealed further population structure in the historical metapopulation (fig. 3; supplementary fig. S7E–I, Supplementary Material online). As K increases, new genetic clusters are identified that correspond to geographical populations defined a priori, in a sequence compatible with their estimated population size and dates of isolation (supplementary fig. S4, Supplementary Material online): Far-Eastern Sierra Morena (K = 3; unsuspected isolation), Central Range (K = 4; <1940), Montes de Toledo-Eastern Sierra Morena (K = 5; 1985), and Vale do Sado-Western Sierra Morena (K = 6; unknown date of isolation) (fig. 3). Genotypes tend thus to cluster by geographical location—even though some populations cover a wide temporal range (e.g., Doñana and Central Range; fig. 3; supplementary tables S1 and S2, Supplementary Material online). However, a temporal pattern is also evident in the data, as the oldest samples from each population tend to cluster together with other populations (fig. 3; supplementary fig. S7E–I, Supplementary Material online). For example, the oldest samples from Montes de Toledo and Eastern Sierra Morena are assigned to the same cluster. This indicates that the two adjacent populations were in fact one panmictic population that progressively became differentiated in recent times, in agreement with our estimates of times of isolation (1985; supplementary fig. S4, Supplementary Material online).
The genetic differentiation between pairs of populations is statistically significant and levels are in accordance with the expectations based on their geographical or temporal distance (supplementary fig. S8A and B, Supplementary Material online). The only nonsignificant pairwise comparison is Western Sierra Morena and the geographically contiguous Vale do Sado, represented by only three samples. The overall level of genetic differentiation is maximal between the two current populations (FST = 0.419) and lower among historical populations (FST = 0.266). Lowest differentiation is observed in the pairwise comparisons involving any of the two larger and more connected populations (Montes de Toledo or Eastern Sierra Morena), reaching a minimum for the comparison between them (FST = 0.025). As expected, comparisons among peripheral populations (Vale do Sado, Central Range and Far-Eastern Sierra Morena) show higher FST values, with the maximum recorded for Doñana versus Central Range (FST = 0.405). A similar pattern is captured with population-specific F values estimated by 2mod, which may better reflect the differential action of genetic drift accumulated in each population. Temporal FST is relatively lower but also significant for comparisons of current versus historical samples of Doñana (FST = 0.044), and Eastern Sierra Morena (FST = 0.071), confirming the occurrence of changes in allele frequency through time in both localities (see also isolation-by-time analyses below).
Regarding mitogenomic variation, haplotype sharing is extensive in ancient samples, with the most extreme example being haplotype 3 occurring as far as in the northeast (Barcelona), southeast (Subbéticas), and southwest (Doñana) of the Iberian Peninsula (supplementary fig. S8A and C, Supplementary Material online). Insufficient sample sizes precluded the estimation of ancient mitogenomic FST values (fig. 4 and supplementary fig. S9, Supplementary Material online). Historical populations show moderate levels of geographical structuring with some haplotypes shared across populations (FST = 0.39; supplementary figs. S8 and S9, Supplementary Material online). Similar to microsatellite patterns, central populations are less differentiated than peripheral ones, including the peripheral Subbéticas population which was absent from the microsatellites data set (supplementary fig. S8B and D, Supplementary Material online). In contrast, current variation is totally structured in remnant Iberian lynx populations with nonoverlapping sets of mitogenomes, as previously observed based on short mitochondrial sequences (Casas-Marce et al. 2013) (FST = 0.78; fig. 4; supplementary fig. S9, Supplementary Material online).
Genetic Diversity
The comparison of current versus historical values of microsatellite and mitogenomic diversity allowed us to estimate the amount of diversity lost through time at the species level (fig. 5; tables 1 and 2). Overall, for microsatellites, expected heterozygosity (HE) dropped moderately from 0.60 ± 0.13 in the historical period to 0.54 ± 0.13 at present (decrease to 90%; S = 75.5, P = 0.003), whereas allelic richness (AR) dropped from 4.95 to 3.67 (74.1%; S = 74.5, P < 0.001) (fig. 5; table 1). A similar decrease was obtained for mitogenomic haplotype diversity (HdHistorical = 0.860 ± 0.021 > HdContemporary = 0.643 ± 0.024; 74.8%; P < 10−11; fig. 5; table 2) and was most dramatic for nucleotide diversity (πHistorical = 0.0005 ± 0.00011 > πContemporary = 0.00018 ± 0.00011; 36%). A higher Hd was estimated for the ancient period (HdAncient = 0.978 ± 0.054), so that the comparison with current is significant (P < 0.01), but the comparison of historical is not, probably due to lack of power conferred by our relatively small ancient sample size (N = 10). Ancient nucleotide diversity is lower than that reported for the historical period (πAncient = 0.00042± 0.00025 < πHistorical = 0.00054 ± 0.00028); smaller sample sizes in the ancient and higher genetic structure in the historical data set may have contributed to this difference. The two extant Iberian lynx populations show extremely low current diversity, especially Doñana (fig. 5; tables 1 and 2; supplementary fig. S9, Supplementary Material online), for both microsatellites and mitogenomes. In particular, current overall mitogenomic diversity is the lowest reported for any mammal, with only three haplotypes defined by six positions, none of them translating to protein sequence variation (supplementary table S6, supplementary figs. S9 and 10, Supplementary Material online), but ancient and historical mitogenomic diversity, although higher, are still among the lowest for any mammal (supplementary figs. S9 and S10, Supplementary Material online).
Fig. 5.

Comparison of genetic diversity across periods. Microsatellite diversity was quantified as the unbiased expected heterozygosity (A), and whole mitochondrial diversity as nucleotide diversity (B). Points represent the average and standard error for each population, whereas the horizontal dashed lines and the corresponding shaded interval represent the same for the pooled samples of each period. Periods are color-coded in shades of grey. See tables 1 and 2 for other diversity and differentiation measures.
Table 1.
