

Abstract
Key points
Coronary wave intensity analysis (WIA) is an emerging technique for assessing upstream and downstream influences on myocardial perfusion.
It is thought that a dominant backward decompression wave (BDWdia) is generated by a distal suction effect, while early‐diastolic forward decompression (FDWdia) and compression (FCWdia) waves originate in the aorta.
We show that wave reflection also makes a substantial contribution to FDWdia, FCWdia and BDWdia, as quantified by a novel method. In 18 sheep, wave reflection accounted for ∼70% of BDWdia, whereas distal suction dominated in a computer model representing a hypertensive human.
Non‐linear addition/subtraction of mechanistically distinct waves (e.g. wave reflection and distal suction) obfuscates the true contribution of upstream and downstream forces on measured waves (the ‘smoke and mirrors’ effect).
The mechanisms underlying coronary WIA are more complex than previously thought and the impact of wave reflection should be considered when interpreting clinical and experimental data.
Abstract
Coronary arterial wave intensity analysis (WIA) is thought to provide clear insight into upstream and downstream forces on coronary flow, with a large early‐diastolic surge in coronary flow accompanied by a prominent backward decompression wave (BDWdia), as well as a forward decompression wave (FDWdia) and forward compression wave (FCWdia). The BDWdia is believed to arise from distal suction due to release of extravascular compression by relaxing myocardium, while FDWdia and FCWdia are thought to be transmitted from the aorta into the coronary arteries. Based on an established multi‐scale computational model and high‐fidelity measurements from the proximal circumflex artery (Cx) of 18 anaesthetized sheep, we present evidence that wave reflection has a major impact on each of these three waves, with a non‐linear addition/subtraction of reflected waves obscuring the true influence of upstream and downstream forces through concealment and exaggeration, i.e. a ‘smoke and mirrors’ effect. We also describe methods, requiring additional measurement of aortic WIA, for unravelling the separate influences of wave reflection versus active upstream/downstream forces on coronary waves. Distal wave reflection accounted for ∼70% of the BDWdia in sheep, but had a lesser influence (∼25%) in the computer model representing a hypertensive human. Negative reflection of the BDWdia at the coronary–aortic junction attenuated the Cx FDWdia (by ∼40% in sheep) and augmented Cx FCWdia (∼5‐fold), relative to the corresponding aortic waves. We conclude that wave reflection has a major influence on early‐diastolic WIA, and thus needs to be considered when interpreting coronary WIA profiles.
Keywords: wave intensity analysis, coronary, haemodynamics, wave reflection, flow waveform, backward expansion wave, suction wave
Key points
Coronary wave intensity analysis (WIA) is an emerging technique for assessing upstream and downstream influences on myocardial perfusion.
It is thought that a dominant backward decompression wave (BDWdia) is generated by a distal suction effect, while early‐diastolic forward decompression (FDWdia) and compression (FCWdia) waves originate in the aorta.
We show that wave reflection also makes a substantial contribution to FDWdia, FCWdia and BDWdia, as quantified by a novel method. In 18 sheep, wave reflection accounted for ∼70% of BDWdia, whereas distal suction dominated in a computer model representing a hypertensive human.
Non‐linear addition/subtraction of mechanistically distinct waves (e.g. wave reflection and distal suction) obfuscates the true contribution of upstream and downstream forces on measured waves (the ‘smoke and mirrors’ effect).
The mechanisms underlying coronary WIA are more complex than previously thought and the impact of wave reflection should be considered when interpreting clinical and experimental data.
Introduction
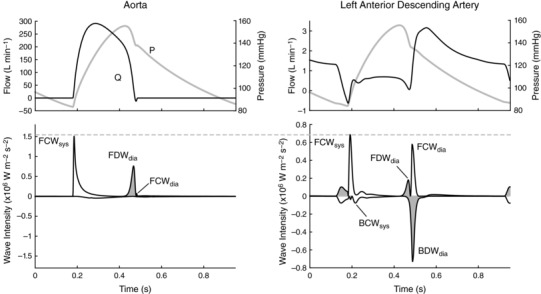
Wave intensity analysis (WIA) has been used to study the upstream and downstream forces contributing to the distinctive pattern of coronary arterial blood flow (Sun et al. 2000, 2004; Davies et al. 2006a; Hadjiloizou et al. 2008; Smolich & Mynard, 2016), with ‘waves’ (i.e. incremental changes in blood pressure and velocity) arising from active forces such as myocardial contraction/relaxation or from wave reflection. Given the diastolic dominance of coronary arterial flow, there has been particular interest in the wave dynamics underlying the early‐diastolic flow surge and how waves around this time are affected by various forms of heart disease and medical intervention (Davies et al. 2006a, 2011; Kyriacou et al. 2012; Lockie et al. 2012; De Silva et al. 2013; Claridge et al. 2015). Three main waves have been consistently identified during this early‐diastolic phase of the cardiac cycle (Fig. 1). [In this paper ‘early‐diastole’ refers to the period soon after the left ventricle starts to relax, and hence encompasses the short period before aortic valve closure, sometimes referred to as protodiastole.] A flow‐increasing backward decompression wave (BDWdia) is believed to arise from the release of extravascular compressive forces on intramyocardial vessels, producing a suction effect that propagates back towards the coronary ostium (Davies et al. 2006a). In addition, two forward waves, a flow‐decreasing forward decompression wave (FDWdia) and a flow‐increasing forward compression wave (FCWdia), are thought to arise from transmission of corresponding aortic waves into the coronary arteries; these aortic waves are generated by the early diastolic fall in left ventricular (LV) cavity pressure and the pressure/flow ‘rebound’ caused by aortic valve closure respectively (Davies et al. 2006a).
Figure 1. Circumflex coronary pressure (P), flow (Q) and wave intensity from an adult sheep.
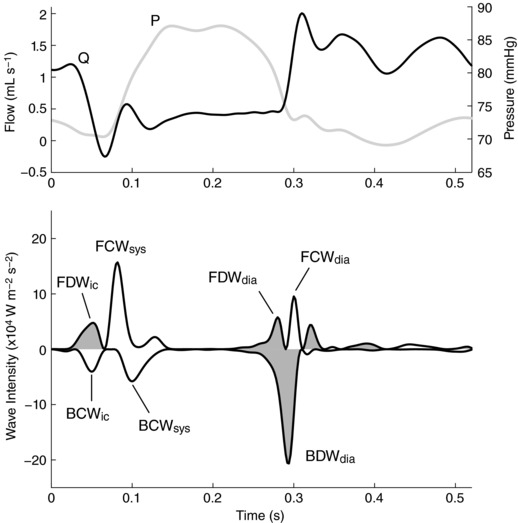
The major waves, which are also present in humans, are labelled according to wave type (FCW, FDW, BCW, BDW; forward/backward compression/decompression wave) and phase of the cardiac cycle (ic, isovolumic contraction; sys, early‐systole; dia, early‐diastole). Decompression (i.e. pressure‐decreasing) waves are shaded.
While the current explanations for early‐diastolic coronary arterial waves are intuitive, they have not been confirmed by any direct approach, while close inspection of published coronary WIA data suggest that the picture is not yet complete. Specifically, although correlations have been observed between BDWdia magnitude and indices of isovolumic relaxation derived from LV cavity pressure, the reported R 2 values (0.21–0.35) suggest that more than 60% of the variability in BDWdia magnitude is currently unexplained (Kyriacou et al. 2012; Ladwiniec et al. 2016). Indeed, it is unclear why the BDWdia is much larger than a backward compression wave occurring during isovolumic contraction (BCWic, Fig. 1), despite published data suggesting that the rate of change of intramyocardial pressure rise (leading to BCWic) and fall (leading to BDWdia) are comparable (Heineman & Grayson, 1985; Stein et al. 1985; Rabbany et al. 1989). In addition, although it is generally assumed that the FDWdia and FCWdia are simply transmitted from the aorta into the coronary arteries (Davies et al. 2006a), several published figures suggest that the FDWdia and FCWdia may be less and more prominent, respectively, in the coronary arteries than aorta (Hughes et al. 2008; Lu et al. 2012), with the basis of such differences yet to be defined.
A potentially significant but largely unexplored factor that may influence the early‐diastolic coronary waves is wave reflection, with backward waves reflecting proximally at the coronary ostium and forward waves reflecting distally in the coronary arterial network. Due to the large impedance mismatch between the coronary arteries and aorta (Davies et al. 2006a), the BCWic is considered to undergo near‐complete negative reflection at the coronary ostium, leading to a subsequent forward decompression wave (FDWic, Fig. 1); however, it is unclear whether the BDWdia also undergoes such reflection and what effect this might have on the FCWdia and FDWdia. In addition, partial reflection of the early‐systolic forward compression wave (FCWsys) in the coronary arterial bed leads to a second backward compression wave (BCWsys), but it is unclear whether the FDWdia is similarly reflected, and whether the resulting reflected decompression wave might contribute to the BDWdia.
