

Abstract
Chemosensory-related gene (CRG) families have been studied extensively in insects, but their evolutionary history across the Arthropoda had remained relatively unexplored. Here, we address current hypotheses and prior conclusions on CRG family evolution using a more comprehensive data set. In particular, odorant receptors were hypothesized to have proliferated during terrestrial colonization by insects (hexapods), but their association with other pancrustacean clades and with independent terrestrial colonizations in other arthropod subphyla have been unclear. We also examine hypotheses on which arthropod CRG family is most ancient. Thus, we reconstructed phylogenies of CRGs, including those from new arthropod genomes and transcriptomes, and mapped CRG gains and losses across arthropod lineages. Our analysis was strengthened by including crustaceans, especially copepods, which reside outside the hexapod/branchiopod clade within the subphylum Pancrustacea. We generated the first high-resolution genome sequence of the copepod Eurytemora affinis and annotated its CRGs. We found odorant receptors and odorant binding proteins present only in hexapods (insects) and absent from all other arthropod lineages, indicating that they are not universal adaptations to land. Gustatory receptors likely represent the oldest chemosensory receptors among CRGs, dating back to the Placozoa. We also clarified and confirmed the evolutionary history of antennal ionotropic receptors across the Arthropoda. All antennal ionotropic receptors in E. affinis were expressed more highly in males than in females, suggestive of an association with male mate-recognition behavior. This study is the most comprehensive comparative analysis to date of CRG family evolution across the largest and most speciose metazoan phylum Arthropoda.
Keywords: chemoreception, chemoreceptor, chemosensory receptors, gene family evolution, Copepoda, Crustacea
Introduction
Chemosensation refers to the physiological responses of sense organs to chemical stimuli, including taste and odor, and is observed across a wide range of taxa from bacteria to humans (Bargmann 2006; Vosshall and Stocker 2007; Nei et al. 2008; Kaupp 2010). Chemosensory systems play critical roles in mediating behavioral responses such as feeding, mating, predator avoidance, and predation. Chemosensing in the phylum Arthropoda is particularly intriguing, given the extraordinary diversity of habitats and ecological niches that arthropods have been able to colonize, spanning marine, brackish, hypersaline, freshwater, terrestrial, and extremely arid environments (Cloudsley-Thompson 1975; Sømme 1989; Glenner et al. 2006; Kelley et al. 2014). These habitat colonizations would have imposed novel challenges and requirements for chemosensation, as the transmission and reception of chemical stimuli become altered in diverse environments, such as in aquatic versus aerial media. Such diverse transmission media would impose varying evolutionary pressures on genes underlying chemosensory responses. Interestingly, the three major subphyla within the Arthropoda, that is, the Pancrustacea (e.g., crustaceans and insects), Myriapoda (e.g., centipede and millipedes), and Chelicerata (e.g., spiders, mites, and scorpions) have colonized freshwater and terrestrial habitats independently (Giribet et al. 2001; Regier et al. 2010; von Reumont et al. 2012; Oakley et al. 2013). Thus, given these independent transitions to land, have chemosensing systems evolved through the same pathways during these parallel but independent colonization events?
Based primarily on the study of the fruit fly Drosophila melanogaster, arthropod chemoreception has been found to be mediated by three different multigene families of chemosensory receptors. These include two gene families of seven transmembrane receptors, namely the gustatory receptors (GRs) (Clyne et al. 2000) and the more derived odorant receptors (ORs) (Clyne et al. 1999; Gao and Chess 1999; Vosshall et al. 1999), which are unrelated to the vertebrate GRs and ORs (Gardiner et al. 2009). More recently, a third family of chemosensory receptors has been discovered in D. melanogaster, namely, the ionotropic receptors (IRs), which are a class within the ancient and highly conserved ionotropic glutamate receptor (iGluR) family of ligand-gated ion channels (Benton et al. 2009; Croset et al. 2010; Abuin et al. 2011; Benton 2015). In addition, two soluble binding protein families, the chemosensory proteins (CSPs) and insect-type odorant binding proteins (OBPs), are known to mediate the transport of ligands to the chemosensory receptors (Pelosi et al. 2006; Laughlin et al. 2008; Vieira and Rozas 2011; Pelosi et al. 2014). In this study, we refer to these five gene families (ORs, GRs, IRs, CSPs, and OBPs) collectively as the “Chemosensory-Related Gene families” (CRGs).
Although CRGs have been studied intensively since the 2000s, little information has been gained regarding these genes in arthropods beyond the insects (Hexapoda), until very recently. Thus, the evolutionary history of CRGs throughout the Arthropoda had remained largely unexplored and poorly understood. Emerging data are beginning to suggest that the major CRGs might have expanded, contracted, or become completely lost throughout the course of arthropod evolution (Robertson and Wanner 2006; Peñalva-Arana et al. 2009; Robertson and Kent 2009; Hansson and Stensmyr 2011; Vieira and Rozas 2011; Zhou et al. 2012; Pelosi et al. 2014; Robertson 2015; Saina et al. 2015).
Some hypotheses have posited a link between CRG family expansion and habitat colonizations. In particular, the expansion of the OR gene family had been hypothesized to be associated with the colonization of land by insects (Hexapoda), to enable the detection of volatile compounds in air (Robertson et al. 2003; Peñalva-Arana et al. 2009; Krång et al. 2012). This hypothesis was consistent with the intriguing absence of ORs and OBPs in the water flea Daphnia pulex, belonging to the crustacean lineage (Branchiopoda) that forms a clade with the insects (Peñalva-Arana et al. 2009; Vieira and Rozas 2011) (fig. 1). Nevertheless, there is some debate regarding whether the expansion of the OR gene family was the result of a terrestrial adaptation (Missbach et al. 2014). In addition, prior studies had not sampled the crustaceans outside of the branchiopod/hexapod clade, preventing resolution on whether the ORs and OBPs are absent from the Daphnia lineage alone or instead absent from all crustaceans outside of the insect clade. Also, unresolved is whether independent colonizations of land in the other arthropod subphyla (i.e., Chelicerata and Myriapoda) also coincided with expansions of the OR gene family (Chipman et al. 2014).
| Placozoa | Trichoplax adhaerensa (Robertson 2015; Saina et al. 2015) |
| Cnidaria | Nematostella vectensisb, Hydra magnipapillatac, Acropora milleporad (Robertson 2015; Saina et al. 2015) |
| Mollusca | Aplysia californicae (Croset et al. 2010; Robertson 2015) |
| Nematoda | Caenorhabditis elegansf (Croset et al. 2010; Robertson 2015) |
| Onychophora | Euperipatoides rowellig |
| Arthropoda | |
| Chelicerata | Ixodes scapularish (Gulia-Nuss et al. 2016), Centruroides exilicaudai, Latrodectus hesperusj, Loxosceles reclusak |
| Myriapoda | Strigamia maritimal (Chipman et al. 2014) |
| Pancrustacea | |
| Copepoda | Eurytemora affinism, Tigriopus californicusn |
| Thecostraca | Amphibalanus amphitrite° |
| Malacostraca | Hyalella aztecapp, Penaeus monodonq |
| Anostraca | Artemia franciscanar |
| Cladocera | Daphnia pulexs (Peñalva-Arana et al. 2009; Croset et al. 2010; Vieira and Rozas 2011) |
| Hexapoda | Drosophila melanogastert, Bombyx moriu, Tribolium castaneumv,Apis melliferaw, Acyrthosiphon pisumx, Pediculus humanusy(Robertson and Wanner 2006; McBride and Arguello 2007; Wanner et al. 2007; Engsontia et al. 2008; Tribolium Genome Sequencing Consortium 2008; Wanner and Robertson 2008; Tanaka et al. 2009; Croset et al. 2010; Kirkness et al. 2010; Vieira and Rozas 2011). |
Fig. 1.

Patterns of chemosensory-related gene (CRG) family evolution in the Arthropoda. Gene family gain events (red plus sign) are shown along the branches. Numbers of GR, IR, CSP, OR, and OBP genes for representative species are shown in the right-hand columns. Letters to the right of the columns indicate the taxa used in the analysis, listed below. Green dots on the phylogeny indicate terrestrial colonization of most members of a lineage. Numbers of CRG genes were obtained from genome sequence data, except for four species (Acropora millepora, Amphibalanus amphitrite, Penaeus monodon, and Artemia franciscana: indicated with underline and gray font letters to the right of the columns), for which CRG genes were obtained from transcriptome data. A consensus of the arthropod phylogeny was obtained from von Reumont et al. (2012), Oakley et al. (2013), and Sasaki et al. (2013). Branching resolution among earliest animal lineages were obtained from Parfrey et al. (2010), Moroz et al. (2014), and Whelan et al. (2015). The gray branches indicate the protistan phyla and the gray-dashed branches are the fungal phyla. Numbers of CRG genes obtained through our analyses are indicated by asterisks to the left of the columns above, whereas references are provided (below) for data obtained from other studies. Species shown in the figure above are as follows:
More generally, the evolutionary histories of CRG families and hypotheses regarding which CRG gene families are the most ancient have been gaining some clarity only recently. An earlier hypothesis had posited that the IRs represent the most ancient arthropod chemoreceptors, dating back to the origin of the Protostomia (Croset et al. 2010). In contrast, more recent studies found GRs to be more ancient, originating early in the evolution of metazoans, given their presence in the eumetazoan phylum Placozoa (Trichoplax adhaerens) (Robertson 2015; Saina et al. 2015). In addition, the analysis of evolutionary histories of IR genes has been based mostly on studies of insects, with relatively little investigation of their presence or absence in other arthropod lineages (Croset et al. 2010).
Addressing the hypotheses above, regarding patterns of CRG evolution across the Arthropoda, requires the analysis of multiple members within the subphylum Pancrustacea beyond the insects (Hexapoda), the inclusion of the arthropod subphyla Myriapoda (e.g., centipedes and millipedes) and Chelicerata (e.g., spiders, mites, and scorpions), as well as the inclusion of outgroup phyla. However, until recently, genomic data beyond the hexapod/branchiopod clade (e.g., insects and Daphnia) had been lacking. Very few comparative analyses of CRG evolution had included the subphyla Chelicerata and Myriapoda (Chipman et al. 2014; Robertson 2015), and only a few molecular evolutionary studies of crustacean CRGs had been performed (Peñalva-Arana et al. 2009; Krång et al. 2012; Corey et al. 2013). Within the Pancrustacea, the critical phylogenetic placement of the Copepoda enables the resolution of CRG family gain or loss in the insects (Hexapoda), as they are outside of the Allotriocarida (Hexapoda/Branchiopoda/Remipedia) clade, yet are often found to be the closest sister group to this clade (von Reumont et al. 2012; Oakley et al. 2013; Sasaki et al. 2013; Eyun 2017). Thus, we focused much attention on the Copepoda, in order to explore patterns of CRG gain or loss in close evolutionary proximity to the clade containing the insects.
In addition to the crucial phylogenetic placement of the Copepoda, their chemoreception is inherently interesting from both ecological and evolutionary perspectives. Copepods occupy an enormous range of habitats in the aquatic realm, from freshwater to hypersaline, and shallow pool to deep sea environments (Hardy 1956; Huys and Boxshall 1991; Martin and Davis 2001). They also form the largest biomass of all animals in the world’s oceans, and possibly on the planet (Hardy 1956; Huys and Boxshall 1991; Humes 1994; Verity and Smetacek 1996). Copepods are particularly known to frequently exhibit cases of cryptic speciation, where large genetic distances and reproductive isolation are accompanied by morphological stasis (Burton 1990; Ganz and Burton 1995; Edmands 1999; Lee 2000; Lee and Frost 2002; Goetze 2003; Grishanin et al. 2006; Rynearson et al. 2006; Eyun et al. 2007; Chen and Hare 2011). In the absence of morphological cues and differentiation, it has been hypothesized that speciation in copepods occurs through rapid evolution of chemical sensing (Snell and Morris 1993).
Thus, the goals of this study were to address the hypotheses above on CRG family evolution across the Arthropoda. Our specific goals were to: 1) determine patterns of gains and losses of CRG families across the phylum Arthropoda, 2) infer the evolutionary origins of the arthropod CRG families, and 3) examine sex-specific differences in CRG family expression in copepods.
In this study, we address current hypotheses and examine prior conclusions regarding GR and OR gene family evolution, as well as explore patterns of IR gene family evolution in greater detail. This study addresses the hypotheses using a more comprehensive data set than in prior studies (Croset et al. 2010; Robertson 2015; Saina et al. 2015). We included all three arthropod subphyla (i.e., the Pancrustacea, Myriapoda, and Chelicerata) and a member of the closest related outgroup phylum, the Onychophora (Euperipatoides rowelli), as well as other outgroup phyla. A unique feature of this study is the inclusion of 14 crustacean genomes and transcriptomes. We additionally introduce the high-quality draft genome of the copepod Eurytemora affinis, as the first published report of a comprehensive copepod genome sequence. The inclusion of multiple crustacean taxa greatly enhances our ability to make inferences regarding patterns and timing of CRG evolution in close phylogenetic proximity to the most heavily studied arthropod clade, the insects (Hexapoda). This study is the most comprehensive comparative analysis to date of CRG family evolution across the largest and most speciose metazoan phylum Arthropoda. As such, this study serves as a critical starting point for generating hypotheses on how different CRGs might have expanded and evolved to adapt to diverse ecological niches.
Results
General Characteristics of the Copepod Eurytemora affinis Genome
We sequenced the full genome of the copepod Eurytemora affinis, as copepods provide a critical phylogenetic outgroup data point to the branchiopod/hexapod clade for analyzing patterns of CRG evolution. The E. affinis genome was sequenced as part of the i5K pilot at the Baylor College of Medicine Human Genome Sequencing Center, a pilot project to investigate large-scale genomic sampling of the arthropods and provide a framework for comparative arthropod genomics. Genome sequencing was performed on an inbred line (see Materials and Methods), with a genome size estimated at 0.6–0.7 pg DNA/cell (∼587–685 Mb) based on Feulgen DNA cytophotometry (Rasch et al. 2004). The draft genome assembly is relatively compact at 495 Mb, smaller than the total genome size due to our inability to assemble highly repetitive heterochromatin from short read sequence data. It is larger than the Daphnia pulex genome (∼200 Mb) (Colbourne et al. 2011), a species selected in part for its small genome size in the age of expensive Sanger sequencing. The genome size of E. affinis is on the lower end of the range observed for copepods (0.14–12 pg) (Gregory 2016), and smaller than most crustaceans, where the average genome size of 6.7 pg has slowed the adoption of genome sequencing of these taxa.
The contiguity was below average with a contig N50 of 5.7 kb with a scaffold N50 of 863 kb, giving us confidence for a high-quality automated annotation (see supplementary table S1 for additional statistics and public repository accession numbers, Supplementary Material online). Automated gene model annotation using a Maker 2.2 pipeline customized for arthropods (Cantarel et al. 2008) generated 29,783 gene models. This number is likely an overestimate due to gene model fragmentation across gaps within and between scaffolds, but is somewhat in-line with the 18,440 gene models in D. pulex (PA42) (Ye et al. 2017) and 29,121 gene models in Daphnia magna, relative to the lower number of ∼15,000 for insects. Of 1,977 control genes expected to be present in all arthropods (Simao et al. 2015), 91.5% were identified in the genome assembly and 86.3% were represented in the automated gene model set. Thus, gene families and most genes were present in the assembly and gene set, but the absence of any particular gene from the assembly could be due to the draft nature of the assembly.