Microsatellite Diversity and Differentiation in Iberian Lynx Populations and Periods.
| Epoch | Population | N | Dates Range | HE | AR (n = 10) | Private AR | FIS | F (2mod) | Fst Global |
|---|---|---|---|---|---|---|---|---|---|
| Current | 210 | 1991–2010 | 0.54 (0.13) | 3.67 | 0.05 | 0.27* | 0.42 | ||
| E. Sierra Morena | 102 | 1991–2010 | 0.51 (0.14) | 2.65 | 0.02 | 0.01 | 0.61 | ||
| Doñana | 110 | 1991–2007 | 0.31 (0.20) | 1.83 | 0.01 | 0.00 | 0.17 | ||
| Historical | 143 | 1856–1990† | 0.60 (0.13) | 4.95 | 1.33 | 0.25* | 0.27 | ||
| Montes de Toledo | 22 | 1939–1977 | 0.58 (0.17) | 3.02 | 0.11 | 0.04 | 0.07 | ||
| E. Sierra Morena | 10 | 1960–1990 | 0.61 (0.17) | 3.25 | 0.13 | 0.14* | 0.03 | ||
| Far-E. Sierra Morena | 13 | 1966–1989 | 0.44 (0.21) | 2.42 | 0.08 | 0.11* | 0.32 | ||
| W. Sierra Morena | 3 | 1970–1972 | 0.51 (0.32) | −0.02 | |||||
| Vale do Sado | 8 | 1881–1956 | 0.55 (0.21) | 2.80 | 0.03 | 0.22* | 0.13 | ||
| Central Range | 18 | 1916–1993† | 0.50 (0.15) | 2.70 | 0.15 | 0.09* | 0.19 | ||
| Doñana | 64 | 1856–1990 | 0.41 (0.20) | 2.21 | 0.06 | 0.01 | 0.37 |
Note.—HE, expected heterozygosity; AR, allelic richness; FIS, population inbreeding coefficient; F2mod, probability of identity by descent estimated by 2mod; FST, genetic differentiation coefficient.
*p < 0.05
†Includes a single sample with uncertain dating outside this range, around 1700.
Table 2.
Mitogenomic Diversity and Differentiation in Iberian Lynx Populations and Periods.
| Epoch | Population | N | Dates Range | Haplotypes | S | Hd (SD) | Pi (SD) (‰) | K | Tajima’s D | Fu and Li’s F | FST Global |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Current | 65 | 1991–2010 | 3 | 6 | 0.64 (0.02) | 0.18 (0.11) | 2.95 | 3.232*** | 2.15 | 0.78 | |
| E. Sierra Morena | 38 | 1991–2010 | 2 | 1 | 0.44 (0.06) | 0.03 (0.03) | 0.44 | 1.253 | 0.88 | ||
| Doñana | 27 | 1991–2007 | 1 | 0 | 0 (0) | 0 (0) | 0.00 | ||||
| Historical | 83 | 1881–1985 | 15 | 32 | 0.86 (0.02) | 0.54 (0.28) | 7.97 | 0.693 | 1.13 | 0.39 | |
| Montes de Toledo | 22 | 1939–1985 | 9 | 23 | 0.81 (0.07) | 0.39 (0.21) | 6.02 | −0.183 | 0.69 | ||
| E. Sierra Morena | 4 | 1960–1972 | 3 | 19 | 0.83 (0.22) | 0.63 (0.44) | 9.83 | −0.541 | 0.05 | ||
| Far-E. S. Morena | 8 | 1964–1971 | 1 | 0 | 0 (0) | 0 (0) | 0.00 | ||||
| W. Sierra Morena | 4 | 1910–1972 | 3 | 17 | 0.83 (0.22) | 0.59 (0.41) | 9.00 | −0.331 | 0.23 | ||
| Vale do Sado | 12 | 1881–1956 | 5 | 21 | 0.79 (0.09) | 0.43 (0.25) | 6.82 | −0.099 | 1.13 | ||
| Central Range | 12 | 1881–1974 | 4 | 20 | 0.65 (0.13) | 0.29 (0.17) | 4.48 | −1.459 | −1.43 | ||
| Doñana | 13 | 1856–1982 | 2 | 5 | 0.54 (0.06) | 0.16 (0.1) | 2.69 | 2.392* | 1.75 | ||
| Subbéticas | 2 | 1874–1880 | 1 | 0 | 0 (0) | 0 (0) | 0.00 | ||||
| Ancient | 10 | 43500–2070 ybp | 9 | 24 | 0.98 (0.05) | 0.42 (0.25) | 6.64 | −1.181 | −1.12 |
Note.—S, number of segregating sites; Hd, haplotype diversity; Pi, nucleotide diversity; K, average number of nucleotide differences; D and F are neutrality indices of Tajima (1989) and Fu and Li (1993), respectively.
*p < 0.05
*p < 0.001
At the population level, historical populations differ widely in their genetic diversity (tables 1 and 2). We found a general trend for higher diversity in central historical populations (e.g., Eastern Sierra Morena and Montes de Toledo) than in peripheral ones (Doñana, Far-E. Sierra Morena; fig. 5). For example, the peripheral Doñana population shows the lowest HE and AR while the central Eastern Sierra Morena and Montes de Toledo always rank as the top two. The microsatellite private allelic richness shows a different, but still parallel pattern. It is high in Montes de Toledo and historical Eastern Sierra Morena, but also in the otherwise derived Central Range, and is low in peripheral populations. This indicates that the peripheral harbour a subset of the diversity present in the central ones. Mitochondrial diversity indices vary similarly from complete uniformity in some of the peripheral populations (Far-Eastern Sierra Morena and Subéticas) to highest values in the central ones (table 2).
When we compare current versus historical genetic diversity within each of the two remnant populations, differences in diversity are also evident between periods. The contemporary population of Doñana harbours only 75% of the HE (S = 84.5, P = 0.0007) and 83% of the AR (S = 103, P < 0.0001) observed at earlier times in this same population. Similar patterns are observed for contemporary Eastern Sierra Morena with ratios of 89% for HE (S = 83.0, P = 0.001) and 81.5% for AR (S = 82.0, P = 0.0012) with respect to its historical variation. Therefore, both populations have progressively lost heterozygosity in the last decades, and they each represent a fraction of the overall historical diversity of the species (fig. 5; table 1).
We further explored the dynamics on genetic variation in each of the populations for which we had a wide temporal range. We plotted the standardized HO of individuals through time and the relatedness of pairs of individuals relative to their temporal distance, the latter used as an indication of the intensity of allelic frequency fluctuations. The contrasting trends range from no changes in either diversity or allelic frequencies through time in Montes de Toledo, to the rather steep decrease and intense fluctuations in Doñana (fig. 6).
Fig. 6.