The foregoing phenomena have particular relevance due to the possibility that a measured wave intensity peak (herein referred to as a ‘wave’) may arise from a combination of factors, such as the simultaneous influence of an active force and wave reflection. Intuitively, the combination of two mechanistically distinct waves would be expected to have an additive effect if both waves are compression waves or both are decompression waves, or a cancelling effect if compression and decompression waves combine. However, the specific nature of such additive or cancelling effects, and a method for unravelling the separate contributions of an active force and wave reflection to a composite wave, have yet to be defined.
The aim of this study was to investigate the extent to which each of the three main early diastolic coronary waves, currently presumed to simply arise from upstream (FDWdia and FCWdia) and downstream (BDWdia) active forces (Davies et al. 2006a), are also influenced by wave reflection. We first demonstrate that there is a non‐linear additive or cancelling effect on wave intensity when two mechanisms contribute to a measured wave. Using a previously described multi‐scale computational model of the coronary circulation (Mynard et al. 2014; Mynard & Smolich, 2016a), we show that wave reflection effects lead to concealment of the coronary FDWdia and augmentation of the BDWdia and FCWdia, which we term a ‘smoke and mirrors’ effect. We also describe methods for unravelling the contributions of active forces and wave reflection underlying measured coronary waves. These principles and methods were initially tested with the computational model and then examined in an experimental model where simultaneous aortic and coronary WIA were obtained under baseline conditions and following an increase in arterial blood pressure, with or without coronary vasoconstriction.
Methods
Ethical approval
Experimental studies (described towards the end of this section) were approved by the Monash University Animal Ethics Committee and conformed to the guidelines of the National Health and Medical Research Council of Australia.
Wave intensity
Wave intensity is defined as the product of incremental changes in pressure and velocity (dPdU) (Parker & Jones, 1990). Since this quantity is sample time‐dependent, a time‐corrected wave intensity is often used, calculated as WI = (dP/dt)(dU/dt) (Ramsey & Sugawara, 1997; Penny et al. 2008). The forward component (WI+) and backward component (WI−) of wave intensity are calculated via WI± = ±(dP/dt ± ρc dU/dt)2/(4ρc), with forward waves having positive WI and backward waves having negative WI. Cumulative wave intensity (CI), related to the energy of a wave (Davies et al. 2006a; Mynard & Smolich, 2016b), is defined as the integral of WI± over the duration of a given wave. Waves that have a pressure‐increasing effect are called ‘compression waves’, whereas waves that have a pressure‐decreasing effect are called ‘decompression waves’ (sometimes also referred to as ‘suction’ or ‘expansion’ waves). The four possible wave types are forward compression (FCW), forward decompression (FDW), backward compression (BCW) and backward decompression (BDW) waves.
Non‐linear addition and subtraction of coronary waves
Since coronary arterial waves may arise from an active mechanism (e.g. a distal suction effect arising in the intramyocardial vessels) and also from wave reflection, it is possible that both phenomena could simultaneously influence the wave intensity profile. In this section, we show that the addition or subtraction of two mechanistically distinct waves is non‐linear (i.e. 1 + 1 ≠ 2). For simplicity of notation, in this section we use the non‐time‐corrected form of wave intensity, but the same principles also apply to the time‐corrected form.
Consider the situation where two waves propagate in the same direction, but one arises via passive reflection of some incident wave (dPdU reflected) and the other is generated from an active process such as a change in extravascular pressure due to myocardial relaxation (dPdU active). If these two mechanistically distinct waves affect pressure and velocity at the same time, the total change in pressure will be dP reflected + dP active and the total change in velocity will be dU reflected + dU active. The wave intensity of the resulting combined wave is therefore
| (1) |
Importantly, we see that these waves do not add linearly, that is
| (2) |
For example, consider two backward decompression waves (BDW1 and BDW2) for which dP and dU have numerical values of −5 and 5 respectively for both waves, with BDW1 arising from wave reflection and BDW2 arising from myocardial relaxation (Fig. 2 A). Wave intensity of each wave in isolation would be −25 (dPdU = −5 × 5). However, if these two waves occur at the same time, the resulting combined BDW will not have an amplitude of −50, but rather dPdU combined = (−5 − 5) × (5 + 5) = −100. This illustrates the principle that when two compression waves or two decompression waves combine, the resultant wave is much larger than the sum of the contributing waves in isolation.
Figure 2. Illustration of non‐linear wave addition and subtraction.
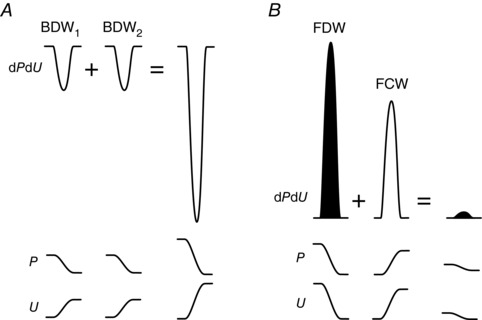
A, when two backward decompression waves (BDW1 and BDW2) combine, the wave intensity (dPdU) of the resultant single combined wave is greater than the sum of the dPdU of the two waves in isolation. B, when a forward decompression wave (FDW) combines with a forward compression wave (FCW), there is a non‐linear cancelling effect. The non‐linear nature of wave intensity addition and subtraction may be understood by assessing the underlying pressure (P) and velocity (U) changes associated with the waves.
Now consider what happens if two waves with opposing pressure effects combine. For example, if an FDW with wave intensity dPdU = −5 × −6 = 30 combines with an FCW with wave intensity dPdU = 4 × 5 = 20, the resulting combined wave does not have an amplitude of 20 + 30 = 50, nor is it 30 − 20 = 10; rather it is dPdU combined = (4 − 5) × (5 − 6) = 1 (Fig. 2 B). Thus, there is a non‐linear cancelling effect when a compression wave combines with a decompression wave.
Coronary reflection and myocardial contraction/relaxation
With these principles of wave addition and subtraction in mind, we therefore tested two main hypotheses:
Hypothesis 1 (Fig. 3 A): The measured early‐diastolic coronary BDW (BDWdia) arises from a combination of two distinct processes, (i) an active suction effect caused by a rapid fall in vascular external compression due to myocardial relaxation (mr), which in isolation would cause a backward decompression wave designated BDWmr; and (ii) passive distal reflection of the early diastolic FDW transmitted from the aorta into the coronary arteries (FDWA→C), which in isolation would cause a wave designated BDWref. The BDWmr and BDWref add non‐linearly to produce the BDWdia, which is thus substantially larger than the sum of the intensities of the individual waves. This hypothesis is illustrated in Fig. 3 A.
Hypothesis 2 (Fig. 3 B): The coronary BDWdia undergoes almost complete negative reflection when it reaches the coronary ostium due to the much lower impedance of the aorta compared with the coronary artery. The resulting reflected FCW (FCWref) has a non‐linear cancelling effect on the aortic FDW transmitted into the coronary arteries (FDWA→C), with the resultant measured coronary wave (FDWdia) being relatively small compared with aortic FDWdia. In addition, the FCWref has a non‐linear additive effect on the FCW related to valve closure that is transmitted from the aorta into the coronary arteries (FDWA→C), with the resultant combined coronary wave (FDWdia) being relatively large compared with aortic FDWdia. This hypothesis is illustrated in Fig. 3 B.
Figure 3. Illustration of the mechanisms underlying early diastolic coronary waves.