Overview of Chemosensory-Related Gene (CRG) Family Evolution
We comprehensively examined gains and losses of CRGs (ORs, GRs, IRs, CSPs, and OBPs), using 33 distinct genomes and transcriptomes across the phylum Arthropoda, as well as multiple metazoan outgroup phyla (Onychophora, Nematoda, Mollusca, Cnideria, Placozoa, Porifera, Ctenophora) and additional fungal and protistan groups (see Materials and Methods; supplementary tables S2–S4, Supplementary Material online). We found fewer chemosensory receptor genes in arthropods (∼12–500; fig. 1), relative to vertebrates (∼1,391 olfactory receptors and >300 vomeronasal receptors in mouse) and nematodes (>1,200 serpentine receptors in Caenorhabditis elegans) (Niimura and Nei 2003; Chen et al. 2005; Bargmann 2006; Robertson and Thomas 2006). While this relatively low number had been known for insects (Nei et al. 2008), we now confirm that this pattern holds generally true across the phylum Arthropoda (fig. 1) (Chipman et al. 2014; Gulia-Nuss et al. 2016).
Our results on patterns of CRG evolution revealed the GRs to be the most ancient of all the eumetazoan CRGs, given the inferred presence of GR or GR-Like genes in the common ancestor between arthropods and the phylum Placozoa (fig. 1; see next section for details). This result was consistent with recent studies (Robertson 2015; Saina et al. 2015) (see Discussion). We also found that GRs are characterized by lineage-specific expansions within the Arthropoda (fig. 2; see below). The IRs date back to at least the emergence of the Protostomes (fig. 1; see below), as found in another study (Croset et al. 2010). This study clarified the evolutionary history of antennal IRs in the Arthropoda (figs. 3 and4; see below) and revealed that the antennal IR76b, previously thought to be insect-specific (Croset et al. 2010), originated prior to the divergence of the insects (fig. 4, see below). Intriguingly, antennal IRs in E. affinis showed higher expression in males than in females, the first such finding for an aquatic animal (fig. 5). These male-biased genes also showed signatures of natural selection (see below). The CSPs were present only in the Arthropoda (figs. 1 and6), as found in another study (Pelosi et al. 2014). Our analysis, which included more pancrustacean taxa and greater sampling of arthropod clades than prior studies, revealed that ORs and OBPs were only present in the Hexapoda (insects), while being absent in other pancrustaceans, other arthropod subphyla, and all outgroup phyla (fig. 1). Our results were consistent with prior studies, while expanding the comparisons with more comprehensive sampling within the Arthropoda (see Discussion).
| Cnideria | Starlet sea anemone Nematostella vectensis (Nvec, green) |
| Mollusca | California sea hare Aplysia californica (Acal, teal) |
| Arthropoda | Branchipoda/Hexapoda |
| Fruit fly Drosophila melanogaster (Dmel, olive) | |
| Waterflea Daphnia pulex (Dpul, blue) | |
| Copepoda (red) | |
| Salmon louse Lepeophtheirus salmonis (Lsal) | |
| Sea louse Caligus rogercressey (Crog) | |
| Freshwater cyclopoid Mesocyclops edax (Meda) | |
| Anchor worm Lernaea cyprinacea (Lcyp) | |
| Tide pool copepod Tigriopus californicus (Tcal) | |
| Asian Pacific copepod Calanus sinicus (Csin) | |
| North Atlantic copepod Calanus finmarchicus (Cfin) | |
| Oceanic shelf copepod Acartia fossae (Afos) | |
| Common estuarine copepod Eurytemora affinis (Eaff) |
Fig. 2.

Phylogeny of the GR gene families from representatives of some major clades within the Arthropoda. Phylogenetic relationships among GR genes of the fruit fly Drosophila melanogaster (Hexapoda, Groups I and II, olive), the waterflea Daphnia pulex (Cladocera, within Branchiopoda, Groups VII–IX, blue), the copepod Eurytemora affinis (Copepoda, Group III, red), the centipede Strigamia maritima (Myriapoda, Group X, magenta), and the black-legged tick Ixodes scapularis (Chelicerata Groups IV–VI, orange). The phylogeny was constructed using maximum likelihood, based on alignments of 1,346 amino acids of GR genes (see Materials and Methods). The numbers at internal branches show bootstrap support values (%) for the maximum-likelihood reconstruction and posterior probabilities (%) for the Bayesian reconstruction. Support values on the major internal branches are shown for values higher than 60%. Groups I–X each represent lineage-specific expansions (of more than three genes) and are supported by > 0.70 posterior probability in the Bayesian reconstruction. The scale bar represents the number of amino acid substitutions per site. See supplementary figure S1, Supplementary Material online, for a more detailed GR amino acid phylogeny using additional taxa.
Fig. 3.

Phylogeny of the iGluR gene families from nine copepod species and four other invertebrate species. All amino acid sequences except for the copepod sequences were taken from Croset et al. (2010). Information on the copepod sequence assemblies are shown in table 1. The phylogeny was constructed using maximum likelihood (see Materials and Methods) based on sequence alignments of 3,211 amino acids. The numbers to the left of the nodes show the bootstrap support values (%) for neighbor-joining and maximum-likelihood reconstructions, and posterior probabilities (%) for the Bayesian analysis, respectively. Support values for the major internal branches are shown only for those higher than 60%. Gene abbreviations for other iGluR members (NMDAR, NMDA receptors; AMPAR, AMPA receptors; KR, Kainate receptors) are adopted from Benton et al. (2009). The NMDAR gene family was used as the outgroup (see Materials and Methods). The inset illustrates a current consensus of the invertebrate phylogeny (Regier et al. 2010; Zwick et al. 2012). Species names were abbreviated according to the following four-letter codes:
Fig. 4.
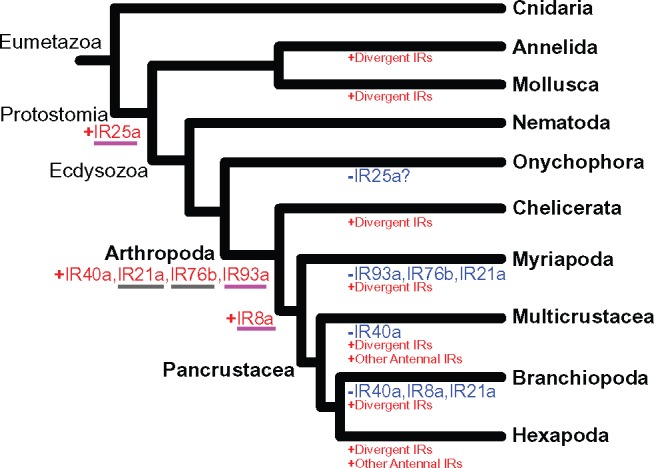
IR gene gains and losses in the Metazoa. IR gene subfamily gains (red color) and losses (blue color) are shown along the branches. The taxa used for this analysis are listed in the Results section (in the section “Origins of Ionotropic Receptors Subfamilies”). The antennal IR genes that show significant differential expression between the sexes in the copepod Eurytemora affinis are underlined in magenta, whereas the IR genes that do not show significant differences are underlined in gray (see fig. 5 and supplementary table S8, Supplementary Material online).
Fig. 5.

Chemosensory-related gene expression levels in males (blue bars) versus females (red bars) of the copepod Eurytemora affinis. Gene expression levels were determined by calculating RPKM values. The average RPKM from two technical replicates were used. Statistical significance of differences in expression levels between male and female samples was analyzed for each gene. Significant P values (<0.001) are marked with an asterisk (*). RPKM and P-values are summarized in the supplementary table S8, Supplementary Material online.
Fig. 6.

Phylogeny of CSPs from 6 copepods and 11 other arthropod species. The phylogeny was constructed using maximum likelihood based on sequence alignments of 402 amino acids. Fourteen CSP sequences from six copepods are included (shown in red). In addition to copepods, 81 CSP sequences are included from 11 representative arthropod species. All amino acid sequences except for the copepod sequences are taken from Vieira and Rozas (2011) and Gu et al. (2012). The asterisks indicate branches with at least one of the phylogenetic reconstruction approaches (maximum-likelihood, neighbor-joining phylogenies, or Bayesian) showing bootstrap values or posterior probabilities greater than 70%. The black arrow on the phylogeny points to the node forming a clade within Pancrustacea, composed of a Daphnia pulex CSP, seven insect CSPs, and copepod CSPs. This clade supports a clear homologous relationship between copepod and insect/branchiopod CSPs. This node is supported by a maximum-likelihood bootstrap value of 62% and a Bayesian posterior probability of 0.78. The following representative species were used in this analysis: the fruit fly Drosophila melanogaster (Diptera, olive), the silkworm moth Bombyx mori (Lepidoptera, pink), the red flour beetle Tribolium castaneum (Coleoptera, brown), the honeybee Apis mellifera (Hymenoptera, dark green), the pea aphid Acyrthosiphon pisum (Hemiptera, cyan), the human body louse Pediculus humanus (Phthiraptera, slate-blue), the waterflea Daphnia pulex (Cladocera, blue), the six copepod species (Copepoda, red), the centipede Strigamia maritima (Myriapoda, magenta), and the black-legged tick Ixodes scapularis (Chelicerata, orange). All other arthropod species are shown in black. The tree is midpoint rooted due to the absence of obvious outgroups. The scale bar represents the number of amino acid substitutions per site.
Origin of Gustatory Receptors (GRs)
Our results place the timing of the origin of GRs to the timing of the most recent common ancestor of the Cnideria/Protostomia clade and the phylum Placozoa (Trichoplax adhaerens) (fig. 1). This timing of the origin of the GRs was based on the presence of GR or GR-like genes in the placozoan T. adhaerens, and the absence of GR gene candidates in the outgroup lineage leading to the animal phylum Porifera (sponge Amphimedon queenslandica) and the more distantly related Ctenophora (comb jelly Mnemiopsis leidyi) (fig. 1). In addition, we did not find GR or GR-like genes in the genomes of any protistan or fungal taxa examined, including members of the protistan phylum Choanozoa (choanoflagellate Monosiga brevicollis), the fungal phyla Ascomycota (Saccharomyces cerevisiae) and Basidiomycota (Sporobolomyces roseus), and the protistan phyla Mycetozoa (slime mold, Dictyostelium purpureum), Percolozoa (amoeboflagellate, Naegleria gruberi), and Metamonada (Giardia intestinalis and Trichomonas vaginalis) (fig. 1; see supplementary table S3 for list of genomes sampled, Supplementary Material online). Although our study was based on sampling of taxa (see supplementary table S4, Supplementary Material online) that was more comprehensive than and distinct from those of two prior studies (Robertson 2015; Saina et al. 2015), our results were consistent with the previous findings.
We found three GR-like genes in the placozoan T. adhaerens, consistent with results from two previous studies (Robertson 2015; Saina et al. 2015). We also identified four GRL genes in the genome of the cnidarian Nematostella vectensis. These genes had been identified previously, two by Saina et al. (2015), NvecGrl1 (KP294348) and NvecGrl2 (KP294349) (located in scaffold_86:815817.816695 and scaffold_91:194748.194002), and two additional genes by Robertson (2015) (jgi|Nemve1|198670 and jgi|Nemve1|214946) found in scaffold_11 (818242.819090) and scaffold_214 (150068.149415). We also found two GR-like genes in a data set of expressed sequence tags of the cnidarian Acropora millepora (fig. 1). On the other hand, our analyses failed to identify GR candidates in another cnidarian genome, that of the polyp hydra, Hydra magnipapillata, consistent with Saina et al. (2015) and Robertson (2015). Additionally, we found three GR fragments (data not shown) in the draft genome of the velvet worm E. rowelli (Onychophora) and GR genes in the Chelicerata (12 GRs in Centruroides exilicauda and 1 GR in Loxosceles reclusa), the Theocostraca (Pancrustacea, one GR in the purple acorn barnacle Amphibalanus amphitrite), and the Copepoda (Pancrustacea, ten GRs in E. affinis and ten GRs in Tigriopus californicus) (fig. 1). Our findings represent the first discovery of GR genes in the Multicrustacea (within the subphylum Pancrustacea) and add to what has been found for other taxa (see fig. 1).
Lineage-Specific Expansions and Contractions of GRs across the Arthropoda
We observed and confirmed the GR gene family to exhibit high levels of lineage-specific gene expansions across the Arthropoda (Chipman et al. 2014; Gulia-Nuss et al. 2016). Our phylogenetic reconstruction suggests that GR genes most likely experienced gene duplications and differentiation following lineage-splitting events, given that we could not resolve orthologous relationships among GR genes from different clades, even among different hexapod orders (fig. 2 and supplementary fig. S1, Supplementary Material online). Based on high-quality full genome sequence data, we found a general pattern of GR gene family expansions in representative members of most major arthropod clades (i.e., Chelicerata, Myriapoda, and Branchiopoda/Hexapoda), but not for the Multicrustacea (e.g., Copepoda and Amphipoda) (figs. 1 and2, and supplementary fig. S1, Supplementary Material online). For instance, based on high-quality genome sequence data, the black-legged tick Ixodes scapularis (Chelicerata) (fig. 2, Groups IV–VI, orange branches), the centipede Strigamia maritima (Myriapoda) (fig. 2, Group X, magenta branches), the waterflea D. pulex (Cladocera, within the Branchiopoda) (fig. 2, Groups VII–IX, blue branches), and the fruit fly D. melanogaster (Hexapoda) (fig. 2, Groups I and II, olive branches) possessed relatively high numbers of GR genes (see Discussion).
In contrast to most arthropod groups (previous paragraph), the multicrustaceans showed a relative lack of a GR gene family expansion (fig. 2, Group III, red branches), typically containing a few or no GR genes within species (fig. 2 and supplementary fig. S1, Supplementary Material online). Based on full genome sequences, we found 10 GR genes in the copepod E. affinis, 10 GR genes in the copepod T. californicus, and 0 GR genes in the amphipod Hyalella azteca (figs. 1 and2). Likewise, based on transcriptome data of additional multicrustacean species (including Thecostraca and Eumalacostraca), which are not fully reliable as GR genes might not be expressed or data sets might be incomplete, we found only one GR gene in the purple acorn barnacle A. amphitrite and two GR genes in the copepod anchor worm Lernaea cyprinacea (supplementary table S5 and file S1, Supplementary Material online). Determining whether the low numbers of GRs are specific to the multicrustaceans, or are also characteristic of other crustacean lineages (such as the Ostracoda), requires further investigation.
Low Homology among GR Gene Candidates
GR sequences share extremely low sequence similarity, even among paralogs within a species and among GR genes of insect species (Robertson et al. 2003; Saina et al. 2015). For example, the amino acid sequence identity among D. melanogaster GR proteins alone drops to as low as 8% (Robertson et al. 2003). Also, there are absolutely no conserved domains among insect GR protein sequences. Because of these characteristics of GRs, homologous relationships are extremely difficult to infer for this protein family. In order to overcome this difficulty, we undertook several analyses (see Materials and Methods). First, we explored the positions of introns, because many intron positions are conserved over extremely long evolutionary time spans (Rogozin et al. 2003). We found that two intron positions were shared even among the highly divergent GR genes of Arthropoda, Cnidarian, and Placozoa (supplementary fig. S2, Supplementary Material online). One intron position (indicated by a pink triangle, supplementary fig. S2, Supplementary Material online) was shared only between Nematostella vectensis Grl1 (NvecGrl1) and Trichoplax adhaerens Grl3 (TadhGrl3), but was absent in arthropods. This finding was consistent with the observation that the ancestral introns have generally been lost in arthropods (Rogozin et al. 2003). In addition, sequence homology was supported by codon phases (supplementary fig. S2, Supplementary Material online). For instance, in the supplementary figure S2, Supplementary Material online, the first matching intron position (indicated by an orange arrow and an asterisk) has phase 0 in all GR sequences except for TadhGrl3, which has phase 2. In this position of TadhGrl3, a non-GT-AG intron was found, indicating either a noncanonical intron or more commonly an error.