Effects of genetic drift in Iberian lynx populations. The effect of drift is illustrated by the decrease in individual standardized observed heterozygosity (HO) (A) and the increase of genetic similarity between pairs of individuals with time (B). The intensity of drift varies in the different populations in agreement with known demographic history, ranging from low in the large and connected Montes de Toledo population to high in the small and isolated Doñana population.
Discussion
Here, we reconstruct the genetic dynamics of the endangered Iberian lynx from ancient to contemporary times in order to shed light into the genetic processes that operated during the species decline toward its almost-extinction.
The Iberian lynx was once widespread in the Mediterranean biogeographic region of the Iberian Peninsula and may have reached Southern France and even Italy in the Pleistocene and the Holocene (Altuna 1972; Vigne and Pascal 2003; Yravedra 2005; Rodríguez-Varela et al. 2015). Taking into account this broad and continuous range, the most likely scenario for its ancestral genetic variation is a single genetically diverse and panmictic population. However, a limited pattern of isolation-by-distance could also have been possible depending on the extent of dispersal in the Iberian lynx. In fact, ancient mitogenomic variation was not highly structured, with haplotypes shared across the Iberian Peninsula, and overall mitogenomic diversity was probably higher than in historical times, but still low when compared with other mammals (fig. 4; supplementary figs. S9 and S10, Supplementary Material online).
Contrastingly, already by the early 20th century, both mitogenomic and nuclear markers revealed a structured metapopulation and locally low genetic diversity (figs. 3–5; supplementary fig. S9, Supplementary Material online). Unfortunately, we could not assess ancient microsatellite diversity, so we cannot be sure whether historical nuclear diversity was lower and structure higher than in more ancient times. A direct extrapolation of mitochondrial patterns is not warranted, because nuclear and mitogenomic patterns and dynamics might differ due to sex-biased dispersal and to their different sensitivity to bottlenecks (Fay and Wu 1999).
Historical genetic structure (fig. 3) could be either the consequence of an ongoing fragmentation and decline process that started earlier than previously thought, or the result of the species’ natural demographic dynamics. Given the evidence for predominance of recent genetic drift in local populations, we favour the view of an almost panmictic ancestral population, which started to contract and fragment before the sampled historical period. Indeed, mitogenomic data indicate the occurrence of a previous bottleneck around 400 years ago. Similarly dated bottlenecks were previously inferred for this species (Casas-Marce et al. 2013; Abascal et al. 2016), and other European carnivores (Breitenmoser 1998; Valdiosera et al. 2008; Schmidt et al. 2011), and are congruent with its contracted and patchy distribution in the period 1572–1897 inferred from historical records (Clavero and Delibes 2013). Although we can only speculate about the causes of this historical decline, they may well be related to an increase in anthropic pressures. The 16th century was a period of intense human population growth in Iberia and across Europe in general, and coincided with the extension of agriculture and the intensification of forest destruction, both of which may have initiated the decline of this Mediterranean shrubland specialist (Ellis et al. 2010). Both ancient and historical bottlenecks have impacted its genetic variation and are in large part responsible for the Iberian lynx being apparently the mammal species with the lowest genome- and species-wide diversity today (Abascal et al. 2016).
During the process of decline, the populations lost diversity and became genetically differentiated due to random fluctuations in allelic frequencies (fig. 6). Such scenario is supported by heterozygosity through time and isolation by time plots (fig. 6), as well as estimates of identity by descent (Column “F2mod” in table 1). These results also reveal differences in the intensity of these processes across the Iberian lynx’s range (fig. 6). The accumulated effects of genetic drift in each population are probably related to the dynamics of the fragmentation and decline process. Genetic drift started earlier and impacted to a greater extent the patches that became isolated sooner at the periphery of the species’ range (fig. 4; Rodríguez and Delibes 2002). For instance, the Doñana population showed the strongest signal of genetic drift, consistent with its peripheral position and its long history of low effective size and genetic isolation, which we have estimated in around 20 individuals and lasting ca. 200 years (around 40 generations, at the very beginning of the 19th century; supplementary fig. S3B, Supplementary Material online). In contrast, we did not observe much genetic drift in Montes de Toledo, and only very recently in Eastern Sierra Morena. Both populations remained large and connected to each other, as well as to other more peripheral populations, until recently (fig. 4; supplementary fig. S3B, Supplementary Material online; Rodríguez and Delibes 2002). Thus, we hypothesize that they played a pivotal role in the metapopulation, acting as a reservoir of genetic diversity and as a source of gene flow to other populations. The major role of effective population size and gene flow in determining genetic patterns following decline and fragmentation is in line with expectations from population genetic theory and with empirical studies in other species (e.g., Méndez et al. 2014).
Despite extensive evidence showing how genetic erosion can negatively affect population viability (Frankham et al. 2010; Allendorf et al. 2013), the amount of drift accumulated in different Iberian lynx populations beared little regard to the final outcome of persistence or extinction (figs. 4 and 6). This study rather suggests that their final fate was generally determined by extrinsic factors. Possibly, the influence of genetics was overridden by increased extrinsic pressures in demographically and genetically healthy populations, and by the protection and active conservation granted to the most severely eroded population. Thus, while Central Range and Far-Eastern Sierra Morena populations did accumulate genetic erosion before their extinction by the late 20th century, the central population of Montes de Toledo remained largely unaffected by genetic drift until its final extirpation. Whereas in the former populations, genetic erosion might have contributed to population decline, this was unlikely to be the case for the latter. Montes de Toledo may have been abruptly pushed from a large and well-connected population to complete extirpation by deterministic factors acting in this area during most of the 20th century, namely landscape homogenization and lasting scarcity of prey (Rodríguez and Delibes 1990). On the other hand, we observe a striking persistence of the Doñana population despite a long history of small population size, isolation, and extreme genetic erosion, which according to recent studies may have affected reproduction and survival (Palomares et al. 2012). The persistence of Doñana could thus be attributed to the protection granted to the area by its designation as a royal hunting reserve during most of the 19th century and to the establishment of Doñana National Park in 1965.
While both remnant populations have progressively lost heterozygosity in the last decades (fig. 6), they have done so to very different extents. In contrast to the highly eroded Doñana population, Eastern Sierra Morena represents the remnant of the large historical core population encompassing Montes de Toledo and Eastern Sierra Morena, and it is the current population genetically closest to the ancestral lynx population (fig. 4). Still, the contemporary diversities of both populations are lower than those reported for demographically healthy populations of its sister species, the Eurasian lynx (Casas-Marce et al. 2013; Ratkiewicz et al. 2014). However, the two contemporary populations conjunctly represent most of the former overall heterozygosity (90%), and also a moderate proportion (74.1%) of the allelic richness of the species in historical times, which were more in line with those observed in the Eurasian lynx (Ratkiewicz et al. 2014).