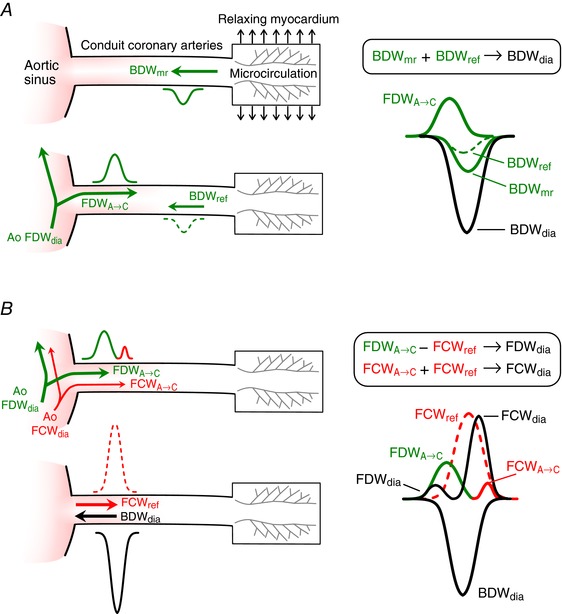
A, the measured BDWdia arises in part from a distal suction effect caused by myocardial relaxation (BDWmr); a second contribution to BDWdia arises from distal reflection (BDWref) of the aortic forward decompression wave (Ao FDWdia) that passes into the coronary arteries (FDWA→C). The BDWmr and BDWref add non‐linearly to produce a prominent BDWdia. B, the BDWdia undergoes near‐complete negative reflection at the coronary ostium, where it encounters a large drop in characteristic impedance. This produces a reflected forward compression wave (FCWref) that non‐linearly subtracts from the FDWA→C and augments the valve closure‐related forward compression wave (FCWA→C) that originates in the aorta (Ao FCWdia), leading to the measured early‐diastolic forward waves (FDWdia and FCWdia). Measured waves are indicated in black while component compression and decompression waves are indicated in red and green respectively (colour in online version only). Waves arising from reflection are indicated with dashed lines. [Color figure can be viewed at wileyonlinelibrary.com]
Distinguishing wave reflection and upstream/downstream forces
Based on the above principles, we propose that the coronary FDWA→C, i.e. the coronary FDWdia that would occur in the absence of BDWdia reflection, may be estimated by assuming that the aortic FCWsys and FDWdia are identically transmitted into the coronary artery, and hence the following ratios (known as transmission coefficients) should also be approximately equal:
| (3) |
This may be rearranged to estimate the coronary FDWA→C, which is the only unknown:
| (4) |
In the same way, the coronary FCWA→C may also be estimated as
| (5) |
If we calculate an early systolic coronary reflection index as BCWsys/FCWsys and assume that the same degree of wave reflection occurs during early diastole (this assumption will be examined in the Results section using the computational model), then the BDWref can be estimated from FDWA→C via
| (6) |
Equations (4) to (6) can be applied to peak wave intensity, cumulative intensity (wave area) or incremental changes in pressure and velocity (dP ± and dU ±). The coronary BDWmr intensity can then be estimated by (i) calculating dP − and dU − associated with BDWref, via eqn (6), (ii) calculating dP − and dU − associated with BDWmr, by subtracting values for BDWref from values for BDWdia, and (iii) calculating BDWmr wave intensity as the product of the resulting BDWmr dP − and dU − values. This may be expressed as
| (7) |
In this equation, dP and dU may be replaced by dP −/dt and dU −/dt for calculation of time‐corrected BDWmr. The contribution of BDWmr to the measured BDWdia may then be quantified as
| (8) |
and expressed as a percentage, noting that this is not the same as BDWmr/BDWdia due to the non‐linear addition effects described above. Note that eqn (7), and hence also eqn (8), depend on instantaneous values of dP and dU and can therefore only be used in relation to peak intensity. This means that the cumulative intensity of the BDWref but not the BDWmr can be estimated.
Model description
The model used in this study is based on our previous work (Mynard et al. 2012, 2014; Mynard & Smolich, 2016a). Aside from some input parameters discussed below, all aspects of the model have been described in these previous papers and we therefore provide only a brief summary below. Model parameters are provided in the Appendix.
First, a one‐dimensional (1D) model of the major right dominant conduit coronary arterial system was derived from measurements obtained in humans by Dodge et al. (1992) (Fig. 4 A). The standard non‐linear 1D equations in these networks were solved as in Mynard & Nithiarasu (2008), making use of a physiologically realistic non‐linear elastic pressure–area relationship (Mynard et al. 2012) and the approximate velocity profile method of Bessems et al. (2007) to estimate the viscous friction term.
Figure 4. Schematics of the model components.
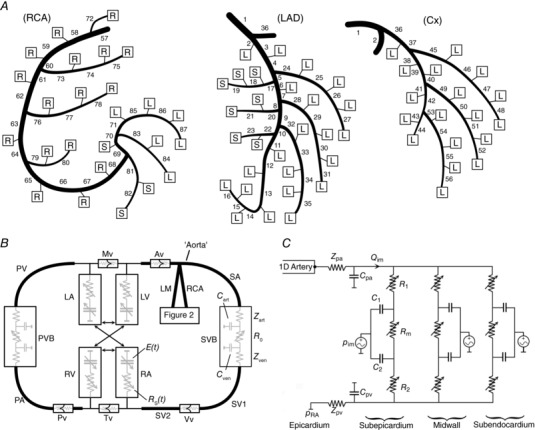
A, one‐dimensional (1D) model of the conduit coronary arteries; B, closed‐loop cardiovascular model within which the coronary model resides; C, the coronary microcirculation model. Boxes in A indicate attachment of terminal 1D segments to instances of the lumped parameter microcirculatory model shown in C, which supply parts of the right ventricular free wall (‘R’ boxes), left ventricular free wall (‘L’ boxes) or septum (‘S’ boxes). Full details of the right dominant coronary network shown in A are provided in Mynard & Smolich (2016a). Note that the proximal end of the SA segment is referred to as the aorta. Abbreviations: Av, aortic valve; Cx, circumflex coronary artery; LA, left atrium; LM, left main coronary artery; LV, left ventricle; Mv, mitral valve; PA, pulmonary artery; Pv, pulmonary valve; PV, pulmonary vein; PVB, pulmonary vascular bed; RA, right atrium; RCA, right coronary artery; RV, right ventricle; SA, systemic artery; SVB, systemic vascular bed; SV, systemic vein; Tv, tricuspid valve; Vv, venous valve.
Second, the coronary model was placed within a closed‐loop model of the cardiovascular system (Mynard et al. 2012) that included the four heart chambers and valves, lumped systemic and pulmonary vascular beds, and single 1D segments approximating each of the major vascular networks (Fig. 4 B); a similar model but with anatomically based vascular networks was described by Mynard and Smolich (2015), but this level of detail is not required for the present study.
Third, the coronary microcirculation was represented by a lumped parameter model (Fig. 4 C), instances of which were inserted at all distal outlets of the 1D conduit artery model (boxes in Fig. 4 A). This microvascular model, based on studies by Bruinsma et al. (1988) and Spaan et al. (2000), was divided into subepicardial, midwall and subendocardial layers. Each layer consisted of two compartments, whose volumes were governed by the compliances C 1 and C 2, approximately corresponding to small arteries and veins respectively in the layer. Each layer also contained three volume‐dependent resistances, R 1 and R 2, which were modulated by the volume in compartments 1 and 2 respectively, and a middle resistance (R m) modulated by the volume in both compartments. Compartment volume depended on the difference between intravascular pressure and intramyocardial pressure (p im). As in Mynard et al. (2014), we assumed that p im arises from (i) cavity‐induced extracellular pressure (CEP), i.e. transmission of ventricular cavity pressure into the ventricular wall, and (ii) shortening‐induced intracellular pressure (SIP), arising from the thickening of shortening myofibrils that leads to compression of adjacent blood vessels (Heineman & Grayson, 1985; Rabbany et al. 1989; Algranati et al. 2010). For a full description of the model, including equations, see Mynard et al. (2014).
Parameters for the cardiovascular model are provided in the Appendix and were chosen to represent an older adult with stage I hypertension, typical of patients encountered in clinical studies of coronary wave intensity (Davies et al. 2006a; Hadjiloizou et al. 2008; Narayan et al. 2015), with aortic pressures of 155/84 mmHg, cardiac output 4.9 L min−1, systemic vascular resistance 1.8 mmHg.s mL−1 and aortic wave speed 9.0 m s−1 (Mitchell et al. 2003). Distributed arterial wave reflection was approximated by introducing linear tapering in the single equivalent systemic artery. Coronary resistance was adjusted using an iterative algorithm (Mynard et al. 2014; Mynard & Smolich, 2016a) to achieve a total coronary flow equal to 4.5% of cardiac output; due to the higher perfusion pressure, a 52–90% increase in intramyocardial resistance was required compared with the original model, depending on the region and transmural layer. Note that the model was not intended to represent the sheep studied in the experimental model; rather, it was used to investigate wave dynamics in a system approximately representative of patients encountered clinically.
Model‐based analysis of coronary wave mechanisms
With the reference simulation of coronary haemodynamics established, we sought to elucidate the mechanisms underlying observed waves with two ‘virtual experiments’.
