
Figure 1.
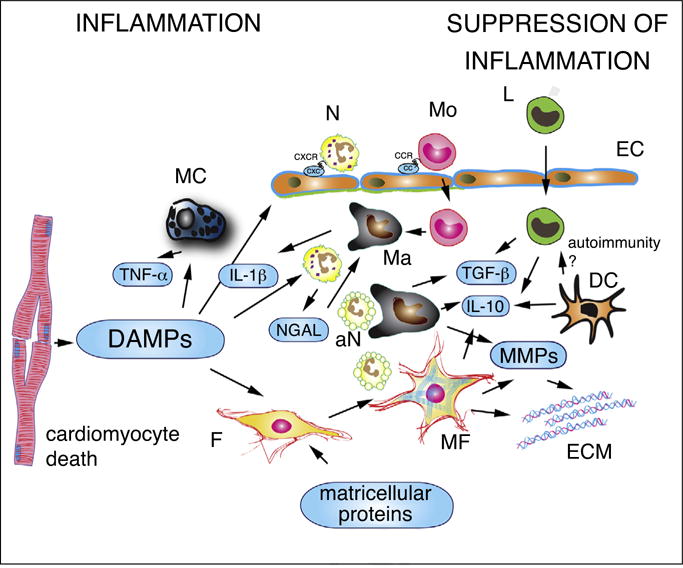
The cell biology of the inflammatory response following myocardial infarction. Dying cardiomyocytes release damage-associated molecular patterns (DAMPs), activating inflammatory signaling in resident macrophages (Ma), mast cells (MC) and fibroblasts (F) and in infiltrating leukocytes. Secretion of pro-inflammatory cytokines (such as IL-1β and TNF-α) stimulates expression of CXC and CC chemokines in endothelial cells (EC), promoting recruitment of neutrophils (N), monocytes (Mo) and lymphocytes (L). In addition to their pro-inflammatory actions, neutrophils may also trigger polarization of reparative macrophages. Phagocytotic macrophages clear the infarct from dead cells (including apoptotic neutrophils, aN) and acquire anti-inflammatory properties, releasing TGF-β and IL-10. Recently published experimental work suggests that myofibroblasts (MF) may also play a role in phagocytosis of dead cells and in negative regulation of the inflammatory reaction. Dendritic cells (DC) are also activated in the infarcted myocardium and have been suggested to inhibit pro-inflammatory signaling by secreting IL-10. Activation of autoimmune pathways by dendritic cells has been suggested, but remains poorly documented in myocardial infarction. Matricellular proteins play an important role in regulating inflammatory and reparative responses in the infarcted myocardium. Matrix metalloproteinases (MMPs) secreted by hematopoietic cells and fibroblasts are involved in extracellular matrix metabolism and regulate inflammation by modulating activity of inflammatory mediators.