It must be noted that at least two sources of bias might have contributed to an underestimation of historical microsatellite diversity, and thus of the amount of diversity lost. First, allelic dropout in historical samples despite the use of replicates will cause heterozygotes to be called as homozygotes. This will reduce the observed population heterozygosity (HO) more than HE, and may thus contribute—together with spatial or temporal structure—to explaining part of the significant heterozygote deficit observed in most historical populations (table 1). Second, the microsatellites used were selected in previous studies (Johnson et al. 2004; Casas-Marce et al. 2013) to be highly variable in the current population, thereby introducing an ascertainment bias that would again lead to an underestimation of historical diversity and thus also of the amount of diversity recently lost.
In summary, our results show that the recent genetic erosion of Iberian lynx has been severe and has affected both microsatellite and mitogenomic diversity. Such erosion has three main components: (1) loss of diversity through time within populations, (2) an increasing differentiation between populations, and (3) extinction of genetically differentiated populations at the edges of the historical distribution.
Conservation Implications
Here, we demonstrate that the species’ genetic makeup has been shaped by a long history of low population size and a sharp population decline around the 16th–17th century AD, well before its last well documented decline in the 20th century that left only two small remnant populations. However, we show that the extremely low genetic diversity of the two remnant populations (Casas-Marce et al. 2013; Abascal et al. 2016) is also the result of intense genetic drift and fragmentation occurring during the last decades. Due to the abrupt and recent nature of these changes, the risk of inbreeding depression in both remaining populations must be explicitly considered and addressed. Our results and conclusions are in contrast to those of a previous study reporting a long-term lack of mitochondrial diversity throughout the Iberian lynx’s history based on short control region sequences, which led to the suggestion that the Iberian lynx’s genetic diversity had always been low and was thus not a threat to its long-term viability (Rodríguez et al. 2011). Evidence for the occurrence of inbreeding depression in the species is accumulating in the form of heterozygosity-fitness correlations of sperm quality, reduced reproductive rates, increased nontraumatic mortality, and high rates of potentially genetic diseases (Peña et al. 2006; Jiménez et al. 2008; Palomares et al. 2012; Ruiz-López et al. 2012; Martínez et al. 2013). At the same time, the lower differentiation in historical and ancient times revealed from our data suggests that the currently observed genetic differentiation between the two remnant populations is mainly the result of genetic drift in recent times and cannot be attributed to independent adaptive evolution over long periods, indicating low risks of outbreeding depression (Frankham et al. 2011). Low-genetic diversity together with higher risks of inbreeding than outbreeding depression support the ongoing admixture of the two genetic pools both in captivity (Godoy et al. 2009) and in the wild through translocations (Simón et al. 2012). Although the positive contribution of admixture and other genetic management to Iberian lynx recovery has not yet been formally evaluated, the observation of an increased reproductive performance of admixed individuals suggests so (Godoy JA, unpublished data). Furthermore, genetic risks call for an intensive genetic monitoring and a sound comprehensive genetic management program for the species, which should also include the ongoing reintroductions. On the other hand, it is likely that the globally low genetic diversity will influence the chances of long-term persistence of the species, especially in a scenario of rapid global change (Fordham et al. 2013). Addressing the possibility of long-term reduced adaptive potential will have to carefully consider risks and benefits of less conventional measures to restore genetic diversity, like facilitated adaptation, genome edition, or assisted introgression (Thomas et al. 2013; Hamilton and Miller 2016).
Conclusions
Over time, species may have experienced complex demographic changes, including earlier anthropogenic impacts, which we cannot directly infer from contemporary genetic patterns and recorded demographic history. In the case of endangered species, disentangling the processes acting upon species throughout their history can help us understand how they became endangered and what we need to do to restore natural and healthy genetic patterns to guarantee their long-term survival. We show that the Iberian lynx has indeed lost a significant part of its already low genetic variation over time due to both recent and unsuspected older demographic declines, and that the contemporary pattern of high genetic differentiation between the remnant populations was caused by genetic drift during the last few centuries. Our retrospective look was only possible due to the availability of extensive ancient, historical, and contemporary samples (Casas-Marce et al. 2012), a careful selection of sampled tissues in historical specimens (Casas-Marce et al. 2010), recent technical advances in the field of ancient DNA (Hofreiter et al. 2015; Orlando et al. 2015), and the combination of nuclear markers and whole mitochondrial genomes.
To our knowledge, our analysis of the Iberian lynx is the most comprehensive retrospective analysis of genetic variation of an endangered species to date. It highlights how the evolutionary history of a species inferred solely from its current genomic makeup can be distorted, misinforming management recommendations. Studying paradigmatic conservation cases like this one is also crucial for deepening our understanding of the demographic and genetic processes occurring during population declines, a much needed input for the effective restoration of endangered species and for the prospective assessment of species viability in a changing world.
Materials and Methods
Samples
We used a combination of modern, historical, and ancient samples (supplementary tables S1–S3, Supplementary Material online). Samples were grouped in a priori populations based on their origin or, when lacking, on results of clustering analyses. We also delimited periods based on recorded date of sampling, using the year 1990 as the limit separating contemporary and historical samples. This is approximately the time of the collapse of all but the two remnant Iberian lynx populations (Ferreras et al. 2010) and the time that roughly separates museum from fresh samples. We extracted DNA for a total of 230 fresh, 296 museum, and 58 archaeological samples.
Microsatellite Genotyping
Microsatellite markers were amplified in contemporary and historical samples. We used 20 microsatellites selected from 36 previously used for the analyses of modern Iberian lynx specimens (Casas-Marce et al. 2013) based on their good performance with degraded DNA. For historical samples we applied a preamplification multiplex approach (Piggott et al. 2004). We obtained good quality genotypes at 20 microsatellite markers for all the fresh samples and for 155 out of the 296 historical samples (52.4%).