First, to identify waves generated from within the myocardium, aortic waves were prevented from passing into the coronary arteries by enforcing a reflection coefficient of −1 at the coronary ostia, achieved by prescribing a constant end‐diastolic pressure at the boundary; this maintained the same reflection conditions for backward‐running coronary waves. To ensure this did not cause any indirect changes to the intramyocardial pump effect, we also prescribed the same time‐varying intramyocardial pressures and resistances as in the reference case.
Second, to elucidate the interactions between waves transmitted from the aorta into the coronary arteries and those originating in the coronary microcirculation, we took the aortic pressure waveform from the reference case and prescribed this at the coronary inlets after applying a phase shift (i.e. time delay) that varied between −110 ms and +110 ms in 10‐ms steps (a total of 23 simulations). This meant that all aortic waves passing into the coronary arteries were variably delayed whereas waves arising from intramyocardial pump effects in the microcirculation were fixed, allowing clear identification of the origin of different waves and the effects of wave addition and subtraction.
We also tested the assumption that the coronary reflection index is similar during early systole and early diastole. Starting with the reference simulation, this was done by (i) removing the intramyocardial pressure source (p im in Fig. 4 C) to ensure backward waves arose only via reflection and (ii) retaining the p im‐related changes in coronary resistance from the reference simulation, noting that changes in resistance are likely to be a key factor determining the instantaneous reflection index. Then, the reflection index (BCWsys/FCWsys) at different times in the cardiac cycle was assessed in the proximal left anterior descending artery (LAD) by introducing delays in the incoming aortic waves. Distal reflection indexes during early systole (BCWsys/FCWsys) and early diastole (BDWdia/FDWdia) were also assessed in all terminal 1D vessels (no delays applied).
Unless otherwise stated, results are presented for the proximal LAD, since this is frequently the subject of clinical investigation, but similar findings were also obtained in other vessels, including the circumflex artery. A sensitivity analysis was performed to evaluate to what extent the contribution of wave reflection versus myocardial relaxation on BDWdia depends on aortic mean and pulse pressures (adjusted via systemic vascular bed resistance and aortic wave speed respectively), intramyocardial resistance (parallel combination of R 1 from all layers, see Fig. 4 C) or compliance (C 1 and C pa), monitoring location along the entire length of the LAD or coronary wave speed.
Experimental preparation
The experimental preparation was similar to that described previously (Penny et al. 2008). Eighteen Border‐Leicester cross ewes weighing 49.6 ± 4.8 kg (mean ± SD) were anaesthetized with an intramuscular injection of ketamine (5 mg kg−1) and xylazine (0.1 mg kg−1) followed by intravenous α‐chloralose (25–50 mg kg−1). Anaesthesia was maintained with intravenous α‐chloralose infused at a rate of 12–25 mg kg h−1. After intubation of the trachea, animals were ventilated with a large animal respirator (model 607; Harvard Apparatus, Dover, MA, USA) with ventilation adjusted to maintain arterial O2 tension at 100–120 mmHg and arterial CO2 tension at 35–40 mmHg. Body temperature was maintained at 39–40°C with a heating pad and towel covering.
The neck was incised in the midline, and polyvinyl catheters were advanced through the left external jugular vein to the superior vena cava for fluid and drug infusion. A 5‐Fr. micromanometer‐tipped catheter (MPC‐500; Millar Instruments, Houston, TX, USA) was inserted into the left common carotid artery, and its tip was advanced into the ascending aorta, just above the leaflets of the aortic valve, to measure high‐fidelity blood pressure. After exposure of the heart and central vessels through a left thoracotomy performed in the fourth intercostal space and incision of the pericardium over the pulmonary trunk and left atrium, cannulae were inserted through purse string sutures in the aortic arch and left atrial appendage, and connected to fluid‐filled polyvinyl tubing. Transit time flow probes (Transonics Systems, Ithaca, NY, USA) were placed around the ascending aorta (20 or 24 mm) and proximal circumflex coronary artery (2 or 4 mm). A second MPC‐500 catheter was inserted through the roof of the left atrium and passed across the mitral valve into the LV cavity.
Experimental protocol
Aortic blood pressure measured via the fluid‐filled catheter was referenced to atmospheric pressure at the level of the mid‐thoracic vertebral spines and calibrated against a manometer before each experiment. Aortic and LV micromanometers were connected to transducer control units (TCB‐500; Millar Instruments), while ascending aortic and coronary flow probes were interfaced with a flowmeter (model T206; Transonic Systems). Animals underwent one of two interventions: (i) in 11 animals, aortic blood pressure and coronary vascular resistance were increased by inhibition of NO synthesis via intravenous infusion of the stereospecific NO synthase inhibitor N ω‐nitro‐l‐arginine (l‐NNA, 25 mg kg−1) over 10 min (Penny & Smolich, 2002); (ii) in seven animals, mean central aortic blood pressure was raised to a similar level as in the l‐NNA studies via constriction of the thoracic descending aorta and brachiocephalic trunk using adjustable snares. Recordings of 20 s duration were digitized at 500 Hz under baseline conditions, and after haemodynamics had stabilized after each intervention. At the end of the study, animals were killed with an overdose of pentobarbitone sodium (100 mg kg−1).
Data analysis
Experimental data were analysed with a custom script written in Spike2 (Cambridge Electronic Design, Cambridge, UK). Signals were low pass filtered (cut‐off 48 Hz) to remove electrical interference and beat onset was defined as the time when LV pressure began to rise, detected via signal curvature according to a validated algorithm (Mynard et al. 2008). Analysis was performed on ensemble‐averaged data derived from at least 10 individual beats.
For WIA, mean blood velocity (U) was estimated from measured flow and cross‐sectional area derived from the flow probe diameter (Hollander et al. 2001; Penny et al. 2008). Proximal circumflex coronary blood pressure (at the location of flow measurement) was assumed to be identical to ascending aortic blood pressure (Pijls et al. 2000; Brosh et al. 2002), aside from a small time delay determined by aligning the early systolic upstroke of pressure and coronary flow. Blood density (ρ) was assumed to be 1.05 g cm−3. Wave speed (c) was calculated via the PU‐loop method for the aorta (Khir et al. 2001) and via the sum of squares method for the circumflex coronary artery (Davies et al. 2006b).
Coronary vascular resistance was calculated as [mean aortic pressure – mean left atrial pressure]/mean circumflex artery (Cx) flow. To define the association between the coronary BDWdia and myocardial relaxation, the peak negative rate of change of LV pressure (LV dP/dt min) and the time constant of relaxation (τ) were calculated (Mirsky, 1984). The latter was obtained by fitting the equation LVP = a 1 exp[−(t − t 0)/τ] + a 0 to LV pressure (LVP), where a 0 and a 1 are fit coefficients, t 0 is the time of dP/dt min and the fit was performed between t 0 and the time at which pressure had dropped to a value of P min + 0.1(P dpdtmin − P min), where P min and P dpdtmin are the minimum diastolic pressure and the pressure at dP/dt min.
Statistical analysis
Statistical analysis of the experimental data was performed with the MATLAB and Statistics Toolbox 2016a (The MathWorks, Inc., Natick, MA, USA). Data were tested for normality using the Lilliefors test and logarithmically transformed when non‐normal. When comparing wave magnitude between aortic and coronary arterial sites, wave intensities were normalized to the peak intensity of the FCWsys in the respective site. Comparisons were performed with repeated measures one‐way analysis of variance. Simple and stepwise multiple linear regression with pooled data was used to test for correlations between BDWdia magnitude and (i) LV dP/dtmin, (ii) LV τ and (iii) aortic FDWdia. Strength of correlation is reported as adjusted R 2 and interpreted as strong (0.5 < R 2 ≤ 1), moderate (0.3 < R 2 ≤ 0.5), weak (0.1 < R 2 ≤ 0.3) or very weak (R 2 ≤ 0.1). Data are reported as mean ± standard deviation, with significance defined as P < 0.05.
Results
Model‐derived data
Simulated pressure, flow and wave intensity signals in the aorta and coronary artery of the virtual older adult human are shown in Fig. 5. The coronary wave intensity profile contains all of the waves commonly identified in clinical studies, and the relative timing and magnitude of the respective waves is representative of published clinical data (Davies et al. 2006a). Importantly, the coronary FDWdia is almost half the size of the aortic FDWdia, relative to the FCWsys, whereas a prominent coronary FCWdia is present despite a negligibly small aortic FCWdia.
Figure 5. Pressure (P), flow (Q) and wave intensity from the aorta and proximal left anterior descending artery (LAD) of the computational model.
Decompression waves are shaded.