In our second approach, we analyzed the domain composition of putative N. vectensis and T. adhaerens GR-like genes to computationally infer their protein family classification (supplementary table S6, Supplementary Material online). All GR-like proteins from N. vectensis and T. adhaerens were related to the GR family or Trehalose receptor family (supplementary table S6, Supplementary Material online). Finally, we confirmed that these GR-like proteins had relatively high sequence similarity with insect GRs (> 31.5% among five representative GRs by local sequence similarity, E-values < 1.2 × 10−3) and that reciprocal blastp results were also supported by the top hit to previously known insect GRs (data not shown). These analyses provided strong support for the identity of the GR gene homologs.
Origins of Ionotropic Receptor (IR) Subfamilies, Particularly the Antennal IRs
Based on localization of gene expression in insects, IRs had been classified into two groups, namely, conserved “antennal IRs” and species-specific “divergent IRs” (Croset et al. 2010) (fig. 3). We found six antennal IR subfamilies present in the Arthropoda, with one (IR25a) stemming back to the origin of the Protostomia, as found by Croset et al. (2010), and five exclusive to the Arthropoda (i.e., IR40a, IR21a, IR76b, IR93a, IR8a). Until recently, four antennal IRs, IR40a, IR21a, IR76b, and IR8a, were thought to be insect-specific (Croset et al. 2010), but recent studies (see Discussion) and our analysis (see below) found many instances of these IR gene subfamilies occurring outside of the insect clade (fig. 4) (Chipman et al. 2014; Groh-Lunow et al. 2015; Gulia-Nuss et al. 2016; Vizueta et al. 2017).
Of the arthropod-specific antennal IR gene subfamilies, our results indicate that the antennal IR93a and IR76b are the most widespread among arthropod lineages, as they were absent only in the Myriapoda (fig. 4), which is represented by only one species (supplementary table S4, Supplementary Material online). We found orthologs of IR93a in the genomes of four chelicerates (I. scapularis, C. exilicauda, Latrodectus hesperus, and L. reclusa), five copepods (Multicrustacea) (Caligus rogercresseyi, T. californicus, Calanus sinicus, Acartia fossae, and E. affinis), and two branchiopods (D. pulex and Artemia franciscana), but absent from the velvet worm Euperipatoides rowelli (phylum Onychophora, immediate outgroup phylum to the Arthropoda) and also absent from all other outgroup phyla.
We are the first to discover orthologs of antennal IR76b occurring in arthropod taxa outside of the insect clade (fig. 4). This antennal IR was previously thought to be insect-specific (Croset et al. 2010). We found orthologs of IR76b in the genomes of the chelicerate bark scorpion C. exilicauda and in two copepod genomes, of E. affinis and L. cyprinacea, and in the genome of the branchipod Daphnia pulex. We confirmed that D. pulex IR304 (EFX75437.1) is the ortholog of IR76b of D. melanogaster. IR76b was absent from the genome of the velvet worm Euperipatoides rowelli (phylum Onychophora) and those of other phyla outside of arthropods.
Of the arthropod-specific antennal IRs, we found that the distributions of IR40a, IR21a, and IR8a were less widespread within the Arthropoda, but still occurring outside of the insect clade (fig. 4). With respect to IR40a, we found an IR40a ortholog (known as SmarIR49) present in the genomes of the myriapod centipede S. maritima (fig. 4). Also, a prior study did find IR40a in a chelicerate (the hunter spider Dysdera silvatica) (Vizueta et al. 2017) (see Discussion). However, our analyses failed to identify IR40a in the Onychophoran velvet worm, chelicerates, and all 14 crustacean species including the branchiopods (D. pulex and A. franciscana) (listed in the supplementary tables S2 and S4, Supplementary Material online).
We found the IR21a genes to be present only in copepods (Caligus rogercresseyi and E. affinis) and hexapods (insects). However, a prior study did find IR21a in present in a chelicerate (the hunter spider D. silvatica) (Vizueta et al. 2017) (see Discussion). Orthologs of IR21a were absent from the genomes of a species of the outgroup phylum Onychophora (velvet worm E. rowelli), four species of Chelicerata (I. scapularis, C. exilicauda, L. hesperus, and L. reclusa), one species of Myriapoda (centipede S. maritima), and two branchiopod species (D. pulex and A. franciscana).
We found IR8a orthologs to be present in the Copepoda (Lepeophtheirus salmonis, C. rogercresseyi, L. cyprinacea, T. californicus, and E. affinis) (supplementary table S7, Supplementary Material online) and also in the Myriapoda (S. maritima) (fig. 4). However, IR8a orthologs were absent from the Chelicerata (D. silvatica, I. scapularis, C. exilicauda, L. hesperus, and L. reclusa) (Vizueta et al. 2017), two branchiopod species (D. pulex and A. franciscana), and the outgroup phylum Onychophora (E. rowelli) (fig. 4).
For the nine copepod species examined (table 1 and supplementary table S2, Supplementary Material online), we identified 33 IRs from seven of the copepod species (fig. 3 and supplementary table S7, Supplementary Material online). Based on sequence similarity and phylogenetic analysis, we were able to classify the 33 copepod IRs into five antennal IR subfamilies (IR25a, IR76b, IR93a, IR8a, and IR21a) and divergent IRs (fig. 3 and supplementary table S7, Supplementary Material online). Interestingly, we observed duplicated IR genes in several copepod species (fig. 3 and supplementary table S7, Supplementary Material online). For instance, two IR8a genes were identified in L. cyprinacea and three in T. californicus, and two IR93a genes were identified each in C. sinicus and E. affinis. These genes (IR93a and IR8a) had not previously been found as duplicated genes in arthropods (Croset et al. 2010). In this study, we were unable to determine the details of the origins of divergent IRs, because divergent IRs showed no one-to-one orthology among Diptera species and exhibited lineage-specific gene duplications (Croset et al. 2010; Chipman et al. 2014) (fig. 4).
Table 1.
Summary of Next-Generation Sequencing Assemblies for Nine Copepod and Three Additional Crustacean Species Used in This Study.
| Order and Family | Species Names | Source | Assembliesa (range; N50) |
|---|---|---|---|
| Siphonostomatoida | |||
| Caligidae | Lepeophtheirus salmonis | Genome | 10,615 (300–827 bp; 366) |
| Caligidae | Caligus rogercresseyi | Transcriptome | 76,788 (301–9,505 bp; 1,414) |
| Cyclopoida | |||
| Cyclopidae | Mesocyclops edax | Genome | 25,442 (300–1,275 bp; 401) |
| Lernaeidae | Lernaea cyprinacea | Transcriptome | 271,824 (301–22,442 bp; 2,266) |
| Harpacticoida | |||
| Harpacticidae | Tigriopus californicus | Genome and Transcriptome | 60,840 (301–8,614 bp; 1,510) |
| Calanoida | |||
| Calanidae | Calanus sinicus | Transcriptome | 29,458 (301–3,923 bp; 513) |
| Calanidae | Calanus finmarchicus | Genome | 8,629 (300–1,067 bp; 347) |
| Acartiidae | Acartia fossae | Transcriptome | 100,383 (301–8,174 bp; 769) |
| Temoridae | Eurytemora affinis | Genome and Transcriptome | 88,104 (301–26,685 bp; 2,), 6,899 (604–7,289,689 bp; 862,645) |
| Branchiopoda, Anostraca | |||
| Artemiidae | Artemia franciscana | Transcriptome | 59,654 (301–48,245 bp; 1,747) |
| Malacostraca, Decapoda | |||
| Penaeidae | Penaeus monodon | Transcriptome | 94,814 (301–17,471 bp; 2,473) |
| Thecostraca, Sessilia | |||
| Balanidae | Amphibalanus amphitrite | Transcriptome | 80,455 (301–8,040 bp; 857) |
The number of contigs (> 300 bp). The NCBI accession numbers and sequencing platforms were summarized in the supplementary tables S1 and S2, Supplementary Material online. The transcriptomes and the genomes were assembled using the software package Trinity and Velvet, respectively (more details in Materials and Methods).
Among the 14 crustacean species examined (table 1 and supplementary table S4, Supplementary Material online), we were unable to find IRs in two copepod species Mesocyclops edax and Calanus finmarchicus. The absence of IRs in these two species might have arisen from very low coverage of whole-genome sequencing. Mesocyclops edax (accession numbers SRX246444 and SRX246445) and C. finmarchicus (accession number SRX456026) were sequenced only to ∼0.22 and ∼0.55 gigabases (∼0.4 and ∼ 3 million reads) by 454 GS FLX Titanium and the Ion Personal Genome Machine sequencer, respectively (supplementary table S2, Supplementary Material online). The N50 length of de novo assemblies in M. edax and C. finmarchicus was shorter than that of other copepod assemblies (401 and 347 bp, respectively; table 1). Therefore, the depth of coverage of sequencing might not have been sufficient to detect any IR sequences.
Sex Differences in Expression Levels of IRs, CSPs, and GRs in the Copepoda
In order to compare expression levels of the GR, IR, and CSP genes between the sexes in the copepod E. affinis, we mapped Illumina RNA-Seq reads to each gene and normalized for sequencing depth and gene length by presenting them in RPKM (reads per kilobase per million mapped reads) values. Most notably, three of the antennal IR genes (EaffIR8a, EaffIR25a, and EaffIR93-1) showed significantly greater expression in the male RNA-Seq samples, relative to the female samples (P < 0.001) (fig. 5 and supplementary table S8, Supplementary Material online). This was the first study to discover IRs with male-biased expression in an aquatic animal.
In contrast to the significant sex-specific differences in expression of the three antennal IR genes, we found no sex-specific difference in five representative housekeeping genes of E. affinis (Cyclophilin-33, Actin 42A, Heat shock protein 83, Glyceraldehyde 3 phosphate dehydrogenase 1, and Ribosomal protein L32) (supplementary table S9, Supplementary Material online). The levels of expression were similar between the sexes for these housekeeping genes, in contrast to the large sex differences in expression we found for three antennal IR genes (EaffIR8a, EaffIR25a, and EaffIR93-1) and one CSP gene (EaffCSP1). Although we had only two replicate samples for each sex, we included ∼220 individual copepods per replicate, and found very low variance between the replicates for both the antennal IRs and CSP gene, as well as for the five housekeeping genes (see standard deviations in the supplementary tables S8 and S9, Supplementary Material online).
In contrast to the male-biased expression of some antennal IR genes, the expression of the E. affinis CSP gene EaffCSP1 was ∼30-fold higher in female RNA-Seq samples (in RPKM reads) than in male samples (P < 0.0001 by edgeR and Prob. = 0.95% by NOISeq; fig. 5 and supplementary table S8, Supplementary Material online) (see Discussion). The other E. affinis CSP genes showed slightly higher, but not significant (P > 0.1643), expression levels in male than in female samples (fig. 5, supplementary table S8, Supplementary Material online).
Six E. affinis GR genes showed no difference in expression between the sexes (fig. 5 and supplementary table S8, Supplementary Material online). In the contrast to relatively high expression levels in antennal IRs, we found that crustacean GRs were generally expressed at very low levels, except for the copepod T. californicus GR7 (TcalGR7) and the barnacle A. amphitrite GR1 (AampGR1) (supplementary tables S5 and S8, Supplementary Material online). In E. affinis, the RPKM values of all six E. affinis GRs from all four samples were lower than 1 (fig. 5 and supplementary table S8, Supplementary Material online).
Signatures of Selection in Antennal IR Genes
When we tested for signatures of natural selection in antennal IR genes, we found significantly stronger signatures of purifying selection in the IR genes showing elevated expression in E. affinis males, relative to IR genes that showed no sex differences in expression (see fig. 5, supplementary fig. S3, Supplementary Material online). Based on expression levels of the antennal IR genes, we classified them into two groups (fig. 5), namely “male-biased expression IRs” (IR25a, IR93a-1, and IR8a), which displayed significantly elevated expression in males, and “unbiased expression IRs” (IR76b and IR21a), which showed no difference in expression between the sexes.
To compare patterns of molecular evolution in the two sets of IR genes, we used the branch model in codeml in the software package PAML (Yang 2007). All the male-biased expressed IR genes (IR25a, IR93a-1, and IR8a) showed significantly stronger signatures of purifying selection relative to the unbiased IR genes (IR76b and IR21a) (supplementary fig. S3, Supplementary Material online). When comparing the average ω (the ratio of nonsynonymous to synonymous substitutions, ω or dN/dS) between the two groups, they both showed signatures of purifying selection (dN/dS < 1) (supplementary fig. S3, Supplementary Material online). However, the ω (dN/dS) of unbiased IRs (ω = 0.0249) was 1.9 times higher than that of the male-biased IRs (ω = 0.0131), and the difference was significant (P = 0.0387; supplementary fig. S3, Supplementary Material online). This lower value of ω (dN/dS) in male-biased IRs indicated that purifying selection has acted more strongly in these genes.
Chemosensory Proteins (CSPs), a Class of CRGs Unique to the Arthropoda
Our results indicated that CSPs are an arthropod-specific gene family that emerged after the divergence between the phyla Arthropoda and Onychophora (698.5 Ma) (fig. 1). CSPs were found in all arthropod taxa we examined (fig. 1 and 6), except for the transcriptome assembly of the barnacle A. amphitrite (Thecostraca) and the genome sequence of the brown recluse spider L. reclusa (Arachnida). In contrast, CSPs were absent in the draft genome of the velvet worm E. rowelli, a member of the outgroup phylum Onychophora, and all other nonarthropod genomes (fig. 1).
CSP gene numbers tended to be low within arthropod genomes, relative to other arthropod CRG families (fig. 1). Within crustaceans, we identified 14 CSPs in six copepod species and seven CSPs in three other crustacean species (Hyalella azteca, Penaeus monodon, and Artemia franciscana) (fig. 1 and supplementary table S10, Supplementary Material online). Our phylogenetic analyses of CSPs showed that subgroups could not be resolved for most of the major nodes due to the low bootstrap values (below 50%) (fig. 6). This result was reflected in the low levels of sequence similarity among all arthropod CSPs (as low as 15.9% among four Drosophila CSP proteins) and the short sequence lengths of CSPs (average length of ∼127 amino acid residues). Our phylogenetic analysis revealed that CSPs from all six copepod species formed a well-supported monophyletic clade (fig. 6, red branches). The copepod CSPs formed a larger clade with a D. pulex CSP and seven insect CSPs (indicated by the arrow in fig. 6, and supported by Bayesian posterior probability of 0.78 and maximum-likelihood bootstrap value of 62%), supporting homology between them.
We found that all arthropod CSPs we examined, including those of D. pulex (Cladocera), S. maritima (Myriapoda), and three chelicerates (I. scapularis, C. exilicauda, and L. hesperus), contained a highly conserved four cysteine motif that is found in insects (Forêt et al. 2007; Liu et al. 2012). Interestingly, copepod CSPs contained this motif (CX6–7CX16–19CX3–4C) and two additional cysteines (supplementary fig. S4, Supplementary Material online). Although this motif in copepods was conserved, it was slightly less conserved than that of insects (CX6CX6–18CX2C) (supplementary fig. S4, Supplementary Material online).