Mitochondrial Genome Sequencing
For contemporary samples, we first used a long-range PCR approach to sequence the whole mitochondrial genome in 12 Iberian lynx (eight from Doñana and four from Eastern Sierra Morena). The amplified products were pooled equimolarly and individually tagged 454 sequencing libraries were prepared following the protocol described by Meyer et al. (2008a; 2008b). In addition, we used available whole-genome shotgun data for 31 different lynx (19 from Eastern Sierra Morena and 12 from Doñana), and shotgun reads from a separate capture-enrichment project targeting a subset of the nuclear genome in 34 individuals (13 from Eastern Sierra Morena and 21 from Doñana). We prepared a total of 111 historical and 20 ancient individually tagged libraries for Illumina or 454 sequencing following Meyer and Kircher (2010) or Maricic et al. (2010) (supplementary table S4, Supplementary Material online). L.pardinus biotinylated capture probes were prepared as described in Maricic et al. (2010) from the products of two overlapping long-rang PCR. Historical and ancient samples were enriched in target mitochondrial sequences by one or two consecutive rounds of hybridization–capture (supplementary table S4, Supplementary Material online). We mapped both merged and unmerged reads to the L.pardinus mitochondrial reference genome (Abascal et al. 2016) using BWA-mem (Li and Durbin 2009) with default parameters. Average coverage per sample ranged from 0.1× to 239.2× (supplementary table S4, Supplementary Material online). Only samples with total coverage over 5.2× were considered for further analyses. Before SNP calling we used mapDamage (Jonsson et al. 2013) on historical and ancient samples to track and quantify DNA damage patterns and rescale quality scores of likely damaged positions accordingly. Bam files generated in this study have been deposited in the European Nucleotide Archive under study accession number PRJEB15462.
Mitochondrial SNP Calling and Annotation
We simultaneously called SNPs on the pooled samples using Freebayes (Garrison and Marth 2012). We visually curated the 57 resulting variants by revising each position called as an SNP for all individuals to identify potential contaminations and artifacts. The number of reference positions not covered by any read was calculated for each individual using the command genomecov in BEDTools (Quinlan and Hall 2010). A consensus for each individual mitochondrial genome was constructed using the FastaAlternateReferenceMaker command in GATK (McKenna et al. 2010) (supplementary table S5, Supplementary Material online). Validated SNPs were annotated as genic/intergenic, transition/transversion, synonymous/nonsynomymous, and aminoacid change using Geneious v. 8 (http://www.geneious.com; last accessed August 25, 2017; Kearse et al. 2012) (supplementary table S6, Supplementary Material online).
Demographic Reconstruction Using Whole Mitochondrial Genomes
Past population dynamics was investigated with a BSP model using BEAST v1.8.1 (Drummond et al. 2012). We calibrated the tree using the divergence of the entire lynx lineage with a mean value of 2.52 My and a standard deviation of 0.4 My to estimate a substitution rate of 1.89×10−8 substitutions/site/year (1.16×10−8–2.78×10−8 substitutions/site/year, 95% HPD). To construct the BSP, we assumed a HKY + G model of evolution and a strict molecular clock, and incorporated the information on the age of the sequences using the sampling date for contemporaneous samples, the date of death for the historical, and the radiocarbon estimated date for the ancient samples (supplementary tables S1–S3, Supplementary Material online).
Estimation of Divergence Parameters with ABC
We used an ABC approach based on coalescent simulations implemented in DIYABC version 2.1.0 (Cornuet et al. 2014) to estimate effective population sizes and divergence times between populations. We used a simple model that assumes constant population sizes and no gene flow between populations after divergence. We used a generation time of 5 years, grouped samples by decades and considered each decade to be separated from the next one by two generations (supplementary fig. S2, Supplementary Material online). Prior distributions for the effective population size of the historical population (N1), contemporary Eastern Sierra Morena (N2), and Doñana (N3) were set based on previous estimates of contemporary sizes (supplementary fig. S3B, Supplementary Material online; Casas-Marce et al. 2013). For the purpose of building the posterior distributions, we used as summary statistics the expected heterozygosity and the number of observed alleles for all samples in the analysis and pair-wise FST values between some of the samples. We ran 1,000,000 simulations and used the 1% closest to our data set to build the posterior distributions using a local linear regression technique (supplementary fig. S3A, Supplementary Material online; Beaumont et al. 2002).
Estimation of Historical Population Sizes and Dates of Isolation
During the period 1950–1990, Iberian lynx abundance was estimated from the textures of relative abundance published by Rodríguez and Delibes (2002) on a 10-km UTM grid. To estimate lynx absolute abundance for a given date, we adopted a three-step process. First, we used percentiles in the frequency distribution of the cumulated number of reports until the map date (Rodríguez and Delibes 2002) as classes of relative abundance. We assumed a nonlinear relationship between relative and absolute densities for the period 1985–1988 (modified from Rodríguez and Delibes 1990). We derived estimates of lynx absolute abundance (N1) from classes of relative abundance (Rodríguez and Delibes 1990). Second, we corrected N1 for the decline in the probability of recording reports with time elapsed since observation. We fitted a generalized linear model of the number of lynx reports per year during the period 1940–1988, using the elapsed time until 1988 as a predictor to estimate, for each year, the expected number of reports we would have obtained had we performed the survey that year. Predicted values were averaged over periods of five consecutive years, and the ratio between predictions for the most recent period (1985–1988) and earlier quinquennia was used as a correction factor. Then we applied the procedure described in step 1, and the corresponding conversion factors, to produce a second estimate of lynx numbers, called N2. Finally, published figures of lynx abundance were computed by combining estimated densities with precisely outlined distribution patches. To make our coarse-grained estimates (on a grid) comparable to published figures, we multiplied the average proportion of the cell area covered by outlined patches of lynx distribution (0.54) by N2 to produce the final estimate of lynx numbers per cell. We employed estimates by Simón et al. (2012) for 2002 (approximated here to year 2000), 2005, and 2010, whereas figures for 2015 were published on the website http://www.iberlince.eu/. (last accessed August 25, 2017) For estimates in 2000 or later, total population size refers to the number of adult females, and juveniles plus the number of adult males assuming a 1:1 sex ratio. Isolation date between genetically similar spatial clusters was defined as the date of loss of physical contact in a 10-km UTM grid.