Figure 6 shows the effects of preventing all aortic waves from passing into the coronary arteries. This abolished both the coronary FCWsys and BCWsys, indicating that the BCWsys arises solely from reflection of the FCWsys. The FDWdia was also abolished as expected, but not the FCWdia, with the remaining wave (designated FCWref) clearly arising from negative reflection of the BDWmr, just as the FDWic arises from negative reflection of the BCWic. The remaining BDWmr peak was 69% smaller than the BDWdia, consistent with a contribution of wave reflection to the BDWdia (keeping in mind that the reflected wave adds non‐linearly to BDWmr).
Figure 6. Results of a virtual experiment in which all aortic waves were prevented from passing into the coronary arteries.
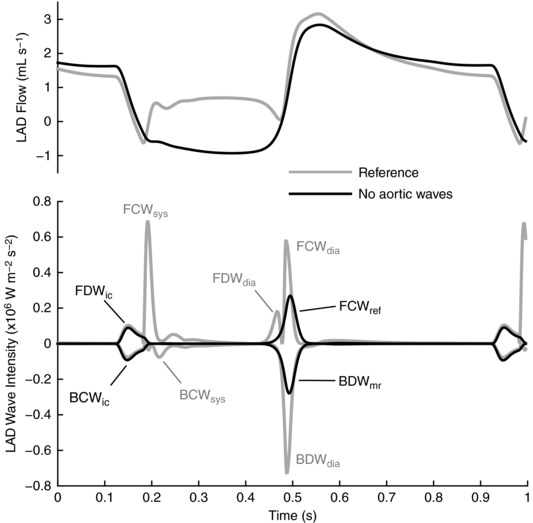
Figure 7 shows wave intensity profiles in a series of simulations in which a variable time delay (t d) was applied to waves entering the coronary arteries from the aorta, where t d = 0 corresponds to the reference simulation (noting that only the late systolic and early‐to‐mid diastolic periods are shown); Fig. 8 shows the corresponding peak intensity and cumulative intensity of observed waves versus t d. With a large negative or positive delay, an ensemble of three waves related to changes in aortic pressure are seen, namely the FDWA→C, which is reflected as a BDWref, then re‐reflected as a forward compression wave, referred to as FCWref1. This wave ensemble shifts with changing t d, whereas the two waves arising distally from myocardial relaxation are fixed (BDWmr and its reflected wave, FCWref2). Importantly, when −20 ≤ t d ≤ 30 ms, the BDWref and BDWmr overlap and only a single combined wave (BDWdia) is apparent, which is substantially larger (peak intensity 0.76 × 106 W m−2 s−2) than the sum of the BDWref and BDWmr waves in isolation (0.44 × 106 W m−2 s−2). Similarly, when the FCWref1 and FCWref2 overlap, the combined single wave (FCWdia) is larger (0.74 × 106 W m−2 s−2) than the sum of the two component waves in isolation (0.31 × 106 W m−2 s−2). Finally, a cancelling effect of FCWref2 on FDWA→C led to a resultant FDWdia that was up to 97% smaller than the FDWA→C. However, even when this cancellation was maximal (at t d = 30 ms), a prominent contribution of BDWref remained (black arrow in Fig. 7).
Figure 7. Results of a virtual experiment in which coronary ostial pressure was shifted with a variable time delay (t d), revealing the mechanistically distinct waves that combine (i.e. add or subtract) non‐linearly when aligned (selected time points are displayed from 23 simulations).
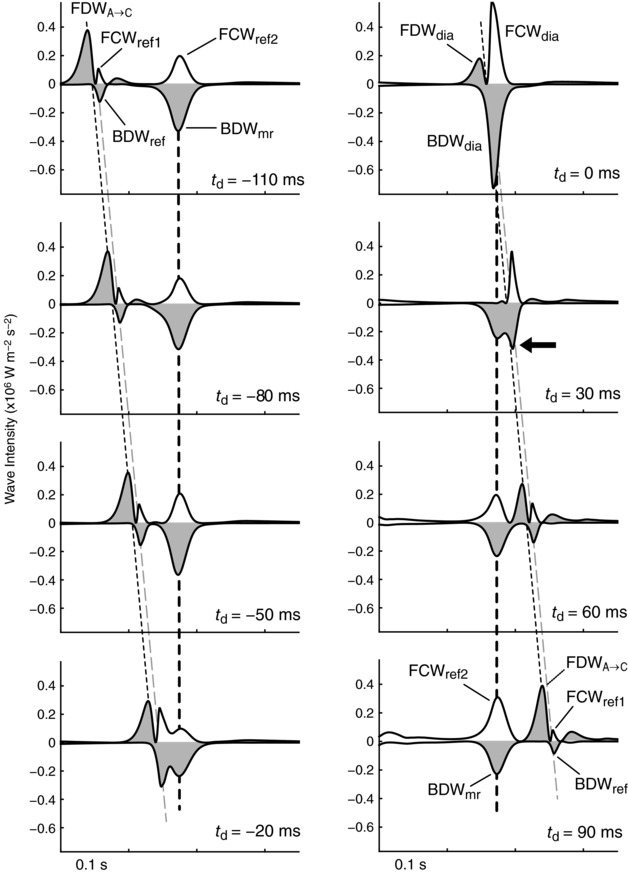
The initial ensemble of three waves seen when t d ≤ −50 ms arise from the passage of a forward decompression wave from the aorta into the coronary arteries (FDWA→C), and its distal reflection (BDWref) and proximal re‐reflection (FCWref1). The BDWmr arises from a distal suction effect and is then reflected proximally (FCWref2). The reference case shown in Fig. 5 corresponds to t d = 0. Dashed lines indicate the times of peak FDWA→C (thin black line), BDWref (grey line) and BDWmr (thick black line). Decompression waves are shaded.
Figure 8. Peak intensity and cumulative intensity of forward and backward waves shown in Fig. 7 versus the delay time (t d).
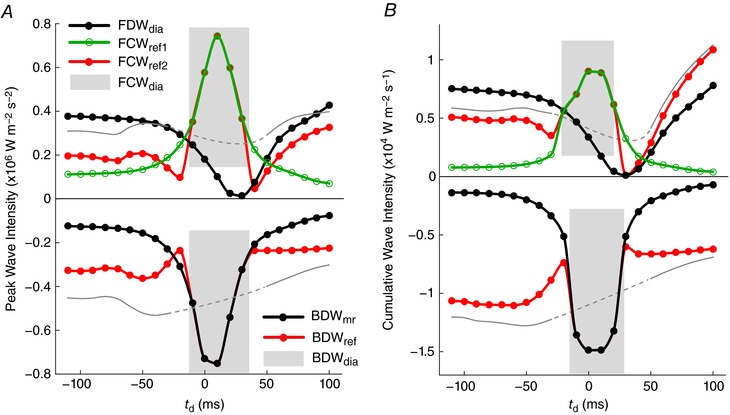
Shaded areas indicated where two waves overlap and appear as a single wave peak. Thin grey lines indicate the linear sum FCWref1 + FCWref2 (top) or BDWmr + BDWref (bottom), while the dashed segments indicate an approximate interpolation; these may be compared with the observed wave magnitude, which results from non‐linear summation. Note that FDWA→C in Fig. 7 refers to FDWdia in the absence of interactions with reflected waves. [Color figure can be viewed at wileyonlinelibrary.com]
Based on the simulations in Fig. 7, BDWmr accounted for 74% of the BDWdia (peak intensity), which was similar to the value of 80% estimated from the reference simulation (t d = 0) via eqn (7); the contribution of wave reflection was thus underestimated by 6%. Figure 9 A shows that coronary reflection index was higher during systole than diastole (difference of 0.08), as expected given the higher coronary impedance during systole. However, this value was almost identical during the early systolic and early diastolic periods in the proximal LAD (0.21 and 0.22 respectively, see shaded bars in Fig. 9 A). Further analysis revealed that the time variation of proximal LAD reflection index was similar to the resistance variation of resistance arteries (R 1) in the intramyocardial circulation (Fig. 9 B, showing data for the terminal LAD segment, where R 1 is the parallel sum from the three transmural layers, cf. Fig. 4 C). Combining data from all terminal arteries supplying the LV free wall and septum, R 1 was 12 ± 3% higher (P < 0.001) during the early diastolic period (‘dia’) than early systolic (‘sys’) period (Fig. 9 C), leading to a 9 ± 8% higher reflection index during early diastole (P < 0.001, Fig. 9 D).
Figure 9. Assessment of the variation in model‐based reflection index over the cardiac cycle, obtained by setting the intramyocardial pressure source (p im in Fig. 4 C) to zero but retaining the p im‐related changes in intramyocardial resistance (R 1, R m, R 2 in Fig. 4 C).