Protein Structural Homology-Modeling and a Potential Conserved Role of IRs and CSPs
To understand the spatial distribution of the ligand-binding amino acid residues, we performed homology-modeling of copepod IR25a and CSP proteins (see Materials and Methods). In IR25a, three ligand-binding amino acid residues (corresponding to the positions 489R, 654A, and 739D in T. californicus IR25a) were proposed (Benton et al. 2009) (supplementary fig. S5, Supplementary Material online). These three potential ligand-binding amino acid residues were located in the extracellular domain, which might play critical roles in ligand recognition (supplementary fig. S6, Supplementary Material online). Furthermore, the potential ligand-binding amino acid residues that we found are identical to those of D. melanogaster (DmelIR25a, ADU79032.1), the waterflea D. pulex (DpulIR25a, EFX86214), and the mollusc Aplysia californica (AcalIR25a, XP_005102425.1) (supplementary fig. S5, Supplementary Material online).
The predicted 3D structural model we constructed for the copepod E. affinis CSP2 protein (EaffCSP2) comprised six α-helices and two pairs of disulphide bridges (supplementary fig. S7, Supplementary Material online). This 3D model was concordant with the X-ray structure of the CSP protein from the cabbage moth (Mamestra brassicae) MbraCSPA6, which appears in a globular shape composed of six amphiphatic α-helices that surround an internal hydrophobic binding pocket (Campanacci et al. 2003). Also, we found that all copepod CSPs, except for one incomplete CSP (LsalCSP: 76 aa), possess the typical six α-helices from the sequence-based secondary structure prediction, but do not have ancient 5-helical structure in arthropods (supplementary table S11, Supplementary Material online) (Kulmuni and Havukainen 2013). Based on the presence of a conserved four-cysteine motif and protein structure similarity, copepod CSPs might have similar functions to those of insects (fig. 6 and supplementary figs. S4 and S7, Supplementary Material online).
Discussion
This study provides the most comprehensive analysis to date of CRG family evolution of the Arthropoda, as well as of some outgroup animal phyla. Our phylogenetic and molecular evolutionary analyses offer a more lucid and comprehensive view of the evolutionary histories of the arthropod CRGs by including more in-depth sampling of arthropod and related taxa. Our study included all the major subphyla within the Arthropoda and representatives from a range of metazoan and protistan phyla. Most notably, this study was the first to include multiple crustacean genomes outside of the branchiopod/insect clade, allowing more detailed inference of evolutionary patterns proximate to the insects. Thus, this more comprehensive analysis provided the strongest case thus far to infer that the ORs and OBPs are unique to insects (Hexapoda) and that CSPs are specific to the Arthropoda, and clarified the evolutionary histories of antennal IR subfamilies (see below). Moreover, we gained insights into general principles governing patterns of multigene family evolution of the CRGs (see below on “Birth-and-Death Model of Multigene Family Evolution”).
Gustatory Receptors (GRs) Are the Most Ancient of the Arthropod CRG Families
Our study confirmed that GRs arose early in the course of metazoan evolution and are the most ancient of the CRGs found in arthropods. Our results revealed the presence of GRs in the metazoan phyla Placozoa and Cnidaria, but not in the phyla Porifera and Ctenophora, or in fungal and protistan phyla (fig. 1). Given that recent phylogenetic studies indicate that lineages leading to the phyla Ctenophora and Porifera branched earlier during metazoan evolution (fig. 1) (Moroz et al. 2014; Whelan et al. 2015), we can infer that the GRs evolved after the emergence of metazoans (850–550 Ma) and during the early stages of animal evolution (fig. 1).
Our finding that places the origin of GRs at the early stages of animal evolution was consistent with results from recent studies (Robertson 2015; Saina et al. 2015). Our results, along with those of Saina et al. (2015) and Robertson (2015), placed the origin of GRs earlier than prior studies, which had placed the origin of GRs dating back either to the Cnideria (Nordström et al. 2011) or to the Ecdysozoa (Robertson et al. 2003; Croset et al. 2010). Our study sampled multiple chelicerates and crustaceans, an immediate outgroup phylum Onychophora, and other basally-branching phyla (including single-cell eukaryotes), making the placement of the evolutionary origins of CRG gene families more certain. This study included five protists and two fungi (supplementary table S3, Supplementary Material online), whereas Robertson (2015) examined two protist species (choanoflagellates Monosiga brevicollis and Salpingoeca rosetta). Our sampling of protists and fungi was similar to that of Saina et al. (2015), but our study included additional invertebrate animal phyla (fig. 1 and supplementary table S4, Supplementary Material online).
The gustatory roles of GRs in noninsect arthropod taxa are poorly understood and require functional studies. In insect models, GRs are known to be typically expressed at low levels in only a few gustatory or olfactory sensory neurons (Wang et al. 2004; Thorne and Amrein 2008). Thus, the low expression levels of E. affinis GRs we found (fig. 5) were consistent with the low levels of expression found in insect GRs. Some Drosophila GR genes are known to be involved in proprioception, hygroreception, light sensing, and other sensory modalities (Thorne and Amrein 2008). The functional roles of GR (or GR-like) genes from the placozoan Trichoplax and cnidarian Nematostella as GRs are still inconclusive, as they have not been confirmed to have obvious chemosensory roles. Our computational protein family classifications strongly support the inference that Trichoplax and Nematostella GRs are homologous to those of arthropods (supplementary table S6, Supplementary Material online). Interestingly, the cnidarian homolog to the insect GR gene, NvecGrl1 (KP294348) in Nematostella, has been found to play a role in early developmental body patterning, rather than in external chemosensation (Saina et al. 2015).
Evolutionary Origins of Ionotropic Receptors (IRs)
The IRs had previously been hypothesized to be most ancient of the arthropod CRGs, dating back to the Protostomia, based on their presence in arthropods, nematodes, and molluscs, but absence in the basally branching metazoan phyla, such as Cnidaria, Placozoa, and Porifera (Croset et al. 2010). Consistent with Croset et al. (2010), our analysis also found IRs present in the protostomes, including arthropods, an onychophoran (velvet worm Euperipatoides rowelli) and a mollusc (California sea slug Aplysia californica), and absent in the basally branching metazoan phyla outside of the protostomes (figs. 1, 3, and4). In contrast to Croset et al.’s (2010) postulation, however, the evolutionary history of IRs is considerably more recent than that of GRs, given that GRs have since been found in several basally branching metazoan phyla (see previous section; fig. 1).
Until recently, only the antennal IRs IR25a and IR93a were thought to occur outside of the insect clade, whereas four others (i.e., IR40a, IR21a, IR76b, and IR8a) were considered to be insect specific (Croset et al. 2010). In addition, more recent studies also have found IR25a and IR93a in the Caribbean hermit crab Coenobita clypeatus (Pancrustacea) (Groh et al. 2014; Groh-Lunow et al. 2015) and in the spider mite Tetranychus urticae (Chelicerata) (Ngoc et al. 2016), and IR93a in the tick Ixodes scapularis (Chelicerata) (Gulia-Nuss et al. 2016). However, recent studies have also uncovered three of the “insect-specific” antennal IRs outside the insect clade, namely IR8a and IR40a in the centipede Strigamia maritima (Myriapoda) (Chipman et al. 2014) and IR21a and IR40a in the hunter spider D. silvatica (Vizueta et al. 2017). With the inclusion of our study we now know that none of the six arthropod antennal IRs are unique to insects (see next paragraph).
Our more comprehensive analysis, including 14 crustacean taxa, revealed patterns of gains and losses of antennal IR subfamilies across the arthropod clades (fig. 4). This study discovered an additional antennal IR gene subfamily occurring outside of the insect (Hexapoda) clade, namely IR76b, which previously had been considered insect-specific (Croset et al. 2010). Our finding of IR76b in the genomes of the chelicerate bark scorpion C. exilicauda and in two copepods, E. affinis and L. cyprinacea, but absent in the velvet worm or some other phyla outside of arthropods, revealed this antennal IR to be more widespread within the Arthropoda than previously thought (fig. 4).
Our results suggest that IR40a emerged in the common ancestor of arthropods, but was subsequently lost from the genomes of some chelicerates (C. exilicauda and L. reclusa) and from all 14 crustacean species we examined, including the multicrustaceans and branchiopods (fig. 4). The insect clade is nested within pancrustaceans, yet they do possess IR40a (fig. 4). Likewise, our finding of IR21a present in copepods and insects and the prior finding of this IR subfamily in a spider (Chelicerata) (Vizueta et al. 2017) suggest that IR21a arose in the common ancestor of arthropods, but was lost in myriapods and branchiopods (fig. 4), although sampling in the myriapods is scant. However, the absence of this gene in two species of branchiopods (D. pulex and A. franciscana) suggests a loss in this clade.
Likewise, we also found IR8a orthologs occurring outside the insect clade, in the Copepoda (see Results; fig. 4), and previous studies found this gene present in the genome of the centipede (Myriapoda) Strigamia maritima (known as SmarIR8a) (Chipman et al. 2014). This antennal IR8a gene was absent in the Onychophora (E. rowelli), five chelicerate species including the spider (Chelicerata) D. silvatica (Vizueta et al. 2017), and two branchiopod species (see Results). Our results suggest that IR8a arose in the myriapod and pancrustacean lineages after their split from the chelicerates, but was subsequently lost in the branchiopods (fig. 4).
IRs of arthropods have been found to be associated with a variety of sensory functions, including taste, olfaction, thermosensation, and hygrosensation (Benton et al. 2009; Croset et al. 2010; Abuin et al. 2011; Zhang et al. 2013; Stewart et al. 2015; Knecht et al. 2016). For example, Knecht et al. (2016) demonstrated that IR93a/IR25a mediates thermosensation and hygrosensation and IR21a/IR25a responds to cool temperatures. In addition, IR76b was found to be expressed in gustatory neurons of D. melanogaster, implicating this IR group in taste detection (Zhang et al. 2013). Antennal IR25a and IR93 have been found to be expressed in the olfactory neurons of antennules of the terrestrial hermit crab Coenobita clypeatus (Pancrustacea) (Groh-Lunow et al. 2015). For the spider Dysdera silvatica (Chelicerata), a homolog of the antennal IR25a/IR8a protein family was found to be overexpressed in the first pair of legs and the palps, which are thought to be olfactory appendages in spiders (Vizueta et al. 2017). These results suggest that some IRs mediate olfactory signaling in a wide range of arthropods. Furthermore, the function of the antennal IR84a might be related to male courtship behavior in D. melanogaster (Grosjean et al. 2011). However, elucidating the functional roles of IRs is still in the very early stages of discovery, and much more remains to be discovered.
Ionotropic Receptors (IRs) Mediating Copepod Chemodetection during Mating?
Examining differences in CRG gene expression profiles between males and females could provide clues regarding the roles of chemical perception in mate-searching. However, few studies have elucidated the molecular mechanisms linking specific genes to specific sexual behaviors (Kopp et al. 2008; Zhou et al. 2009). In D. melanogaster, the expression of OR, GR, and OBP genes is more extensive in males than in females, but other receptors (4 GRs and 12 ORs) show altered expression in females after mating (Zhou et al. 2009). In this study, several intriguing patterns emerged regarding the expression and incidence of the antennal IRs in copepods, suggestive of a role in mating. In particular, we found that three antennal IR genes (IR8a, IR25a, IR93a-1) showed significantly greater expression in males of the copepod E. affinis, relative to females (fig. 5 and supplementary table S8, Supplementary Material online). In contrast, expression levels of all six GR genes in E. affinis showed no difference between the sexes (supplementary table S8, Supplementary Material online). Our findings are notable in being the first to find sex-specific differences in expression of CRGs in an aquatic organism.
Interestingly, two of the antennal IR genes that exhibited male-biased expression (IR8a and IR93a) have also experienced gene duplications in several copepod species (supplementary table S7, Supplementary Material online). These gene duplicates of male-biased antennal IRs might serve to increase expression of antennal IR proteins even further. The duplications of IR8a and IR93a we found in copepods are notable, given that the IR93a and IR8a subfamilies have generally not been found as duplicated genes in arthropods (Croset et al. 2010).
Most notably, the same antennal IR genes showing male-biased expression (fig. 5, IR8a, IR25a, IR93a-1) also exhibited stronger purifying selection than the unbiased IR genes (supplementary fig. S3, Supplementary Material online). This result suggests that the antennal IR genes showing elevated expression in males are subjected to greater functional evolutionary constraints. Such functional conservation is consistent with our protein structure model of the copepod T. californicus IR25a, where the potential ligand-binding amino acid residues were found to be identical to those of D. melanogaster, D. pulex, and the mollusc A. californica (see Results; supplementary fig. S6, Supplementary Material online). This result suggests that ligand-binding functions of IR25a are conserved across protostomian species (Benton et al. 2009; Liang et al. 2016). Whether these conserved ligand-binding regions serve an important role in male behavior or other functions would be worth investigating.
Our results, as well as results from other studies, suggest that antennal IRs might have functions related to the chemically mediated mate-recognition behavior observed in male copepods. For instance, in the fruit fly D. melanogaster, mutational knockdown of the antennal IR84a markedly reduces male courtship behavior (Grosjean et al. 2011). In three Drosophila sibling species, IR genes are differentially expressed among species and between the sexes (Shiao et al. 2015). IR76a shows significantly higher expression in female D. simulans, but no significant difference between the sexes in D. melanogaster and D. sechellia (Shiao et al. 2015). Also, IR25a shows slightly greater expression in the females than males in all three Drosophila sibling species (supplementary table S4 in Shiao et al. 2015). This result differs from ours, as we found no antennal IR gene where female expression was significantly higher than that of males (fig. 5). Interestingly, in the hover fly Scaeva pyrastri, only one IR gene (SpyrIR84a) exhibits significant sex differences in expression, with male-biased expression in the antennae (Li et al. 2016).
The male-biased elevated antennal IR expression we found (fig. 5) might possibly be localized in the antennal tissue, and might be involved in functions related to mating. We speculate that the expression of these antennal IRs is localized in the antennae based on anatomical studies of this species, where chemosensory palps are localized heavily in the antennae, especially of the male copepod (Katona 1973; Griffiths and Frost 1976; Snell and Morris 1993). IR25a, which we found to be highly expressed in males (fig. 5), was also found localized in olfactory organs of a hermit crab (Groh-Lunow et al. 2015) and a spider (Vizueta et al. 2017).
In copepods, studies have shown evidence of chemosensation by males during initial mating, such as the detection and tracking of females from a distance (Gauld 1957; Katona 1973; Friedman and Strickler 1975; Snell and Morris 1993; Doall et al. 1998; Heuch et al. 2007; Yen et al. 2011). During mating, the male copepod grips the female with his first antenna (see supplementary movies S1 and S2, Supplementary Material online) (Katona 1973; Snell and Morris 1993), consistent with the potential importance of antennal IRs in mating. The possible roles of antennal IRs in mediating the mating behavior of males might have imposed functional evolutionary constraints, possibly imposed by coevolution between female ligand/pheromone and male IRs. Such coevolutionary constraints might be reflected in the signatures of purifying selection we found in the male-biased antennal IR genes (supplementary fig. S3, Supplementary Material online). Elucidating the actual functions of these male-biased antennal IRs, and whether they are localized in the copepod antennae and are involved in mating, requires further investigation.
Chemosensory Proteins (CSPs) Occur in the Arthropoda Only
Our analysis revealed that CSPs are unique to the phylum Arthropoda, and are present in all the major arthropod lineages, including in the chelicerates, myriapods, and pancrustaceans (crustaceans and insects) (fig. 1). Our results were consistent with a prior study that found CSPs only in arthropods (Vieira and Rozas 2011; Pelosi et al. 2014). However, our study differed from this prior study in that we included 14 crustacean taxa beyond the Branchiopoda/Hexapoda (Allotriocarida) clade (table 1 and supplementary tables S2 and S4, Supplementary Material online), and also incorporated many additional invertebrate animal phyla (fig. 1; supplementary table S4, Supplementary Material online), making the conclusion more robust. Given that we did find CSP genes in all the major crustacean taxa examined, the widespread occurrence of CSPs across the Arthropoda is more strongly substantiated. Also, the lack of CSPs in the other invertebrate phyla (fig. 1) strengthened the conclusion that CSPs occur in arthropods only.