Population-Based Microsatellite Data Analyses
In order to assess structure through time and across space, we used the Bayesian clustering method implemented in STRUCTURE (Pritchard et al. 2000; Falush et al. 2003) to infer the number of genetic clusters in our data set, and to obtain assignment values for each individual in each cluster (Q). We also performed FCA as implemented in Genetix (Belkhir et al. 1996–2004) and plotted them with the threejs R package (https://github.com/bwlewis/rthreejs/blob/master/R/threejs.R; last accessed August 25, 2017) to visualize overall patterns of genetic structure in our data. We calculated diversity parameters for each of the geographical groups and temporal periods using Genetix 4.05 (Belkhir et al. 1996–2004) and HP-Rare (Kalinowski 2005). Statistical comparisons in diversity between populations or periods were performed using nonparametric Wilcoxon signed rank tests. In order to evaluate which of the scenarios is the most plausible to explain the genetic differentiation between the populations, pure genetic drift or drift-migration equilibrium, we used a Bayesian–MCMC approach implemented in 2mod (Ciofi et al. 1999) to estimate the relative likelihood of each model.
Mitogenome Sequence Analyses
Consensus mitogenome sequences were aligned and collapsed to distinct haplotypes using pegas R package (Paradis 2010). Mitogenomic consensus sequences for each sample have been deposited in GeneBank under accession numbers KX911255–KX911412. Diversity and differentiation indices were estimated with PopGenome (Pfeifer et al. 2014) and ape (Paradis et al. 2004) R. Empirical distributions were generated to determine sampling variance and standard deviations. We tested whether haplotype diversity had declined over time using the conservative double-testing implemented in TEST_H_DIFF R (http://www.ucl.ac.uk/tcga/software/; last accessed August 25, 2017). Phylogenetic relationships among sequence haplotypes were inferred by constructing a median-joining haplotype network (Bandelt et al. 1999) using pegas R package (Paradis 2010).
Individual-Based Microsatellite Data Analyses
To assess changes in microsatellite genetic diversity over time, we calculated individuals’ standardized observed heterozygosity with IRmacroN4 (Amos et al. 2001) and assessed its relationship with time using linear regression. Changes in allelic frequencies were assessed by testing an isolation-by-time hypothesis, that is, we assessed the correlation between a genetic distance matrix—linearized inter-individual pairwise distance described as â by Rousset (2000), as implemented in SPAGeDi (Hardy and Vekemans 2002)—and a temporal distance matrix in years. Significance of the regression slopes was assessed with Mantel tests. We performed this analysis with the four populations for which we had enough samples from a wide enough temporal range (Doñana, Central Range, Montes de Toledo and Eastern Sierra Morena).
For a more detailed version of methods, see supplementary methods, Supplementary Material online.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This research was funded by the Spanish Dirección General de Investigación Científica y Técnica, through projects CGL2006-10853/BOS, CGL2010-21540/BOS, and CGL2013-47755-P of the Spanish Ministerio de Ciencia e Innovación (MICINN) to J.A.G., and by the Fundación BBVA through a project on ancient lynx genetics to M.D. M.C. and E.M. received a JAE predoctoral grant from CSIC (Spanish National Research Council), M.L.P. was supported by PhD contracts from Programa Internacional de Becas “La Caixa-Severo Ochoa.” J.N. received financial support through projects HAR2014-55131 from the Ministerio de Ciencia e Innovación (MICINN) and SGR2014-108 from Generalitat de Catalunya. We thank the following people and institutions for contributing specimens information and/or samples: Estación Biológica de Doñana—CSIC, Swedish Natural History Museum, Natural History Museum of London, Smithsonian Natural History Museum, Museo Nacional de Ciencias Naturales—CSIC, Colegio Nuestra Señora de los Reyes, Instituto Aguilar y Eslava, Instituto Padre Luís Coloma, Museo de Ciencias del Instituto Padre Suárez, Facultad de Ciencias de Granada, Museo de Historia Natural de Viso del Marqués, Sociedad Aranzadi, Palacio Real de Riofrío, Museu da Faculdade de Ciências do Porto, Museu Zoológico de Coimbra, Museu Nacional de História Natural (Portugal), Museu Municipal de Penamacor, José Antonio Riquelme, Grupo MIDAS III Milenio, Universidad de Huelva, Ayuntamiento de Úbeda, Instituto Português de Arqueologia, Tapada Nacional de Mafra, Clube Português de Canicultura, Museu Municipal de Moià, Museu do Instituto Geológico e Mineiro (Portugal), Museo Arqueológico Nacional, Prof. Ana Margarida Arruda, Câmara Municipal de Lagos, Ostrava Zoo (Check Republic) and Zoo de Córdoba (Spain). We equally acknowledge all the anonymous owners of lynx specimens who allowed us to sample them; many of these were located with the help of Juan C. del Olmo, Javier N. Guzmán and Margarida Fernandes. We thank Ana Martínez Fontúrbel and Margarida Fernandes for field assistance. Contemporary samples were received under agreements with the Consejería de Medio Ambiente de la Junta de Andalucía, and were contributed by the LIFE projects LIFE-NAT 06/E/000209 and LIFE10 NAT/E/000570, the Iberian lynx Ex Situ Conservation Program, or by Alejandro Rodríguez, Francisco Palomares and José V. López-Bao.
Author Contributions
J.A.G, A.R., E.R., and M.D. designed the study. M.C.-M., E.M. M.L.P., and J.A.G wrote the manuscript. J.A.G, M.S., and M.H. directed the experimental work. M.C.-M., E.M., L.S., and M.L.P. produced the molecular data. M.C.-M., E.M., L.S., B.M.-C., and J.A.G. analyzed the data. A.R. quantified demographic changes. F.N., A.R.-H., A.C., J.N. C.D., E.B.-S., C.F.-R., and M.P.-R. excavated the ancient samples and provided archaeological expertise. All authors contributed to the final version of the manuscript.
References
- Abascal F, Corvelo A, Cruz F, Villanueva-Canas JL, Vlasova A, Marcet-Houben M, Martinez-Cruz B, Cheng JY, Prieto P, Quesada V.. 2016. Extreme genomic erosion after recurrent demographic bottlenecks in the highly endangered Iberian lynx. Genome Biol. 17(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf FW, Luikart G.. 2007. Conservation and the genetics of populations. Malden (MA: ): Blackwell. [Google Scholar]
- Allendorf FW, Luikart G, Aitken SN.. 2013. Conservation and the genetics of populations. Hoboken: John Wiley & Sons. [Google Scholar]
- Altuna J. 1972. Fauna de Mamíferos de los Yacimiento Prehistórico de Guipúzcoa. Munibe 24:1–472. [Google Scholar]
- Amos W, Wilmer JW, Fullard K, Burg TM, Croxall JP, Bloch D, Coulson T.. 2001. The influence of parental relatedness on reproductive success. Proc R Soc B Biol Sci. 268(1480):2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A.. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 16(1):37–48. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Zhang WY, Balding DJ.. 2002. Approximate Bayesian computation in population genetics. Genetics 162(4):2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F.. 1996–2004. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations: Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier (France).