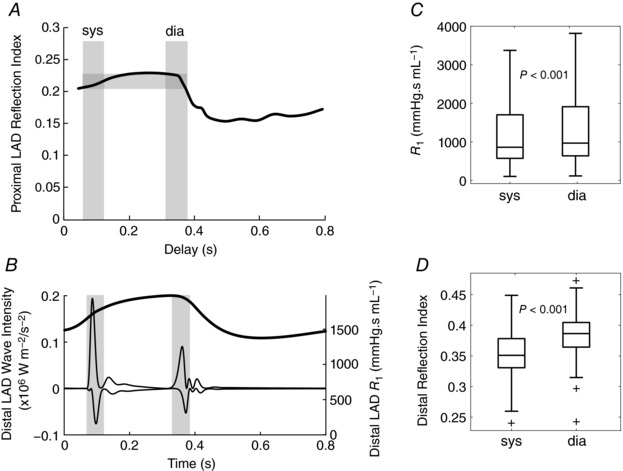
A, reflection index calculated from BCWsys/FCWsys cumulative intensity ratio in the proximal left anterior descending artery (LAD), with intra‐cycle variations obtained by introducing delays in coronary ostial pressure and hence the timing of incident waves (delay time based on peak BCWsys). Grey bars indicate the early systolic period (sys) and early diastolic period (dia), which span from the start of the incident wave to the end of the reflected wave. B, wave intensity at the end of the terminal LAD segment and time‐varying R 1 (parallel combination from all layers) in the connecting 0D microcirculation compartment. C and D, time‐averaged R 1 during the ‘sys’ and ‘dia’ periods (C), and reflection index calculated from the local incident and reflected waves (D), obtained from all terminal 1D segments supplying the left ventricular free wall and septum.
As shown in Fig. 10, the contribution of myocardial relaxation to BCWdia displayed minor sensitivity to changes in aortic mean pressure, intramyocardial compliance, coronary wave speed and monitoring location along the LAD; moderate sensitivity to aortic pulse pressure; and high sensitivity to intramyocardial resistance (R 1). Overall, estimation of this contribution via eqn (7) was 6 ± 5% higher than the actual contribution obtained via the wave shifting approach. These results support the applicability of eqn (7) and suggest that small differences in reflection index between early systole and early diastole are likely to cause minor overestimation and underestimation of the contributions of myocardial relaxation and wave reflection respectively.
Figure 10. Model sensitivity analysis of key variables to the percentage contribution of myocardial relaxation to the BCWdia .
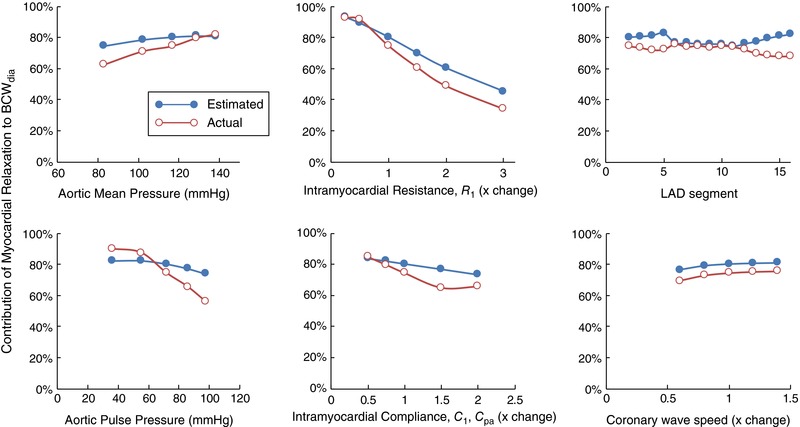
Aortic mean and pulse pressures were adjusted by varying systemic vascular resistance and aortic wave speed respectively. Intramyocardial resistance (R 1) and compliances (C 1 and C pa) are indicated in Fig. 4 C. ‘LAD segment’ refers to the 1D segment numbers shown in Fig. 4 A. ‘Estimated’ refers to values estimated via eqn (7) and ‘actual’ refers to values obtained by shifting ostial pressure by −60 ms, which separates the BDWmr and BDWref (cf. Fig. 7). [Color figure can be viewed at wileyonlinelibrary.com]
The difference between true coronary FDWA→C (based on simulations in Fig. 7) and that estimated from eqn (4) (when t d = 0) was less than 5%. True coronary FCWA→C could not be ascertained for comparison with eqn (5) due to re‐reflection of the BDWref at the coronary ostium.
In vivo data
Haemodynamics
A summary of relevant haemodynamic data is provided in Table 1. Heart rate and cardiac output decreased after l‐NNA (P < 0.05) but did not change with aortic constriction. Systolic blood pressure increased by a similar amount with l‐NNA and aortic constriction (P = 0.13) but was related to an increase in mean pressure in the former and pulse pressure in the latter. Cx flow was similar in both groups at baseline (P = 0.7) but increased more with aortic constriction than l‐NNA (by 26.5 ± 9.8 vs. 13.7 ± 12.3 mL min−1, P = 0.03) despite a higher diastolic pressure with l‐NNA (P = 0.03, Table 1). Coronary vascular resistance increased by ∼30% after l‐NNA (P = 0.04) but did not change with aortic constriction (P = 0.28). LV dP/dt min and τ were similar in both groups at baseline (P > 0.5) and displayed similar (P > 0.3) changes with afterload increase.
Table 1.
Haemodynamic data for the in vivo experiments
| l‐NNA group (n = 11) | Constriction group (n = 7) | |||
|---|---|---|---|---|
| Baseline | l‐NNA | Baseline | Aortic constriction | |
| Heart rate (bpm) | 110 ± 14 | 100 ± 14a | 105 ± 17 | 111 ± 17 |
| Cardiac output (L min−1) | 3.12 ± 0.68 | 2.56 ± 0.44a | 3.07 ± 0.65 | 3.67 ± 0.88 |
| Cx flow (mL min−1) | 29.5 ± 11.6 | 43.2 ± 17.7b | 32.1 ± 16.1 | 58.6 ± 19.9c |
| Cx effective vascular resistance (mmHg.min mL−1) | 2.44 ± 1.18 | 3.13 ± 1.60a | 2.37 ± 0.99 | 2.13 ± 0.80 |
| Ao systolic BP (mmHg) | 75.5 ± 7.7 | 134.5 ± 11.9c | 79.5 ± 7.6 | 147.4 ± 7.6c |
| Ao mean BP (mmHg) | 66.3 ± 9.1 | 123.9 ± 11.2c | 70.5 ± 9.2 | 121.8 ± 8.8c |
| Ao diastolic BP (mmHg) | 58.1 ± 10.0 | 115.5 ± 10.6c | 62.4 ± 10.5 | 103.0 ± 11.4c |
| Ao pulse BP (mmHg) | 17.4 ± 3.9 | 19.0 ± 2.6 | 17.0 ± 4.3 | 44.3 ± 9.9c |
| LV dP/dtmin (mmHg s−1) | −1195 ± 191 | −1878 ± 370c | −1271 ±294 | −2126 ± 443c |
| LV τ (ms) | 27.8 ± 4.7 | 19.3 ± 4.6c | 29.1 ± 5.9 | 21.5 ± 8.7a |
Ao, aortic; BP, blood pressure; Cx, circumflex coronary artery; dP/dtmin, maximal rate of pressure fall; LV, left ventricular; τ, time constant of isovolumic relaxation.
a P < 0.05, b P ≤ 0.01, c P < 0.001 compared with baseline.
Baseline variables between l‐NNA and aortic constriction groups were not significantly different.
Systolic waves
Wave intensity data are summarized in Table 2 and a representative example is shown in Fig. 11. Results are here discussed in relation to peak intensity unless otherwise stated, but similar findings pertain to cumulative intensity (Table 3). Aortic (Ao) and Cx FCWsys decreased substantially with l‐NNA (P < 0.004) but did not change with aortic constriction (P = 0.7). The concept that Cx BCWsys arises from reflection of the Cx FCWsys was supported by a strong correlation between these waves (R 2 = 0.62; P < 0.001), along with a relatively consistent delay of 23.9 ± 6.6 ms that did not change with increased afterload (P > 0.4). Moreover, the reflection index Cx BCWsys/FCWsys (0.26 ± 0.20 at baseline) more than doubled after l‐NNA infusion (P < 0.001), but was unaffected by aortic constriction (P = 0.8).
Table 2.