Insect CSP genes have been linked to a variety of feeding, mating, and other behaviors (Gu et al. 2012; Liu et al. 2012; Pelosi et al. 2014). For example, in the Oriental migratory locust Locusta migratoria manilensis, the CSP gene LmigCSP91 was highly expressed only in adult male testicles and adult female accessory glands, but was absent in male accessory glands and ovaries, as well as in sensory organs (Zhou et al. 2013). In the tsetse fly, Glossina morsitans morsitans, GmmCSP2 was proposed to be associated with female host-seeking behavior, because this gene was mainly expressed in the female antennae and their transcript levels increased markedly after a blood meal (Liu et al. 2012). In addition, in the alfalfa plant bug, Adelphocoris lineolatus, three antennae-biased CSPs might mediate host plant recognition (Gu et al. 2012). These genes showed higher expression levels in the antennae than in the head, legs, and wings.
Interestingly, the copepod E. affinis CSP gene EaffCSP1 showed significantly higher expression in female RNA-Seq samples relative to male samples (P < 0.0001 by edgeR and Prob. = 0.95% by NOISeq) (fig. 5). Based on this pattern, we speculate that the EaffCSP1 gene might be involved in mate recognition. It would be worth exploring the functions of this gene in future studies, especially with respect to its role in mating behavior and interaction with sex pheromone compounds.
Odorant Receptors (ORs) and Odorant Binding Proteins (OBPs) Are in Insects Only
We found ORs and OBPs present only in the insects (Hexapoda), and completely lacking in all other arthropod taxa, including the nonhexapod pancrustaceans, chelicerates and myriapods (fig. 1). Our study more conclusively revealed that ORs and OBPs are specific to the insects alone (fig. 1), given that our analysis was the first to examine genomes of multiple crustacean taxa outside of the Branchiopoda/Hexapoda clade, including the genomes and transcriptomes of 13 crustacean species (supplementary table S4, Supplementary Material online). This inclusion of multiple crustacean taxa was critically important for discerning the uniqueness of ORs and OBPs to the insects, because the insects are nested within the pancrustacean clade (von Reumont et al. 2012; Oakley et al. 2013; Sasaki et al. 2013). With our more intensive sampling within the Arthropoda and of outgroup phyla (fig. 1 and supplementary tables S2–S4, Supplementary Material online), our study showed more definitively than prior studies that the ORs and OBPs are present in the Hexapoda alone. In addition, our results indicated that OR genes are not universally associated with terrestrial invasions by arthropods, given the absence of these genes in the terrestrial chelicerates and myriapods (fig. 1).
Although, Vizueta et al. (2017) found two novel candidate chemosensory gene families in the hunter spider D. silvatica, one of them being distantly related to the canonical insect OBPs (i.e., three copies of OBP-like proteins) and the other encoding 12 copies (not related to OBPs). Some of these genes are expressed in the putative chemosensory appendages of these spiders, and show typical characteristics of secreted chemosensory proteins, such as a conserved cysteine pattern and the presence of a clear signal peptide. However, the specific functional roles of these putative chemosensory related genes are unknown, and further studies are required to determine whether they do function similarly as insect OBPs.
Our more comprehensive analysis is consistent with, and considerably extends, results from prior studies, which did not include the crustaceans beyond the branchiopod/hexapod clade (Peñalva-Arana et al. 2009; Missbach et al. 2014). Our analysis was consistent with the hypothesis, first stated by Robertson et al. (2003), that the ORs arose after the emergence of the Hexapoda from within the Pancrustacea (∼470 Ma), and expanded greatly in the hexapod lineage. Prior studies found that the genome of the water flea D. pulex (Branchiopoda) and the transcriptome of the Caribbean hermit crab Coenobita clypeatus (Pancrustacea, Malacostraca) lacked ORs and OBPs (Peñalva-Arana et al. 2009; Vieira and Rozas 2011; Groh et al. 2014). Recent studies also found ORs and OBPs to be lacking in the genomes of several species from the arthropod subphyla Chelicerata and Myriapoda, such as the myriapod centipede (S. maritima) (Chipman et al. 2014) and three chelicerate spider mites (Tetranychus urticae, Tetranychus evansi, and Tetranychus lintearius) (Phuong 2013). We confirmed the absence of ORs and OBPs in four additional chelicerates (black-legged tick I. scapularis, bark scorpion C. exilicauda, black widow spider L. hesperus, and brown recluse spider L. reclusa).
Existing data from the literature indicate that OBPs evolved earlier in the evolution of the insects, whereas ORs are thought to have emerged long after the establishment of a terrestrial lifestyle, with their appearance correlated with the emergence of winged insects (Missbach et al. 2014). For instance, recent studies focusing on basally branching insects, such as members of the orders Archaeognatha, Zygentoma, and Phasmatodea, demonstrate that the jumping bristletail Lepismachilis y-signata (Archaeognatha, wingless insects) possesses OBPs, but does not have ORs (Missbach et al. 2014, 2015). In contrast, OR repertoires (including Orco) were found in the firebrat Thermobia domestica (Zygentoma) and the leaf insect Phyllium siccifolium (Phasmatodea) that do have wings, indicating that they arose after the emergence of wings (Missbach et al. 2014). However, these studies examined transcriptome sequences of insects, and more thorough analyses of comprehensive genome data would clarify the evolutionary history of the emergence of OR and OBP gene families within the insects.
Although our study provided much added support for the exclusivity of ORs and OBPs to the insects (Hexapoda), one taxonomic group remains to be examined. No study has yet examined the other member of the Allotriocarida clade, the class Remipedia, which are also crustaceans closely related to the Hexapoda. Thus, we cannot yet conclude definitively that ORs and OBPs are exclusive to the Hexapoda (fig. 1).
The absence of ORs and OBPs in noninsect arthropod clades raises the interesting question of what chemosensing system the noninsect terrestrial arthropods (i.e., Chelicerata, Myriapoda) are using to detect volatile ligands in air. Insect ORs respond to various volatile odorants and pheromonal molecules that diffuse in air (Hallem et al. 2004). So then have the terrestrial chelicerates and myriapods co-opted an existing system that has been described to perform this function? Or are they using some other gene family that has not yet been discovered? The newly discovered CCPs and OBP-like genes found in a spider (chelicerate) (Vizueta et al. 2017) might fulfill this role in terrestrial habitats, though the functions of these genes are not yet known. The chemosensing systems of the noninsect terrestrial arthropods would be worth exploring.
Most Arthropod CRG Families Follow the Birth-and-Death Model of Multigene Family Evolution
The patterns we found of frequent gene losses and gains by the GR gene families and the lack of orthologous GR genes among different arthropod orders (supplementary fig. S1, Supplementary Material online) suggest that these genes have been evolving according to the “birth-and-death” model of multigene family evolution (Nei and Hughes 1992; Sánchez-Gracia et al. 2011). A few prior studies have also found patterns of CRG evolution consistent with this model (see below) (Vieira et al. 2007; Sánchez-Gracia et al. 2009, 2011). Under this model, new genes are created by gene duplication. Then, after the divergence of major lineages, some of the genes are retained in the genome for a long time as functional genes, whereas others become nonfunctional through deleterious mutations or are eliminated from the genome (Nei and Rooney 2005). This model was first proposed as an alternative to the previously well-accepted model of concerted evolution (Nei and Rooney 2005), in order to explain the high degree of polymorphism found at MHC loci in mammals (Nei and Hughes 1992).
One line of support for the “birth-and-death” model of gene family evolution would be that different lineages have undergone unique gene family expansions or contractions. We see such patterns of lineage-specific expansions or contractions in multiple arthropod lineages (fig. 1 and2 and supplementary fig. S1, Supplementary Material online). For instance, most of the insect, copepod, and chelicerate GRs generally formed distinct clades without clear orthology to one another (Groups I–IX in fig. 2, >0.73 posterior probability in the Bayesian phylogeny). Likewise, there was an expansion of 61 GRs in the myriapod (S. maritima) genome, forming a distinct monophyletic clade (Group X in fig. 2 and supplementary fig. S1, Supplementary Material online) (Chipman et al. 2014). Within the Hymenoptera, the wasp Nasonia vitripennis genome had an expansion of 58 GRs (Robertson et al. 2010). In contrast, the honeybee (Apis mellifera) genome had only ten GRs (Robertson and Wanner 2006), suggesting a lineage-specific GR gene family contraction in this lineage (supplementary fig. S1, Supplementary Material online).
Also in support of the “birth-and-death” model is the fact that we observed large genetic divergences between GR gene families in closely related clades (supplementary fig. S1, Supplementary Material online). For the GR proteins within the purported Allotriocarida (Branchiopoda/Hexapoda) clade, the closest sequence similarity between D. melanogaster and D. pulex was 43.4% (by local alignment between DmelGR64b and DpulGR56). A consequence of the large divergences between the clades is the fact that different orders of arthropods lack truly orthologous GR genes (fig. 2 and supplementary fig. S1, Supplementary Material online). For example, D. melanogaster and the silkworm moth Bombyx mori represent two closely related orders (see the inset of supplementary fig. S1, Supplementary Material online). However, GR orthologs cannot be identified between the two species, except for the carbon dioxide receptors and sugar receptors (Wanner and Robertson 2008), which are relatively conserved within insects (supplementary fig. S1, Supplementary Material online).
We also observed patterns consistent with the birth-and-death model in other CRG members. For instance, divergent IRs displayed patterns consistent with this model, such as large genetic divergences and no orthology between divergent IRs of D. melanogaster and B. mori (Croset et al. 2010). Additionally, insect ORs formed a large and highly divergent gene family with no close orthologs, such as between ORs of D. melanogaster and B. mori, except for Orco (Hansson and Stensmyr 2011). Patterns consistent with the birth-and-death model have also been reported for CSPs and OBPs (Vieira et al. 2007; Sánchez-Gracia et al. 2009; Hansson and Stensmyr 2011).
In contrast, antennal IRs are quite conserved in sequence within the Arthropoda (fig. 3), and did not conform to the birth-and-death model. The antennal IRs showed clear orthologous relationships even among distantly related species, such as between D. melanogaster and copepod species (fig. 3). Many of the antennal IRs have generally remained as single-copy genes (fig. 3). These genes have remained highly conserved and retained homologous structures across all protostomian species (fig. 4 and supplementary figs. S5 and S6, Supplementary Material online).
Overall, the evolutionary patterns we observed here are consistent with the birth-and-death model being a major mechanism of molecular evolution in all the CRG families except for the antennal IRs. Thus, we speculate that such a birth-and-death process of CRG evolution might reflect a common process of rapid diversification associated with adaptation to diverse environments (Hayden et al. 2010), resulting in high rates of gene family gains and losses.
Conclusions and Future Studies
Elucidating patterns of CRG family evolution provides an important step toward understanding the interactions between organisms and their environments, as CRGs are fundamental to sensing the environment and adapting to various ecological niches (Hayden et al. 2010). For instance, the fact that most CRG families appear to be evolving under the birth-and-death model, with rapid species-specific gene duplications, suggests rapid species-specific adaptations to their environments. Several of our results are suggestive of some CRGs found in this study playing functional roles in mating. For instance, this study is the first to find sex differences in expression of CRGs in an aquatic organism. Most notably, we found that three of the antennal IRs were highly expressed in male copepods of the species Eurytemora affinis, and that these male-biased antennal IRs showed significantly stronger signatures of purifying selection than nonsex-biased IRs. It would be worth exploring the role of antennal IRs in mating behavior, and how molecular evolutionary changes in antennal IR proteins might correspond to changes in mating behavior. In addition, prior studies on CRGs’ roles in mating have focused predominantly on terrestrial organisms. As the physics of chemosensing and the diffusion of ligands would differ between water and air, the role of CRGs in mating and other functions would be worth exploring in the aquatic realm.
Overall, our results have generated several intriguing hypotheses that should be further explored with functional studies. A comparative functional evolutionary approach that included diverse arthropod taxa, especially beyond the insect clade and from multiple habitat types, would provide key insights into the evolutionary history and functions of CRG families.
Materials and Methods
Eurytemora affinis Genome Sequencing
Sample Preparation Genome Sequencing
To generate the comprehensive genome sequence for the copepod E. affinis, an inbred line (VA-1) derived from a saline population in Baie de L’Isle Verte, St. Lawrence marsh, Quebec, Canada (48°00′14″N, 69°25′31″W) was used (Lee 1999, 2000; Winkler et al. 2008). The inbred line was generated through full-sibling mating for 30 generations (2.5 years), in order to reduce problems posed by heterozygosity during genome assembly and annotation. Only egg sacs were used for genome sequencing to avoid including the rich microbiome associated with the copepod. Prior to DNA extraction, the culture was treated with a series of antibiotics to greatly reduce bacterial contamination, including Primaxin (20 mg/l), Voriconazole (0.5 mg/l for at least 2 weeks prior to DNA extraction), d-amino acids to reduce biofilm (10 µM d-methionine, d-tryptophan, d-leucine, and 5 µM d-tyrosine, for at least for 2 weeks prior to DNA extraction). In addition, to remove bacterial contamination from our sample, egg sacs with 10% bleach for ∼1 min were bleached. Our method was verified for drastically reducing the bacterial load using qPCR. In total, DNA from ∼4,000 egg sacs was extracted for genome sequencing, using the QIAGEN QIAamp DNA Mini Kit (catalog #51304) with 4 µl of RNase A (100 mg/ml).
High Throughput Genome and Transcriptome Sequencing and Genome Assembly
The copepod E. affinis was one of 30 arthropod species sequenced as a part of the pilot project for the i5K Arthropod Genomes Project at the Baylor College of Medicine Human Genome Sequencing Center (https://www.hgsc.bcm.edu/arthropods; last accessed May 4, 2017). Supplementary table S1, Supplementary Material online, provides details of all sequences generated for the copepod E. affinis and their National Center for Biotechnology Information (NCBI) accession numbers, as well as assembly and annotation statistics and their NCBI accessions. The primary NCBI BioProject for the genome sequencing, annotation, and assembly of E. affinis is PRJNA203087.
An enhanced Illumina-ALLPATHS-LG sequencing and assembly strategy enabled multiple species to be approached in parallel at reduced costs. For E. affinis, four libraries of nominal insert sizes 180 bp, 500 bp, 3 kb, and 8 kb at genome coverages of 28.1×, 21.2×, 16.6× and 9.0×, respectively, for a total of 75× genome coverage were sequenced. Libraries were prepared using standard methods as described previously (Anstead et al. 2015). Sequencing was performed on Illumina HiSeq2000 platforms generating 100-bp paired-end reads. These raw sequences have been deposited in the NCBI SRA, accessions are shown in the supplementary table S1, Supplementary Material online, BioSample ID: SAMN02302763. Additionally, three RNAseq libraries were prepared from separated samples of adult males, females, and mixed sex juvenile stages and sequenced using standard techniques (Anstead et al. 2015). These transcriptome sequences were used to support automated and manual annotation.
The genomic sequence of the copepod E. affinis was assembled using ALLPATHS-LG (release 3‐35218) (Gnerre et al. 2011) and further scaffolded and gap-filled using in-house tools Atlas-Link (v.1.0) and Atlas gap-fill (ver.2.2) (https://www.hgsc.bcm.edu/software; last accessed May 4, 2017). This yielded an assembly of size 494.8 Mb including gaps within scaffolds, with contig N50 of 5.7 kb and scaffold N50 of 862.6 kb. The assembly has been deposited in the NCBI (BioProject PRJNA203087).