- Breitenmoser U. 1998. Large predators in the Alps: the fall and rise of man's competitors. Biol Conserv. 83(3):279–289. [Google Scholar]
- Casas-Marce M, Revilla E, Fernandes M, Rodríguez A, Delibes M, Godoy JA.. 2012. The value of hidden scientific resources: preserved animal specimens from private collections and small museums. Bioscience 62(12):1077–1082. [Google Scholar]
- Casas-Marce M, Revilla E, Godoy JA.. 2010. Searching for DNA in museum specimens: a comparison of sources in a mammal species. Mol Ecol Resour. 10(3):502–507. [DOI] [PubMed] [Google Scholar]
- Casas-Marce M, Soriano L, López-Bao JV, Godoy JA.. 2013. Genetics at the verge of extinction: insights from the Iberian lynx. Mol Ecol. 22(22):5503–5515. [DOI] [PubMed] [Google Scholar]
- Ceballos G, Ehrlich PR, Barnosky AD, García A, Pringle RM, Palmer TM.. 2015. Accelerated modern human-induced species losses: entering the sixth mass extinction. Sci Adv. 1(5):e1400253.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofi C, Beaumont MA, Swingland IR, Bruford MW.. 1999. Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc R Soc B Biol Sci. 266(1435):2269–2274. [Google Scholar]
- Clavero M, Delibes M.. 2013. Using historical accounts to set conservation baselines: the case of lynx species in Spain. Biodiv Conserv. 22(8):1691–1702. [Google Scholar]
- Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A.. 2014. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30(8):1187–1189. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29(8):1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis EC, Goldewijk KK, Siebert S, Lightman D, Ramankutty N.. 2010. Anthropogenic transformation of the biomes, 1700 to 2000. Glob Ecol Biogeogr 19:589–606. [Google Scholar]
- Falush D, Stephens M, Pritchard JK.. 2007. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7(4):574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK.. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI.. 1999. A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol. 16(7):1003–1005. [DOI] [PubMed] [Google Scholar]
- Ferreras P, Rodríguez A, Palomares F, Delibes M.. 2010. Iberian lynx: the uncertain future of a critically endangered cat In: MacDonald DW, Loveridge AJ, editors. The biology and conservation of wild felids. Oxford: Oxford University Press; p. 507–520. [Google Scholar]
- Fordham DA, Akcakaya R, Brook BW, Rodriguez A, Alves PC, Civantos E, Trivino M, Watts MJ, Araujo MB.. 2013. Adapted conservation measures are required to save the Iberian lynx in a changing climate. Nat Clim Change 3(10):899–903. [Google Scholar]
- Frankham R, Ballou JD, Briscoe DA.. 2010. Introduction to conservation genetics. Cambridge (NY: ): Cambridge University Press. [Google Scholar]
- Frankham R, Ballou JD, Briscoe DA.. 2002. Introduction to conservation genetics. Cambridge (NY: ): Cambridge University Press. [Google Scholar]
- Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR, Fenster CB.. 2011. Predicting the probability of outbreeding depression. Conserv Biol. 25(3):465–475. [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH.. 1993. Statistical Tests of Neutrality of Mutations. Genetics 133:693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G.. 2012. Haplotype-based variant detection from short-read sequencing. In: arXiv. [Google Scholar]
- Godoy JA, Casas-Marcé M, Fernández J.. 2009. Genetic issues in the implementation of the Iberian lynx Ex situ Conservation Programme. In: Vargas A, Breitenmoser C, Breitenmoser U, editors. Iberian lynx ex situ conservation: an interdisciplinary approach. Madrid: Fundación Biodiversidad in collaboration with: International Union for Conservation of Nature, Species Survival Commission (IUCN/SSC) Cat Specialist Group. p. 86–99.
- Hamilton JA, Miller JM.. 2016. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv Biol. 30(1):33–41. [DOI] [PubMed] [Google Scholar]
- Hardy OJ, Vekemans X.. 2002. SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2(4):618–620. [Google Scholar]
- Hedrick PW. 2001. Conservation genetics: where are we now? Trends Ecol Evol. 16(11):629–636. [Google Scholar]
- Hofman CA, Rick TC, Fleischer RC, Maldonado JE.. 2015. Conservation archaeogenomics: ancient DNA and biodiversity in the Anthropocene. Trends Ecol Evol. 30(9):540–549. [DOI] [PubMed] [Google Scholar]
- Hofreiter M, Barlow A, Paijmans J, Westbury M.. 2015. Genomic analyses from highly degraded DNA. Genome 58: 228–228. [Google Scholar]
- Jiménez MA, Sánchez B, Alenza MDP, García P, López JV, Rodríguez A, Muñoz A, Martínez F, Vargas A, Peña L.. 2008. Membranous glomerulonephritis in the Iberian lynx (Lynx pardinus). Vet Immunol Immunopathol. 121(1–2):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Godoy JA, Palomares F, Delibes M, Fernandes M, Revilla E, O'Brien SJ.. 2004. Phylogenetic and phylogeographic analysis of Iberian lynx populations. J Hered. 95(1):19–28. [DOI] [PubMed] [Google Scholar]
- Jonsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L.. 2013. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29:1682–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski ST. 2005. HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5(1):187–189. [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JA. 2008. Ancient DNA applications for wildlife conservation. Mol Ecol. 17(19):4186–4196. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricic T, Whitten M, Paabo S.. 2010. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE 5(11):e14004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez F, Manteca X, Pastor J.. 2013. Retrospective study of morbidity and mortality of captive Iberian lynx (Lynx pardinus) in the ex situ conservation programme (2004–June 2010). J Zoo Wildl Med. 44(4):845–852. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan R. 2014. Living planet report 2014 In: McLellan R, editor. Living planet report: World Wildlife Fund; p. 178. [Google Scholar]