Peak wave intensity for the in vivo study
| Baseline | l‐NNA | Baseline | Aortic constriction | |
|---|---|---|---|---|
| Systolic waves | ||||
| Ao FCWsys | 81.8 ± 55.9 | 19.1 ± 4.5b | 86.5 ± 56.7 | 95.6 ± 43.7 |
| Cx FCWsys | 14.6 ± 8.0 | 2.0 ± 1.1c | 18.6 ± 11.1 | 20.7 ± 13.6 |
| Cx BCWsys | −3.1 ± 2.2 | −1.1 ± 0.4b | −6.0 ± 5.1 | −6.6 ± 5.8 |
| Cx | BCWsys/FCWsys | | 0.21 ± 0.08 | 0.64 ± 0.31c | 0.35 ± 0.30 | 0.32 ± 0.14 |
| Diastolic waves | ||||
| Ao FDWdia | 74.3 ± 18.3 | 106.6 ± 30.9c | 86.8 ± 25.9 | 152.6 ± 81.2a |
| Ao FCWdia | 21.9 ± 14.1 | 28.0 ± 16.8 | 30.2 ± 29.6 | 27.7 ± 51.4 |
| Cx FDWdia | 6.0 ± 3.0 | 5.3 ± 2.6 | 5.5 ± 2.3 | 9.2 ± 4.8 |
| Cx FDWA → C (eqn 4) | 14.9 ± 6.4 | 10.5 ± 5.5 | 17.9 ± 5.3 | 31.9 ± 22.2 |
| Cx FCWdia | 13.1 ± 5.7 | 16.0 ± 10.3 | 18.7 ± 13.5 | 15.8 ± 12.7 |
| Cx FCWA → C (eqn 5) | 3.9 ± 2.2 | 3.0 ± 2.6 | 6.7 ± 7.1 | 4.0 ± 5.4 |
| Cx BDWdia | −8.5 ± 6.2 | −10.3 ± 5.5 | −13.2 ± 7.7 | −22.8 ± 17.1 |
| Cx BDWref (eqn 6) | −3.2 ± 1.9 | −5.7 ± 1.9 b | −5.5 ± 4.1 | −9.7 ± 7.5 |
| Cx BDWmr (eqn 7) | −2.2 ± 3.3 | −1.1 ± 1.4 | −2.5 ± 2.7 | −3.2 ± 2.6 |
| Cx BDWmr /(BDWmr + BDWref) | 30 ± 34% | 13%± 16% | 31 ± 27% | 24 ± 21% |
Time‐corrected wave intensity units: 104 W m−2 s−2.
a P < 0.05, b P ≤ 0.01, c P < 0.001 compared with the respective baseline.
Items in italics refer to component waves that were estimated via the indicated equation. There were no significant differences between baselines of l‐NNA and aortic constriction groups for any quantity.
Figure 11. Representative example of simultaneously acquired wave intensity in the sheep aortic trunk (left) and circumflex coronary artery (right) in relation to blood pressure (P) and flow (Q) waveforms.

High‐fidelity pressure was measured in the aortic trunk and was assumed to be the same as circumflex coronary pressure (aside from a time offset applied to align early‐systolic pressure and flow upstrokes). Wave abbreviations: BCW, backward compression wave; BDW, backward decompression wave; FCW, forward compression wave; FDW, forward decompression wave. Decompression waves are shaded. The main waves occur during early systole (‘sys’) and early diastole (‘dia’).
Table 3.
Cumulative wave intensity (i.e. area under the wave)
| Baseline | l‐NNA | Baseline | Aortic constriction | |
|---|---|---|---|---|
| Systolic waves | ||||
| Ao FCWsys | 22.46 ± 12.86 | 9.81 ± 3.10b | 24.93 ± 14.14 | 27.44 ± 9.02 |
| Cx FCWsys | 2.83 ± 1.61 | 0.86 ± 0.40b | 3.80 ± 2.08 | 4.23 ± 3.25 |
| Cx BCWsys | −0.81 ± 0.56 | −0.53 ± 0.28 | −1.42 ± 1.11 | −1.47 ± 1.11 |
| Cx | BCWsys/FCWsys | | 0.29 ± 0.11 | 0.61 ± 0.20c | 0.39 ± 0.26 | 0.39 ± 0.20 |
| Diastolic waves | ||||
| Ao FDWdia | 14.38 ± 3.52 | 18.57 ± 4.33b | 16.86 ± 3.38 | 31.72 ± 11.27b |
| Ao FCWdia | 2.42 ± 1.51 | 2.69 ± 1.65 | 3.62 ± 3.41 | 2.36 ± 4.64 |
| Cx FDWdia | 1.00 ± 0.51 | 0.70 ± 0.31a | 0.92 ± 0.48 | 1.72 ± 1.16 |
| Cx FDWA → C (eqn 4) | 1.93 ± 0.85 | 1.60 ± 0.56 a | 2.62 ± 1.30 | 4.68 ± 2.72 |
| Cx FCWdia | 1.62 ± 0.68 | 1.77 ± 1.11 | 2.32 ± 1.84 | 1.62 ± 1.38 |
| Cx FCWA → C (eqn 5) | 0.30 ± 0.17 | 0.22 ± 0.17 | 0.64 ± 0.79 | 0.24 ± 0.37 |
| Cx BDWdia | −1.95 ± 0.83 | −2.10 ± 0.87 | −2.60 ± 1.15 | −4.80 ± 2.42a |
| Cx BDWref (eqn 6) | −0.57 ± 0.32 | −0.95 ± 0.34 a | −0.87 ± 0.51 | −1.63 ± 1.16 |
Cumulative wave intensity units: 103 W m−2 s−1.
a P < 0.05, b P ≤ 0.01, c P < 0.001 compared with the respective baseline.
Items in italics refer to component waves that were estimated via the indicated equation. There were no significant differences between baseline of Group 1 and baseline of Group 2 for any quantity. Note that BDWmr cumulative intensity cannot be calculated as for peak intensity.
Diastolic waves
Large differences in the relative intensity of early‐diastolic forward waves were found when comparing Ao and Cx sites (Fig. 12). After normalization to FCWsys, Cx FDWdia was smaller than Ao FDWdia in every recording (by 40 ± 16% overall), whereas Cx FCWdia was always much larger than Ao FCWdia (by 512 ± 373%).
Figure 12. Comparison of early diastolic forward wave magnitude in the ascending aorta vs. the circumflex coronary artery at (A) baseline (all animals, n = 18), (B) after aortic constriction (n = 7) and (C) after administration of l‐NNA (n = 11, note the difference in y‐axis scale).
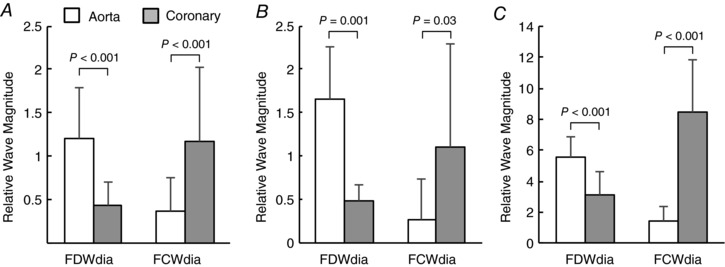
Peak wave intensities of FDWdia and FCWdia are expressed relative to that of FCWsys in the respective measurement location.
The estimated Cx BDWref almost doubled with l‐NNA (P = 0.007) but did not increase significantly with aortic constriction (P = 0.12). Cx BDWmr accounted for only ∼30% of the measured BDWdia at baseline (eqn 8), with a trend for this contribution to decrease after LNNA (to 13%, P = 0.09, Table 2).
With simple linear regression, no correlation was detected between Cx BDWdia and LV τ (R 2 < 0.1, P > 0.05, Fig. 13, left panels), but a weak‐to‐moderate correlation was found between normalized Cx BDWdia magnitude and LV dP/dtmin (R 2 = 0.3, P ≤ 0.001, Fig. 13, middle panels). Conversely, a strong correlation was found between Cx BDWdia and Ao FDWdia (R 2 > 0.6, P < 10−7, Fig. 13, right panels). Upon multiple linear regression analysis, LV dP/dtmin did not provide additional predictive value over Ao FDWdia (P = 0.4), again suggesting that reflection of FDWA→C made a greater contributor to BDWdia than distal suction effects.
Figure 13. Linear regression analyses investigating relationships between the circumflex coronary (Cx) BDWdia and LV isovolumic time constant (τ), LV dP/dt min and aortic (Ao) FDWdia, with wave magnitude quantified via peak wave intensity (top panels) and cumulative intensity (bottom panels) normalized to the respective FCWsys .