Automated Gene Annotation Using a Maker 2.0 Pipeline Tuned for Arthropods
The copepod E. affinis was one of 30 i5K pilot genome assemblies subjected to automatic gene annotation using a Maker 2.0 annotation pipeline tuned specifically for arthropods. The pipeline was designed to be systematic, providing a single consistent procedure for the species in the pilot study. Also, the pipeline was scalable to handle hundreds of genome assemblies, evidence-guided (using both protein and RNA-Seq evidence to guide gene models), and targeted to utilize extant information on arthropod gene sets. The core of the pipeline was a Maker 2 instance, modified slightly to enable efficient running on our computational resources (Holt and Yandell 2011). The genome assembly was first subjected to de novo repeat prediction and CEGMA analysis to generate gene models for initial training of the ab initio gene predictors. Three rounds of training of the Augustus (Stanke et al. 2008) and SNAP (Korf 2004) gene predictors within Maker were used to bootstrap to a high-quality training set. Input protein data included 1 million peptides from a nonredundant reduction (90% identity) of Uniprot Ecdysozoa (1.25 million peptides), supplemented with proteomes from 18 additional species (S. maritima, Tetranychus urticae, C. elegans, Loa loa, T. adhaerens, A. queenslandica, Strongylocentrotus purpuratus, N. vectensis, Branchiostoma floridae, Ciona intestinalis, Ciona savignyi, Homo sapiens, Mus musculus, Capitella teleta, Helobdella robusta, Crassostrea gigas, L. gigantea, and Schistosoma mansoni), leading to a final nr peptide evidence set of 1.03 million peptides. RNA-Seq reads from E. affinis adult males and females were used judiciously to identify exon–intron boundaries, but with a heuristic script to identify and split erroneously joined gene models. CEGMA models for QC purposes were used: for E. affinis, of 1,977 CEGMA single-copy ortholog gene models, 1,808 were found in the assembly, and 1,707 in the final predicted gene set. Finally, the pipeline used a nine-way homology prediction with human, Drosophila and C. elegans, and InterPro Scan5 to allocate gene names. The automated gene set is available at the BCM-HGSC website (https://www.hgsc.bcm.edu/arthropods/bed-bug-genome-project) and at the National Agricultural Library (https://i5k.nal.usda.gov).
Sample Preparation for Transcriptome Sequencing
Eurytemora affinis transcriptomes were generated using RNA-Seq strand-specific paired-end Illumina sequencing with one sample per HiSeq2000 channel at the Institute for Genome Sciences in the University of Maryland School of Medicine. To compare relative expression of CRG families in males versus females of the copepod E. affinis, three different types of samples were sequenced: Female, male, and mixed female + male samples, with two replicates each and ∼220 individual copepods per replicate sample. Females and males from an inbred line (line VA-30-1, 30 generations of full-sib mating) derived from the same population of E. affinis used for genome sequencing (described above) were used. Inbred copepods were reared under controlled laboratory conditions, at 13 °C, 15 PSU (practical salinity unit ≈ parts per thousand) salinity, and on a 15L:9D photoperiod, until the copepods reached adulthood. The copepods were fed with saltwater algae Rhodomonas salina. To prevent bacterial infection, copepods were treated with the antibiotics Primaxin (20 mg/l), d-amino acid cocktail (10 µM of d-methionine, d-leucine, d-tryptophan, and 5 µM d-tyrosine), and Voriconazole (0.5 mg/l) every 3–4 days. The d-amino acids we used (d-methionine, d- tryptophan, d-leucine, and 5 µM d-tyrosine) were found to induce negligible responses in the insect chemoreceptors tested (D. melanogaster IRs) (Croset et al. 2016).
Two days prior to RNA extraction, males and females were separated into different beakers, and treated them with an antibiotic cocktail in order to minimize contamination of copepod RNA with bacterial RNA. The separated female and male samples (two replicates per sex) received a full antibiotic cocktail (described below), whereas the mixed males + female samples (two replicates) received the regular antibiotic cocktail used in culturing (with only Primaxin, d-amino acids, and Voriconazole). qPCR of the bacterial 16S rRNA gene on the copepod RNA samples was performed before and after administering varying combinations of antibiotic recipes to devise a full antibiotic cocktail that effectively removed nearly all bacterial contamination. All the antibiotics used had been tested for toxicity on E. affinis in prior experiments. Our full antibiotic cocktail consisted of: Primaxin (20 mg/l), Voriconazole (0.5 mg/l), d-amino acids (10 µM d-methionine, d-tryptophan, d-leucine, and 5 µM d-tyrosine), Sitofloxacin (10 mg/l, increased to 20 mg/l in last 24 h), Rifaximin (3 mg/l, increased to 10 mg/l in last 24 h), Phosophomycin (20 mg/l), Daptomycin (3 mg/l), and Metronidazole (15 mg/l). In order to clear the guts, the copepods were starved and treated with 120 µl/l of 6.0-µm copolymer microsphere beads (Thermo Scientific cat# 7505A, Fremont, CA) for the last 24 h before RNA extraction. Total RNA was extracted with Trizol reagent (Ambion RNA, Carlsbad, CA) and then purified with Qiagen RNeasy Mini Kit (Qiagen cat# 74104, Valencia CA), following the protocol described by Lopez and Bohuski (2007).
Assembly of Crustacean Transcriptomes and Genomes
Data Source and De Novo Assembly for 12 Crustacean Species
De novo genome and transcriptome assemblies were performed for 12 publicly available crustacean species (table 1). Next-generation sequence data for the 12 crustacean species were obtained from the NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra) (supplementary table S1, Supplementary Material online); For the copepod species, five transcriptome (C. rogercresseyi, L. cyprinacea, T. californicus, C. sinicus, and A. fossae) and three genome sequences (L. salmonis, M. edax, and C. finmarchicus) were downloaded from NCBI SRA. Three additional crustacean species were also included: The giant tiger prawn P. monodon (Malacostraca: Penaeidae), the purple barnacle A. amphitrite (Cirripedia: Balanidae), and the brine shrimp A. franciscana (Branchiopoda: Artemiidae) from NCBI SRA. Their NCBI accession numbers and sequencing platforms are summarized in the supplementary table S2, Supplementary Material online.
A stringent quality filter process was applied because sequencing errors can cause difficulties for the assembly algorithm (Martin and Wang 2011). For 454 reads, the adapter and poly(A/T) sequences were trimmed using PRINSEQ (Schmieder and Edwards 2011). In total, 454 reads that had abnormal read length (<50 or >1,000 bp) or that had average quality score of less than 20 were removed. Illumina reads that did not have the minimum quality score of 20 per base across the whole read were removed using PRINSEQ (Schmieder and Edwards 2011). The quality scores of 20 (Q20) correspond to 1% expected error rates. Also, Illumina reads that had any unknown nucleotide “N” were removed.
After the filtering process, de novo assemblies of the crustacean transcriptome sequences using the software package Trinity (release 2013-11-10) were performed (Haas et al. 2013). The Trinity assembly algorithm uses the minimum contig length set to 300 bp with a fixed k-mer size of 25. Note that Zhao et al. (2011) showed that Trinity had the highest accuracy in mapping reads to the reference genome among methods specialized in de novo transcriptome assemblies, including SOAPdenovo (Li et al. 2009), ABySS (Birol et al. 2009), Velvet/Oasis (ver. 1.2.03) (Zerbino and Birney 2008), and Mira (ver. 3.4.0) (Chevreux et al. 2004). Also, Eyun et al. (2014) showed that fractions of contigs that had highly significant hits to the UniProt protein database (E-value ≤ 10−100) were larger with Trinity than with Velvet/Oasis or Mira. To assemble the three copepod genome sequences (L. salmonis, M. edax, and C. finmarchicus), their sequences were assembled using Velvet with minimum contig length set to 300 bp and using multiple k-mer sizes (data not shown).
Sources of Additional Genome Sequences
In addition to the 12 crustacean species mentioned above (table 1), several publicly released genome assemblies were added to this study. For the most closely related outgroup phylum Onychophora, the current version of genome for velvet worm Euperipatoides rowelli (BioProject accession number: PRJNA203089) was downloaded from https://www.hgsc.bcm.edu. Five genomes were downloaded from the i5K project (https://i5k.nal.usda.gov), namely those of two crustaceans, a copepod (T. californicus, Harpacticoida) and an amphipod (H. azteca), and three chelicerates (C. exilicauda, L. hesperus, and L. reclusa). In addition, genomes of four basal metazoan phyla (Cnideria, Placozoa, Porifera, and Ctenophora), two fungi (Ascomycota and Basidiomycota), and four single-celled eukaryotic (protist) phyla (Choanozoa, Mycetozoa, Percolozoa, and Metamonada) were analyzed (see supplementary tables S2 and S3 for lists of genomes sampled, Supplementary Material online).
Chemosensory Gene Search and Homology
Query Sequences and Gene Mining
Previously reported insect CRG sequences were used as search queries to identify the putative CRG genes in 33 species (supplementary tables S2 and S3, Supplementary Material online). Insect-type OR and GR sequences were obtained from Robertson and Wanner (2006), McBride and Arguello (2007), Wanner et al. (2007), Engsontia et al. (2008), Peñalva-Arana et al. (2009), Chipman et al. (2014), and Gulia-Nuss et al. (2016). The carbon dioxide receptors were obtained from Jones et al. (2007). IR sequences were obtained from Benton et al. (2009) and Croset et al. (2010).
Using these sequences as initial queries, chemosensory gene candidates were mined from our transcriptomes and genomes (supplementary table S4, Supplementary Material online) using NCBI BLAST (standalone tblastn, ver. 2.2.28+) (Altschul et al. 1997; Camacho et al. 2009). The initial E-value threshold used (of 60 for GRs and ORs and 1 × 10−6 for the other CRGs) was rather lenient and chosen to avoid missing all true positives, even though some false positives from nontarget genes could be included. In order to obtain comparable E-values, the database size of 1.4 × 1010 (using the “-dbsize” option) was set to be equivalent to the size of the Nucleotide collection (nr/nt) database at NCBI. The putative CRG genes were verified by conducting searches using blastp against the NCBI NR protein database and with phylogenetic analyses. A putative protein was designated as a CRG candidate if the top hit from the blastp search was previously identified as a CRG. The newly identified gene candidates were subsequently used as queries against their transcriptomes or genomes again to find any additional candidates. These steps were performed recursively until no other gene candidate sequences were detected from each assembly. The candidate genes were manually curated using JBrowse in Web Apollo for the copepod E. affinis. Reading frames and intron/exon boundaries were determined using GeneWise (ver. 2.2) (Birney et al. 2004) for all our genomes and were manually adjusted using the multiple alignments of the homologs.
For a more sensitive search, profile hidden Markov models (HMM) were constructed with insect-type ORs and GR protein sequences from D. melanogaster, Tribolium castaneum, A. mellifera, D. pulex, I. scapularis, and S. maritima. Sequences from D. pulex, I. scapularis, and S. maritima were used only for the GR models. Each assembly was searched using the hmmbuild and hmmsearch programs of the HMMER package (ver. 3.0) (Eddy 2011) for building and calibrating HMMs. Customized profile HMMs were also used with only the most conserved regions (near the seventh transmembrane to the C-terminus).
Protein Family Classification and Transmembrane Protein Topology Prediction
To perform computational analysis of protein family classification of GRs, three different algorithms, namely the Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) (Marchler-Bauer et al. 2015), the PANTHER system (http://www.pantherdb.org) (Mi et al. 2013), and HHpred (http://toolkit.tuebingen.mpg.de/hhpred) (Söding et al. 2005) were used.
To predict the transmembrane protein topology of GRs, HMMTOP (ver. 2.1) (Tusnady and Simon 2001) and Phobius (ver. 1.01) (Kall et al. 2007) were used. These analyses were included in N-terminal and C-terminal regions and the number of transmembrane. These results were summarized in the supplementary table S6, Supplementary Material online.
Gene Nomenclature
The newly designated gene names were represented by a four-letter species abbreviation combined with the name of the D. melanogaster orthologs. The species abbreviations consisted of an uppercase initial letter from the genus name and three lowercase initial letters from the species name. For example, Eaff refers to Eurytemora affinis. Genes orthologous to those in Drosophila followed the unified nomenclature system of the Drosophila receptors, according to Drosophila OR and GR gene families (Drosophila Odorant Receptor Nomenclature Committee 2000; Benton et al. 2009). For multiple gene duplicates, each copy was designated by a dash and a number (e.g., TcalIR8a-1, TcalIR8a-2, and TcalIR8a-3). Unfortunately, many CRG genes could not be named using this approach, as they showed no clear orthology to Drosophila counterparts. In such cases, the genes were given names that indicated the species and the CRG family, such as EaffGR1 and EaffGR2.
Multiple Sequence Alignments
Multiple alignments of GR, IR, and CSP protein sequences were generated using MAFFT (ver. 7.149b) (Katoh and Standley 2013) with the L-INS-i algorithm (1,000 maxiterate and 100 retree). This algorithm uses a consistency-based objective function and local pairwise alignment with affine gap costs. We also employed the alignment programs ProbCons (ver. 1.12) (Do et al. 2005) and PRALINE (Pirovano et al. 2008) using the default parameters for the comparison. Alignments were adjusted manually when necessary. All CRG sequences and alignments are available at the Dryad Digital Repository, http://dx.doi.org/10.5061/dryad.ts747.
Phylogenetic Analysis of CRG Families
Phylogenetic relationships of GR, IR, and CSP gene families were reconstructed using maximum-likelihood with the PROTGAMMAJTT model using the software package RAxML (ver. 8.1.3) (Stamatakis 2014). Neighbor-joining phylogenies (Saitou and Nei 1987) were reconstructed using neighbor in the software package PHYLIP (ver. 3.67) (Felsenstein 2005). Protein distances were estimated using protdist with the JTT (Jones, Taylor, and Thornton) substitution model in the PHYLIP package, while accounting for gamma-distributed rate variation among amino acid sites (α = 3.2253 for GRs, α = 1.7133 for IRs, and α = 1.5826 for CSPs) (Yang 1994) estimated using maximum-likelihood with RAxML. Nonparametric bootstrapping with 1,000 pseudoreplicates (Felsenstein 1985) was used to estimate the confidence of branching topology for the maximum-likelihood and neighbor-joining phylogenies. Bayesian phylogenetic inference was performed using MrBayes (v3.2.3) (Ronquist and Huelsenbeck 2003) with the JTT substitution model with a gamma-distributed rate variation. A Markov Chain Monte Carlo search was run for 5 × 106 generations, with a sampling frequency of 102, using three heated and one cold chain and with a burn-in of 102 trees. The homolog of iGluRs is present in plants, namely the plant glutamate-like receptors (GLRs) (Croset et al. 2010; Price et al. 2012). Among iGluRs, the NMDAR subfamily is the closest gene family to GLRs, indicating that the NMDAR subfamily is the most ancient. Thus, all the iGluR trees were rooted using the NMDAR subfamily (Croset et al. 2010). Graphical presentation of the phylogenies was performed using FigTree (ver. 1.4.2) (http://tree.bio.ed.ac.uk/software/figtree).
Differential Gene Expression Analysis of CRGs between the Sexes
To compare relative expression of CRG families in males versus females of the copepod E. affinis, sex-specific expression levels of the GR, IR, and CSP genes were determined in transcriptome sequences of female and male samples, with two replicates each and ∼220 individual copepods per replicate sample. The approaches used for transcriptome sequencing are described above. As controls, we also examined differential expression of five representative housekeeping genes (Cyclophilin-33, Actin 42A, Heat shock protein 83, Glyceraldehyde 3 phosphate dehydrogenase 1, and Ribosomal protein L32) (supplementary table S9, Supplementary Material online) in males and female samples. These controls were used to verify that there was no general sex-specific bias in gene expression in these samples.