- Méndez M, Vögeli M, Tella JL, Godoy JA.. 2014. Joint effects of population size and isolation on genetic erosion in fragmented populations: finding fragmentation thresholds for management. Evol Appl. 7(4):506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Briggs AW, Maricic T, Hober B, Hoffner BH, Krause J, Weihmann A, Paabo S, Hofreiter M.. 2008a. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic Acids Res. 36(1):e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M.. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protoc. 2010(6):pdb.prot5448.. [DOI] [PubMed] [Google Scholar]
- Meyer M, Stenzel U, Hofreiter M.. 2008b. Parallel tagged sequencing on the 454 platform. Nat Protoc. 3(2):267–278. [DOI] [PubMed] [Google Scholar]
- Nowell K, Jackson P.. 1996. Wild cats: status survey and conservation action plan. Gland, Switzerland: IUCN. [Google Scholar]
- Orlando L, Gilbert MTP, Willerslev E.. 2015. Applications of next-generation sequencing reconstructing ancient genomes and epigenomes. Nat Rev Genet. 16(7):395–408. [DOI] [PubMed] [Google Scholar]
- Palomares F, Godoy JA, López-Bao JV, Rodríguez A, Roques S, Casas-Marce M, Revilla E, Delibes M.. 2012. Possible extinction vortex for a population of Iberian lynx on the verge of extirpation. Conserv Biol. 26(4):689–697. [DOI] [PubMed] [Google Scholar]
- Palomares F, Rodríguez A, Revilla E, López-Bao JV, Calzada J.. 2011. Assessment of the conservation efforts to prevent extinction of the Iberian lynx. Conserv Biol. 25(1):4–8. [DOI] [PubMed] [Google Scholar]
- Paradis E. 2010. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26(3):419–420. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290. [DOI] [PubMed] [Google Scholar]
- Peña L, García P, Jiménez MA, Benito A, Alenza MDP, Sánchez B.. 2006. Histopathological and immunohistochemical findings in lymphoid tissues of the endangered Iberian lynx (Lynx pardinus). Compar Immunol Microbiol Infect Dis 29(2–3):114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B, Wittelsburger U, Ramos-Onsins SE, Lercher MJ.. 2014. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol. 31(7):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott MP, Bellemain E, Taberlet P, Taylor AC.. 2004. A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv Genet. 5(3):417–420. [Google Scholar]
- Pimm SL, Jenkins CN, Abell R, Brooks TM, Gittleman JL, Joppa LN, Raven PH, Roberts CM, Sexton JO.. 2014. The biodiversity of species and their rates of extinction, distribution, and protection. Science 344(6187):1246752.. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratkiewicz M, Matosiuk M, Saveljev AP, Sidorovich V, Ozolins J, Mannil P, Balciauskas L, Kojola I, Okarma H, Kowalczyk R, et al. 2014. Long-range gene flow and the effects of climatic and ecological factors on genetic structuring in a large, solitary carnivore: the Eurasian lynx. PLoS ONE 9(12):e115160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A, Calzada J.. 2015. Lynx pardinus. The IUCN Red List of Threatened Species 2015. doi:e.T12520A50655794.
- Rodríguez A, Delibes M.. 1992. Current range and status of the Iberian lynx Felis-Pardina Temminck, 1824 in Spain. Biol Conserv. 61(3):189–196. [Google Scholar]
- Rodríguez A, Delibes M.. 1990. El lince ibérico (Lynx pardina) en España: distribución y problemas de conservación. Madrid, Spain: Instituto Nacional para la Conservación de la Naturaleza.
- Rodríguez A, Delibes M.. 2002. Internal structure and patterns of contraction in the geographic range of the Iberian lynx. Ecography 25(3):314–328. [Google Scholar]
- Rodríguez R, Ramírez O, Valdiosera CE, García N, Alda F, Madurell-Malapeira J, Marmi J, Doadrio I, Willerslev E, Gotherstrom A, et al. 2011. 50,000 years of genetic uniformity in the critically endangered Iberian lynx. Mol Ecol. 20(18): 3785–3795. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Varela R, Tagliacozzo A, Ureña I, García N, Crégut-Bonnoure E, Mannino MA, Arsuaga JL, Valdiosera C.. 2015. Ancient DNA evidence of Iberian lynx palaeoendemism. Q Sci Rev. 112:172–180. [Google Scholar]
- Rousset F. (647 co-authors). 2000. Genetic differentiation between individuals. J. Evol. Biol. 13:58–62. [Google Scholar]
- Ruiz-López MJ, Gañán N, Godoy JA, Del Olmo A, Garde J, Espeso G, Vargas A, Martínez F, Roldán ERS, Gomendio M.. 2012. Heterozygosity-fitness correlations and inbreeding depression in two critically endangered mammals. Conserv Biol. 26(6):1121–1129. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Ratkiewicz M, Konopiński MK.. 2011. The importance of genetic variability and population differentiation in the Eurasian lynx Lynx lynx for conservation, in the context of habitat and climate change. Mammal Rev. 41(2):112–124. [Google Scholar]
- Simón M, Gil-Sánchez J, Ruiz G, Garrote G, McCain E, Fernández L, López-Parra M, Rojas E, Arenas-Rojas R, Rey T, et al. 2012. Reverse of the decline of the endangered Iberian lynx. Conserv Biol 26(4):731–736. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical-Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CD, Williamson M.. 2012. Extinction and climate change. Nature 482(7386):E4–E5. [DOI] [PubMed] [Google Scholar]
- Thomas MA, Roemer GW, Donlan CJ, Dickson BG, Matocq M, Malaney J.. 2013. Gene tweaking for conservation. Nature 501(7468):485–486. [DOI] [PubMed] [Google Scholar]
- Valdiosera CE, Garcia-Garitagoitia JL, Garcia N, Doadrio I, Thomas MG, Hanni C, Arsuaga JL, Barnes I, Hofreiter M, Orlando L, et al. 2008. Surprising migration and population size dynamics in ancient Iberian brown bears (Ursus arctos). Proc Natl Acad Sci U S A. 105(13):5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne J-D, Pascal M.. 2003. Le Lynx pardelle: Lynxpardinus (Temminck, 1827) In: Pascal M, Lorvelec O, Vigne J-D, Keith P, Clergeau P, editors. Évolution holocène de la faune de Vertébrés de France: invasions et disparitions. Paris: Institut National de la Recherche Agronomique, Centre National de la Recherche Scientifique, Muséum National d'Histoire Naturelle; p. 87–88. [Google Scholar]
- Yravedra J. 2005. Aprovechamiento cárnico de lince (Lynx pardina) durante el Pleistoceno Superior en el interior de la Península Ibérica. Munibe 75:303–311. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.