Single-end reads were mapped onto our assembled transcriptomes using Bowtie (ver. 1.0.1) with 0 mismatches (Langmead et al. 2009; Katz et al. 2010). We checked the raw Illumina sequences corresponding to the IR genes and confirmed their identities using Integrative Genomics Viewer (Thorvaldsdóttir et al. 2013). Numerical count data were transformed into RPKM to normalize for the number of sequencing reads and total read length (Mortazavi et al. 2008). RPKM values above 0.3 were used as the threshold for gene expression (Ramsköld et al. 2009). The statistical differences in gene expression levels between male and female samples were determined using both parametric (edgeR) (Zhou et al. 2014) and nonparametric (NOISeq) (ver. 2.8.0) (Tarazona et al. 2011) approaches in the R Bioconductor package (http://www.bioconductor.org) (ver. 3.1.2). Genes were considered to be differentially expressed if they had P-values less than 0.05 using edgeR or had probability values above 0.70 using NOISeq (supplementary table S8, Supplementary Material online).
Testing for Signatures of Selection in Antennal IR Genes
To test whether the antennal IR genes show differing patterns of molecular evolution between the male-biased expression IRs versus the unbiased expression IRs, we used the “branch models” implemented in codeml in the software package PAML (Phylogenetic Analysis by Maximum Likelihood, version 4.8) (Yang 2007). To estimate the average ω (the ratio of nonsynonymous to synonymous divergences, dN/dS), we performed likelihood ratio tests (LRTs) with df = 1 between a one-ratio model (R1; the same ω for all branches) and a two-ratio model (R2; two independent ω’s) (Yang and Nielsen 2002). As illustrated in the supplementary figure S3, Supplementary Material online, each test was set up to compare antennal IR genes more highly expressed in males (male-biased expression IR genes) (IR25a, IR93a, and IR8a) against antennal IR genes that showed no sex differences in expression (unbiased expression IR genes) (IR76b and IR21a). All PAML analyses were performed using the F3X4 model of codon frequency (Goldman and Yang 1994). The level of significance (P) for the LRTs was estimated using a χ2 distribution with given degrees of freedom (df). The test statistic was calculated as twice the difference in log-likelihood between the models (2Δln L= 2[ln L1 − ln L0] where L1 and L0 are the likelihoods of the alternative and null models, respectively).
Protein Structural Homology Modeling
Homology modeling of the copepod Tigriopus californicus IR25a and the copepod Eurytemora affinis CSP2 protein structures was performed using the SWISS-MODEL Web server (http://swissmodel.expasy.org) (Arnold et al. 2006) (supplementary figs. S6 and S7, Supplementary Material online). For the T. californicus IR25a, the B-chain of the rat (Rattus norvegicus) iGluR (PDB: 1YAE, NP_062182.1) was selected as the template (E-value: 2.5 × 10−36; sequence similarity: 53.4%). The QMEAN4 Z-score given by SWISS-MODEL was −5.48 (raw score is 0.439). The N-terminal 401 amino acids (aa) and the C-terminal 96 aa were excluded from the modeling due to insufficient sequence similarity. For the E. affinis CSP2, the moth M. brassicae CSP (CSPMbraA6, PDB: 1N8V, AAF71289.1) was used as the template. The sequence similarity against CSPMbraA6 was 25.93% and the QMEAN4 Z-score given by SWISS-MODEL was −1.62. The graphical representations of the protein structures for the T. californicus TcalIR25a and E. affinis CSP2 structure were created using PyMOL (version 1.3) (DeLanoScientific, San Carlos, CA).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
Sample preparation, genome sequencing, assembly, transcriptome sequencing, and automated genome annotation of the copepod Eurytemora affinis were funded by grants from the National Human Genome Research Institute (NHGRI) U54 HG003273 to R.A.G. and National Science Foundation OCE-1046372 to C.E.L. (the latter funded all the labor and cost for copepod inbreeding, rearing, and preparation). Additional transcriptome sequencing was funded by National Science Foundation grants OCE-1046372 to C.E.L. and OCE-1046371 to J.C.S., and DEB-1050565 and DEB-0745828 to C.E.L. This work was additionally supported in part by the Nebraska Research Initiative (to S.E.) and Chonnam National University, Korea (CNU 2013 to H.Y.S.). Hugh M. Robertson (University of Illinois at Urbana-Champaign, USA) provided the Ixodes scapularis gustatory receptor sequences and valuable comments. Richard Benton (University of Lausanne, Switzerland) provided the I. scapularis ionotropic receptors. Susumu Ohtsuka (Hiroshima University, Japan) and members of Carol Lee’s laboratory provided helpful comments and suggestions on drafts of this manuscript.
References
- Abuin L, Bargeton B, Ulbrich MH, Isacoff EY, Kellenberger S, Benton R.. 2011. Functional architecture of olfactory ionotropic glutamate receptors. Neuron 69:44–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ.. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstead CA, Korhonen PK, Young ND, Hall RS, Jex AR, Murali SC, Hughes DST, Lee SF, Perry T, Stroehlein AJ, et al. 2015. Lucilia cuprina genome unlocks parasitic fly biology to underpin future interventions. Nat Commun. 6:7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T.. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. 2006. Comparative chemosensation from receptors to ecology. Nature 444:295–301. [DOI] [PubMed] [Google Scholar]
- Benton R. 2015. Multigene family evolution: perspectives from insect chemoreceptors. Trends Ecol Evol. 30:590–600. [DOI] [PubMed] [Google Scholar]
- Benton R, Vannice KS, Gomez-Diaz C, Vosshall LB.. 2009. Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell 136:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R.. 2004. GeneWise and genomewise. Genome Res. 14:988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics 25:2872–2877. [DOI] [PubMed] [Google Scholar]
- Burton RS. 1990. Hybrid breakdown in developmental time in the copepod Tigriopus californicus. Evolution 44:1814–1822. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL.. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanacci V, Lartigue A, Hällberg BM, Jones TA, Giudici-Orticoni M-T, Tegoni M, Cambillau C.. 2003. Moth chemosensory protein exhibits drastic conformational changes and cooperativity on ligand binding. Proc Natl Acad Sci U S A. 100:5069–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M.. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao TNP. 2013. Genome annotation and evolution of chemosensory receptors in spider mites [PhD dissertation]. Ghent, Belgium: Ghent University.
- Chen G, Hare MP.. 2011. Cryptic diversity and comparative phylogeography of the estuarine copepod Acartia tonsa on the US Atlantic coast. Mol Ecol. 20:2425–2441. [DOI] [PubMed] [Google Scholar]
- Chen N, Pai S, Zhao Z, Mah A, Newbury R, Johnsen RC, Altun Z, Moerman DG, Baillie DL, Stein LD.. 2005. Identification of a nematode chemosensory gene family. Proc Natl Acad Sci U S A. 102:146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S.. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipman AD, Ferrier DEK, Brena C, Qu J, Hughes DST, Schröder R, Torres-Oliva M, Znassi N, Jiang H, Almeida FC, et al. 2014. The first myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol. 12:e1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloudsley-Thompson JL. 1975. Adaptations of arthropods to arid environments. Annu Rev Entomol. 20:261–283. [DOI] [PubMed] [Google Scholar]
- Clyne PJ, Warr CG, Carlson JR.. 2000. Candidate taste receptors in Drosophila. Science 287:1830–1834. [DOI] [PubMed] [Google Scholar]
- Clyne PJ, Warr CG, Freeman MR, Lessing D, Kim JH, Carlson JR.. 1999. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila. Neuron 22:327–338. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. 2011. The ecoresponsive genome of Daphnia pulex. Science 331:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey EA, Bobkov Y, Ukhanov K, Ache BW.. 2013. Ionotropic crustacean olfactory receptors. PLoS ONE 8:e60551.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset V, Rytz R, Cummins SF, Budd A, Brawand D, Kaessmann H, Gibson TJ, Benton R.. 2010. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 6:e1001064.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset V, Schleyer M, Arguello JR, Gerber B, Benton R.. 2016. A molecular and neuronal basis for amino acid sensing in the Drosophila larva. Sci Rep. 6:34871.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S.. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doall MH, Colin SP, Strickler JR, Yen J.. 1998. Locating a mate in 3D: the case of Temora longicornis. Philos Trans R Soc Lond B Biol Sci. 353:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila Odorant Receptor Nomenclature Committee. 2000. A unified nomenclature system for the Drosophila odorant receptors. Cell 102:145–146. [DOI] [PubMed] [Google Scholar]
- Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol. 7:e1002195.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmands S. 1999. Heterosis and outbreeding depression in interpopulation crosses spanning a wide range of divergence. Evolution 53:1757–1768. [DOI] [PubMed] [Google Scholar]
- Engsontia P, Sanderson AP, Cobb M, Walden KKO, Robertson HM, Brown S.. 2008. The red flour beetle's large nose: an expanded odorant receptor gene family in Tribolium castaneum. Insect Biochem Mol Biol. 38:387–397. [DOI] [PubMed] [Google Scholar]
- Eyun S. 2017. Phylogenomic analysis of Copepoda (Arthropoda, Crustacea) reveals unexpected similarities with earlier proposed morphological phylogenies. BMC Evol Biol. 17:23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyun S, Lee Y-H, Suh H-L, Kim S, Soh HY.. 2007. Genetic identification and molecular phylogeny of Pseudodiaptomus species (Calanoida, Pseudodiaptomidae) in Korean waters. Zool Sci. 24:265–271. [DOI] [PubMed] [Google Scholar]
- Eyun S, Wang H, Pauchet Y, ffrench-Constant RH, Benson AK, Valencia-Jiménez A, Moriyama EN, Siegfried BD.. 2014. Molecular evolution of glycoside hydrolase genes in the western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 9:e94052.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. [Seattle (WA)]: Department of Genome Sciences, University of Washington.
- Forêt S, Wanner KW, Maleszka R.. 2007. Chemosensory proteins in the honey bee: insights from the annotated genome, comparative analyses and expressional profiling. Insect Biochem Mol Biol. 37:19–28. [DOI] [PubMed] [Google Scholar]
- Friedman MM, Strickler JR.. 1975. Chemoreceptors and feeding in calanoid copepods (Arthropoda: Crustacea). Proc Natl Acad Sci U S A. 72:4185–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz HH, Burton RS.. 1995. Genetic differentiation and reproductive incompatibility among Baja California populations of the copepod Tigriopus californicus. Mar Biol. 123:821–827. [Google Scholar]
- Gao Q, Chess A.. 1999. Identification of candidate Drosophila olfactory receptors from genomic DNA sequence. Genomics 60:31–39. [DOI] [PubMed] [Google Scholar]
- Gardiner A, Butlin RK, Jordan WC, Ritchie MG.. 2009. Sites of evolutionary divergence differ between olfactory and gustatory receptors of Drosophila. Biol Lett. 5:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauld DT. 1957. Copulation in calanoid copepods. Nature 180:510–510.13464867 [Google Scholar]
- Giribet G, Edgecombe GD, Wheeler WC.. 2001. Arthropod phylogeny based on eight molecular loci and morphology. Nature 413:157–161. [DOI] [PubMed] [Google Scholar]
- Glenner H, Thomsen PF, Hebsgaard MB, Sørensen MV, Willerslev E.. 2006. The origin of insects. Science 314:1883–1884. [DOI] [PubMed] [Google Scholar]
- Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetze E. 2003. Cryptic speciation on the high seas; global phylogenetics of the copepod family Eucalanidae. Proc R Soc Lond B Biol Sci. 270:2321–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z.. 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 11:725–736. [DOI] [PubMed] [Google Scholar]
- Gregory TR. 2016. Animal genome size database. Available from: http://www.genomesize.com.
- Griffiths AM, Frost BW.. 1976. Chemical communication in the marine planktonic copepods Calanus pacificus and Pseudocalanus sp. Crustaceana 30:1–8. [Google Scholar]
- Grishanin AK, Rasch EM, Dodson SI, Wyngaard GA.. 2006. Genetic architecture of the cryptic species complex of Acanthocyclops vernalis (Crustacea: Copepoda). II. Crossbreeding experiments, cytogenetics, and a model of chromosomal evolution. Evolution 60:247–256. [PubMed] [Google Scholar]
- Groh-Lunow KC, Getahun MN, Grosse-Wilde E, Hansson BS.. 2015. Expression of ionotropic receptors in terrestrial hermit crab’s olfactory sensory neurons. Front Cell Neurosci. 8:448.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh K, Vogel H, Stensmyr M, Grosse-Wilde E, Hansson BS.. 2014. The hermit crab’s nose-antennal transcriptomics. Front Neurosci. 7:266.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean Y, Rytz R, Farine J-P, Abuin L, Cortot J, Jefferis GSXE, Benton R.. 2011. An olfactory receptor for food-derived odours promotes male courtship in Drosophila. Nature 478:236–240. [DOI] [PubMed] [Google Scholar]
- Gu S-H, Wang S-Y, Zhang X-Y, Ji P, Liu J-T, Wang G-R, Wu K-M, Guo Y-Y, Zhou J-J, Zhang Y-J.. 2012. Functional characterizations of chemosensory proteins of the alfalfa plant bug Adelphocoris lineolatus indicate their involvement in host recognition. PLoS ONE 7:e42871.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulia-Nuss M, Nuss AB, Meyer JM, Sonenshine DE, Roe RM, Waterhouse RM, Sattelle DB, de la Fuente J, Ribeiro JM, Megy K, et al. 2016. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat Commun. 7:10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallem EA, Ho MG, Carlson JR.. 2004. The molecular basis of odor coding in the Drosophila antenna. Cell 117:965–979. [DOI] [PubMed] [Google Scholar]
- Hansson BS, Stensmyr MC.. 2011. Evolution of insect olfaction. Neuron 72:698–711. [DOI] [PubMed] [Google Scholar]
- Hardy A. 1956. The open sea. It's natural history: the world of plankton. London: Collins. [Google Scholar]
- Hayden S, Bekaert M, Crider TA, Mariani S, Murphy WJ, Teeling EC.. 2010. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 20:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuch PA, Doall MH, Yen J.. 2007. Water flow around a fish mimic attracts a parasitic and deters a planktonic copepod. J Plankton Res. 29:i3–i16. [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humes AG. 1994. How many copepods? Hydrobiologia 292/293:1–7. [Google Scholar]
- Huys R, Boxshall GA.. 1991. Copepod evolution. London: The Ray Society. [Google Scholar]
- Jones WD, Cayirlioglu P, Kadow IG, Vosshall LB.. 2007. Two chemosensory receptors together mediate carbon dioxide detection in Drosophila. Nature 445:86–90. [DOI] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL.. 2007. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 35:W429–W432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona SK. 1973. Evidence for sex pheromones in planktonic copepods. Limnol Oceanogr. 18:574–583. [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB.. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 7:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB. 2010. Olfactory signalling in vertebrates and insects: differences and commonalities. Nat Rev Neurosci. 11:188–200. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Peyton JT, Fiston-Lavier A-S, Teets NM, Yee M-C, Johnston JS, Bustamante CD, Lee RE, Denlinger DL.. 2014. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat Commun. 5:4611.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkness EF, Haas BJ, Sun W, Braig HR, Perotti MA, Clark JM, Lee SH, Robertson HM, Kennedy RC, Elhaik E, et al. 2010. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc Natl Acad Sci U S A. 107:12168–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht ZA, Silbering AF, Ni L, Klein M, Budelli G, Bell R, Abuin L, Ferrer AJ, Samuel ADT, Benton R, et al. 2016. Distinct combinations of variant ionotropic glutamate receptors mediate thermosensation and hygrosensation in Drosophila. eLife 5:e17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp A, Barmina O, Hamilton AM, Higgins L, McIntyre LM, Jones CD.. 2008. Evolution of gene expression in the Drosophila olfactory system. Mol Biol Evol 25:1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:59.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krång A-S, Knaden M, Steck K, Hansson BS.. 2012. Transition from sea to land: olfactory function and constraints in the terrestrial hermit crab Coenobita clypeatus. Proc R Soc Lond B Biol Sci. 279:3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmuni J, Havukainen H.. 2013. Insights into the evolution of the CSP gene family through the integration of evolutionary analysis and comparative protein modeling. PLoS ONE 8:e63688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S.. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin JD, Ha TS, Jones DNM, Smith DP.. 2008. Activation of pheromone-sensitive neurons is mediated by conformational activation of pheromone-binding protein. Cell 133:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CE. 1999. Rapid and repeated invasions of fresh water by the saltwater copepod Eurytemora affinis. Evolution 53:1423–1434. [DOI] [PubMed] [Google Scholar]
- Lee CE. 2000. Global phylogeography of a cryptic copepod species complex and reproductive isolation between genetically proximate “populations.” Evolution 54:2014–2027. [DOI] [PubMed] [Google Scholar]
- Lee CE, Frost BW.. 2002. Morphological stasis in the Eurytemora affinis species complex (Copepoda: Temoridae). Hydrobiologia 480:111–128. [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J.. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967. [DOI] [PubMed] [Google Scholar]
- Li XM, Zhu XY, He P, Xu L, Sun L, Chen L, Wang ZQ, Deng DG, Zhang YN.. 2016. Molecular characterization and sex distribution of chemosensory receptor gene family based on transcriptome analysis of Scaeva pyrastri. PLoS ONE 11:e0155323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Wang T, Rotgans BA, McManus DP, Cummins SF.. 2016. Ionotropic receptors identified within the tentacle of the freshwater snail Biomphalaria glabrata, an intermediate host of Schistosoma mansoni. PLoS ONE 11:e0156380.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, He X, Lehane S, Lehane M, Hertz-Fowler C, Berriman M, Field LM, Zhou JJ.. 2012. Expression of chemosensory proteins in the tsetse fly Glossina morsitans morsitans is related to female host-seeking behaviour. Insect Mol Biol. 21:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez JA, Bohuski E.. 2007. Total RNA extraction with Trizol Reagent and purification with Qiagen RNeasy Mini Kit. ©DGC, Bloomington, IN: Indiana University.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res. 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z.. 2011. Next-generation transcriptome assembly. Nat Rev Genet. 12:671–682. [DOI] [PubMed] [Google Scholar]
- Martin JW, Davis GE.. 2001. An updated classification of the recent Crustacea. Natural History Museum of Los Angeles County, Los Angeles, California: Science Series No; 39:1–124. [Google Scholar]
- McBride CS, Arguello JR.. 2007. Five Drosophila genomes reveal nonneutral evolution and the signature of host specialization in the chemoreceptor superfamily. Genetics 177:1395–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD.. 2013. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41:D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missbach C, Dweck HK, Vogel H, Vilcinskas A, Stensmyr MC, Hansson BS, Grosse-Wilde E.. 2014. Evolution of insect olfactory receptors. eLife 3:e02115.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missbach C, Vogel H, Hansson BS, Groβe-Wilde E.. 2015. Identification of odorant binding proteins and chemosensory proteins in antennal transcriptomes of the jumping bristletail Lepismachilis y-signata and the firebrat Thermobia domestica: evidence for an independent OBP-OR origin. Chem Senses. 40:615–626. [DOI] [PubMed] [Google Scholar]
- Moroz LL, Kocot KM, Citarella MR, Dosung S, Norekian TP, Povolotskaya IS, Grigorenko AP, Dailey C, Berezikov E, Buckley KM, et al. 2014. The ctenophore genome and the evolutionary origins of neural systems. Nature 510:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B.. 2008. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 5:621–628. [DOI] [PubMed] [Google Scholar]
- Nei M, Hughes A.. 1992. Balanced polymorphism and evolution by the birth-and-death process in the MHC loci In: Tsuji K, Aizawa M, Sasazuki T, editors. Proceedings of the Eleventh International Histocompatibility Workshop and Conference, held in Yokohama, Japan 1991. Oxford: Oxford University Press; p. 27–38. [Google Scholar]
- Nei M, Niimura Y, Nozawa M.. 2008. The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet. 9:951–963. [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP.. 2005. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 39:121–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoc PCT, Greenhalgh R, Dermauw W, Rombauts S, Bajda S, Zhurov V, Grbić M, Van de Peer Y, Van Leeuwen T, Rouzé P, et al. 2016. Complex evolutionary dynamics of massively expanded chemosensory receptor families in an extreme generalist chelicerate herbivore. Genome Biol Evol. 8:3323–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Nei M.. 2003. Evolution of olfactory receptor genes in the human genome. Proc Natl Acad Sci U S A. 100:12235–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström KJV, Sällman Almén M, Edstam MM, Fredriksson R, Schiöth HB.. 2011. Independent HHsearch, Needleman-Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol. 28:2471–2480. [DOI] [PubMed] [Google Scholar]
- Oakley TH, Wolfe JM, Lindgren AR, Zaharoff AK.. 2013. Phylotranscriptomics to bring the understudied into the fold: monophyletic ostracoda, fossil placement, and pancrustacean phylogeny. Mol Biol Evol. 30:215–233. [DOI] [PubMed] [Google Scholar]
- Parfrey LW, Grant J, Tekle YI, Lasek-Nesselquist E, Morrison HG, Sogin ML, Patterson DJ, Katz LA.. 2010. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst Biol. 59:518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi P, Iovinella I, Felicioli A, Dani FR.. 2014. Soluble proteins of chemical communication: an overview across arthropods. Front Physiol. 5:320.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi P, Zhou JJ, Ban LP, Calvello M.. 2006. Soluble proteins in insect chemical communication. Cell Mol Life Sci. 63:1658–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñalva-Arana DC, Lynch M, Robertson HM.. 2009. The chemoreceptor genes of the waterflea Daphnia pulex: many Grs but no Ors. BMC Evol Biol. 9:79.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirovano W, Feenstra KA, Heringa J.. 2008. PRALINE™: a strategy for improved multiple alignment of transmembrane proteins. Bioinformatics 24:492–497. [DOI] [PubMed] [Google Scholar]
- Price MB, Jelesko J, Okumoto S.. 2012. Glutamate receptor homologs in plants: functions and evolutionary origins. Front Plant Sci. 3:235.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Wang ET, Burge CB, Sandberg R.. 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 5:e1000598.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch EM, Lee CE, Wyngaard GA.. 2004. DNA-Feulgen cytophotometric determination of genome size for the freshwater-invading copepod Eurytemora affinis. Genome 47:559–564. [DOI] [PubMed] [Google Scholar]
- Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW.. 2010. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463:1079–1083. [DOI] [PubMed] [Google Scholar]
- Robertson HM. 2015. The insect chemoreceptor superfamily is ancient in animals. Chem Senses. 40:609–614. [DOI] [PubMed] [Google Scholar]
- Robertson HM, Gadau J, Wanner KW.. 2010. The insect chemoreceptor superfamily of the parasitoid jewel wasp Nasonia vitripennis. Insect Mol Biol. 19:121–136. [DOI] [PubMed] [Google Scholar]
- Robertson HM, Kent LB.. 2009. Evolution of the gene lineage encoding the carbon dioxide receptor in insects. J Insect Sci. 9:19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Thomas JH.. 2006. The putative chemoreceptor families of C. elegans.WormBook. http://www.wormbook.org/chapters/www_putativechemoreceptorfam/putativechemoreceptorfam.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Wanner KW.. 2006. The chemoreceptor superfamily in the honey bee, Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res. 16:1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Warr CG, Carlson JR.. 2003. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc Natl Acad Sci U S A. 100(Suppl 2):14537–14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV.. 2003. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol. 13:1512–1517. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP.. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. [DOI] [PubMed] [Google Scholar]
- Rynearson TA, Newton JA, Armbrust EV.. 2006. Spring bloom development, genetic variation, and population succession in the planktonic diatom Ditylum brightwellii. Limnol Oceanogr. 51:1249–1261. [Google Scholar]
- Saina M, Busengdal H, Sinigaglia C, Petrone L, Oliveri P, Rentzsch F, Benton R.. 2015. A cnidarian homologue of an insect gustatory receptor functions in developmental body patterning. Nat Commun. 6:6243.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M.. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gracia A, Vieira FG, Almeida FC, Rozas J.. 2011. Comparative genomics of the major chemosensory gene families in arthropods. Encycl Life Sci. 3:476–490. [Google Scholar]
- Sánchez-Gracia A, Vieira FG, Rozas J.. 2009. Molecular evolution of the major chemosensory gene families in insects. Heredity 103:208–216. [DOI] [PubMed] [Google Scholar]
- Sasaki G, Ishiwata K, Machida R, Miyata T, Su ZH.. 2013. Molecular phylogenetic analyses support the monophyly of Hexapoda and suggest the paraphyly of Entognatha. BMC Evol Biol. 13:236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R.. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao M-S, Chang J-M, Fan W-L, Lu M-YJ, Notredame C, Fang S, Kondo R, Li W-H.. 2015. Expression divergence of chemosensory genes between Drosophila sechellia and its sibling species and its implications for host shift. Genome Biol Evol. 7:2843–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Snell T, Morris P.. 1993. Sexual communication in copepods and rotifers. Hydrobiologia 255–256:109–116. [Google Scholar]
- Söding J, Biegert A, Lupas AN.. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sømme L. 1989. Adaptations of terrestrial arthropods to the alpine environment. Biol Rev. 64:367–407. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D.. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24:637–644. [DOI] [PubMed] [Google Scholar]
- Stewart S, Koh TW, Ghosh AC, Carlson JR.. 2015. Candidate ionotropic taste receptors in the Drosophila larva. Proc Natl Acad Sci U S A. 112:4195–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Uda Y, Ono Y, Nakagawa T, Suwa M, Yamaoka R, Touhara K.. 2009. Highly selective tuning of a silkworm olfactory receptor to a key mulberry leaf volatile. Curr Biol. 19:881–890. [DOI] [PubMed] [Google Scholar]
- Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A.. 2011. Differential expression in RNA-seq: a matter of depth. Genome Res. 21:2213–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N, Amrein H.. 2008. Atypical expression of Drosophila gustatory receptor genes in sensory and central neurons. J Comp Neurol. 506:548–568. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP.. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribolium Genome Sequencing Consortium. 2008. The genome of the model beetle and pest Tribolium castaneum. Nature 452:949–955. [DOI] [PubMed] [Google Scholar]
- Tusnady GE, Simon I.. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. [DOI] [PubMed] [Google Scholar]
- Verity P, Smetacek V.. 1996. Organism life cycles, predation, and the structure of marine pelagic ecosystems. Mar Ecol Prog Ser. 130:277–293. [Google Scholar]
- Vieira FG, Rozas J.. 2011. Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: origin and evolutionary history of the chemosensory system. Genome Biol Evol. 3:476–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira FG, Sánchez-Gracia A, Rozas J.. 2007. Comparative genomic analysis of the odorant-binding protein family in 12 Drosophila genomes: purifying selection and birth-and-death evolution. Genome Biol. 8:R235.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizueta J, Frías-López C, Macías-Hernández N, Arnedo MA, Sánchez-Gracia A, Rozas J.. 2017. Evolution of chemosensory gene families in arthropods: insight from the first inclusive comparative transcriptome analysis across spider appendages. Genome Biol Evol. 9:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reumont BM, Jenner RA, Wills MA, Dell’Ampio E, Pass G, Ebersberger I, Meyer B, Koenemann S, Iliffe TM, Stamatakis A, et al. 2012. Pancrustacean phylogeny in the light of new phylogenomic data: support for Remipedia as the possible sister group of Hexapoda. Mol Biol Evol. 29:1031–1045. [DOI] [PubMed] [Google Scholar]
- Vosshall LB, Amrein H, Morozov PS, Rzhetsky A, Axel R.. 1999. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell 96:725–736. [DOI] [PubMed] [Google Scholar]
- Vosshall LB, Stocker RE.. 2007. Molecular architecture of smell and taste in Drosophila. Annu Rev Neurosci. 30:505–533. [DOI] [PubMed] [Google Scholar]
- Wang Z, Singhvi A, Kong P, Scott K.. 2004. Taste representations in the Drosophila brain. Cell 117:981–991. [DOI] [PubMed] [Google Scholar]
- Wanner KW, Anderson AR, Trowell SC, Theilmann DA, Robertson HM, Newcomb RD.. 2007. Female-biased expression of odourant receptor genes in the adult antennae of the silkworm, Bombyx mori. Insect Mol Biol. 16:107–119. [DOI] [PubMed] [Google Scholar]
- Wanner KW, Robertson HM.. 2008. The gustatory receptor family in the silkworm moth Bombyx mori is characterized by a large expansion of a single lineage of putative bitter receptors. Insect Mol Biol. 17:621–629. [DOI] [PubMed] [Google Scholar]
- Whelan NV, Kocot KM, Moroz LL, Halanych KM.. 2015. Error, signal, and the placement of Ctenophora sister to all other animals. Proc Natl Acad Sci U S A. 112:5773–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler G, Dodson JJ, Lee CE.. 2008. Heterogeneity within the native range: population genetic analyses of sympatric invasive and noninvasive clades of the freshwater invading copepod Eurytemora affinis. Mol Ecol. 17:415–430. [DOI] [PubMed] [Google Scholar]
- Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 39:306–314. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R.. 2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 19:908–917. [DOI] [PubMed] [Google Scholar]
- Ye Z, Xu S, Spitze K, Asselman J, Jiang X, Ackerman MS, Lopez J, Harker B, Raborn RT, Thomas WK, et al. 2017. A new reference genome assembly for the microcrustacean Daphnia pulex. G3 7:1405–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen J, Sehn JK, Catton K, Kramer A, Sarnelle O.. 2011. Pheromone trail following in three dimensions by the freshwater copepod Hesperodiaptomus shoshone. J Plankton Res. 33:907–916. [Google Scholar]
- Zerbino DR, Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YV, Ni J, Montell C.. 2013. The molecular basis for attractive salt-taste coding in Drosophila. Science 340:1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P.. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics 12:S2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Stone EA, Mackay TFC, Anholt RRH.. 2009. Plasticity of the chemoreceptor repertoire in Drosophila melanogaster. PLoS Genet. 5:e1000681.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lindsay H, Robinson MD.. 2014. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 42:e91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Slone JD, Rokas A, Berger SL, Liebig J, Ray A, Reinberg D, Zwiebel LJ.. 2012. Phylogenetic and transcriptomic analysis of chemosensory receptors in a pair of divergent ant species reveals sex-specific signatures of odor coding. PLoS Genet. 8:e1002930.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XH, Ban LP, Iovinella I, Zhao LJ, Gao Q, Felicioli A, Sagona S, Pieraccini G, Pelosi P, Zhang L, et al. 2013. Diversity, abundance, and sex-specific expression of chemosensory proteins in the reproductive organs of the locust Locusta migratoria manilensis. Biol Chem. 394:43–54. [DOI] [PubMed] [Google Scholar]
- Zwick A, Regier JC, Zwickl DJ.. 2012. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS ONE 7:e47450.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.