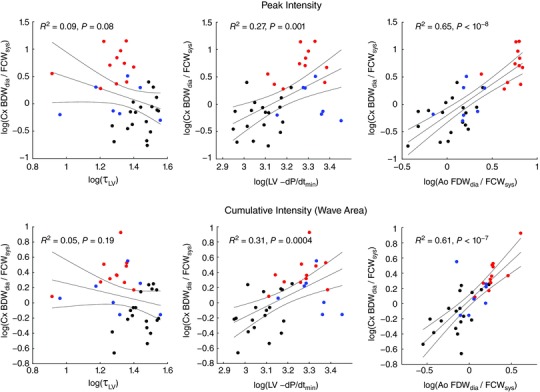
Data point colours (online version only) refer to baseline (black, both groups), l‐NNA infusion (red) and aortic constriction (blue). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
A skilled magician uses smoke and mirrors to conceal, exaggerate and mislead. This study uncovered a phenomenon whereby coronary wave reflection acts in a similar way, obscuring the true influence of active upstream and downstream forces on early diastolic waves in conduit arteries. Specifically, although distal myocardial relaxation generates a backward‐running decompression (or ‘suction’) wave, as is currently believed (Davies et al. 2006a), distal reflection of a forward decompression wave can have a major ‘exaggerating’ effect on the measured backward wave (BDWdia), due to a non‐linear wave addition phenomenon. Moreover, near‐complete negative reflection of the BDWdia at the coronary ostium produces a forward compression wave (FCWref) that conceals the aortic FDWdia and exaggerates the aortic FCWdia when viewed from the coronary arteries.
While an oft‐stated benefit of coronary wave intensity is that it allows clear assessment of upstream and downstream processes (Siebes et al. 2009; Lu et al. 2011; Kyriacou et al. 2012; Rolandi et al. 2012; Claridge et al. 2015; Lee et al. 2016; Raphael et al. 2016), a key conclusion of this study is that, mechanistically, both upstream and downstream processes can have a significant effect on the magnitude of both forward and backward waves (directly and via wave reflection). However, with additional knowledge of aortic wave intensity, we have described a novel method to unmask the effect of wave reflection and recover information about upstream and downstream forces (such as distal suction) on early‐diastolic coronary waves.
Wave reflection in coronary arteries
Limited information exists about wave reflection in coronary arteries. Rumberger et al. (1979) posited wave reflection as an explanation for prominent pressure and velocity oscillations observed in distal coronary arteries of the horse, while Arts et al. (1979) concluded that, in dogs, frequencies below 7 Hz are reflected at the coronary peripheral resistance, whereas higher frequencies undergo minimal reflection. The study by Arts et al. (1979), and in particular a recent detailed morphometric and theoretical study by Rivolo et al. (2016), suggested that the major conduit coronary arteries are relatively well matched in the forward direction, as was assumed in the design of our 1D model. This implies that wave reflection occurs mainly in the small resistance arteries.
Davies et al. (2006a) proposed reflection of FCWsys as a likely mechanism underlying the BCWsys. This was supported by our modelling study, in which blocking the aortic FCWsys from passing into the coronary arteries abolished the BCWsys (Fig. 6). Further support for this interpretation was provided by experimental data showing a strong correlation between BCWsys and FCWsys, a consistent delay between these waves and a wave intensity‐based reflection index that increased dramatically with coronary vasoconstriction.
Along with distal reflection, Davies et al. (2006a) also proposed that backward‐running waves arriving at the coronary ostium are likely to undergo near‐complete negative reflection, due to the large impedance decrease at this junction. Hence, during isovolumic contraction, an FDWic was postulated to arise from negative reflection of the BCWic; this explanation was supported by our virtual experiments in which preventing aortic waves from passing into the coronary arteries did not affect FDWic (Fig. 6).
In contrast to its recognized major influence during early systole, however, the potential role of wave reflection in modulating the coronary wave intensity profile during early diastole has largely been ignored until the present study.
Early‐diastolic backward decompression wave (BDWdia)
The BDWdia is considered the most important coronary wave physiologically, as it appears to be the primary actuator of diastolic coronary blood flow and is influenced by factors such as ageing, exercise, aortic stenosis, LV hypertrophy, coronary stenosis and myocardial resynchronization therapy (Davies et al. 2006a; Kyriacou et al. 2012; Lockie et al. 2012; De Silva et al. 2013; Claridge et al. 2015; Narayan et al. 2015). The BDWdia is currently interpreted as arising solely from a suction effect within the coronary microcirculation, as the relaxing myocardium releases the external (intramyocardial) pressure developed during systole (Davies et al. 2006a; Kyriacou et al. 2012; Narayan et al. 2015). Lee et al. (2016) investigated the sensitivity of the major coronary waves to several key parameters in their model and found a dependence of BDWdia on myocardial relaxation rate and degree of dysynchrony, consistent with the distal suction mechanism; however, a possible contribution of wave reflection was not explored.
Our experimental and computational data provide strong evidence that wave reflection makes an important contribution to the BDWdia. In the computational model, preventing aortic waves from passing into the coronary arteries caused BDWdia amplitude to fall by 69%, suggesting a contribution of reflection to this wave. In the experimental data, a strong correlation was found between the size of the incident wave (FDWA→C) and BDWdia (R 2 > 0.6), but there was no correlation with LV τ and only a weak‐to‐moderate correlation with LV dP/dt min (R 2 = 0.3) that lost significance in a multivariate model. Previous studies have reported R 2 values of 0.21−0.35 between LV dP/dt min and the coronary BDWdia, in agreement with our findings (Kyriacou et al. 2012; Ladwiniec et al. 2016), although Ladwiniec and colleagues also reported a correlation (R 2 = 0.35) between LV τ and BDWdia (Ladwiniec et al. 2016).
A surprising finding of our study was that the distal suction mechanism (i.e. BDWmr) accounted for only 30% of the measured BDWdia in the experimental study, and hence wave reflection appeared to be the dominant mechanism. Moreover, coronary vasoconstriction with l‐NNA increased the reflection‐related wave (BDWref) and tended to reduce the contribution of distal suction (see Table 2). However, these data in healthy, young sheep contrasted with the computational model representing an older, hypertensive human in which the distal suction mechanism was dominant, accounting for ∼75% of the BDWdia. The contribution of distal suction to BDWdia may therefore be quite variable and influenced by a variety of (patho)physiological factors; these should be investigated in more detail in future studies.
We showed for the first time that, due to the definition of wave intensity as dPdU, the addition of two mechanistically distinct waves is non‐linear. In other words, when BDWref and BDWmr combine, the resultant wave (BDWdia) is substantially greater than simply BDWref + BDWmr (by ∼60% at baseline in our sheep studies). This may partly explain why the BDWdia is so prominent in many published figures, much more so than the BCWic despite in vivo data suggesting that intramyocardial pressure rises (during isovolumic contraction) and falls (during isovolumic relaxation) at a similar rate (Rabbany et al. 1989). An important implication of this non‐linear addition principle is that any delay between BDWref and BDWmr may substantially influence the intensity of the combined or partially combined wave (e.g. compare t d = −20 ms and t d = 0 ms in Fig. 7). That the BDWref and BDWmr may not always be aligned is suggested by numerous published figures in which multiple BDW peaks are evident in the early diastolic period, although this could also be due to other factors such as dyssynchronous relaxation (Hadjiloizou et al. 2008; Davies et al. 2011; Kyriacou et al. 2012; Lu et al. 2012; De Silva et al. 2014; Broyd et al. 2016; Raphael et al. 2016).
Early‐diastolic forward decompression wave (FDWdia)
Based on the seminal work of Parker et al. (1988), the FDWdia is widely regarded as the second major wave in the aortic wave intensity profile and is associated with a fall in pressure and flow prior to valve closure. An FDWdia also appears in the coronary wave intensity profile and is believed to arise from transmission of the aortic FDWdia into the coronary arteries (Davies et al. 2006a). However, we found that the coronary FDWdia was ∼60% smaller (peak intensity, after normalization to FCWsys) than the aortic FDWdia. Closer inspection revealed that the FDW entering the coronary arteries from the aorta (FDWA→C) is partially cancelled by negative reflection of the BDWdia at the coronary ostium (see Fig. 3 B). Such reflection produces a forward compression wave (FCWref) which has a cancelling/concealing effect on the FDWA→C.
Introduction of a variable delay between aortic‐originating waves and distally generated waves in a virtual experiment indicated that this cancelling effect can cause the FDWA→C to essentially disappear under certain circumstances (Fig. 7). It is tempting to conclude that if the measured coronary FDWdia is small or even absent, wave reflection must make a negligible contribution to the BDWdia. However, we found that a prominent BDWref may still be present in such circumstances, apparently because the BDWref arises from reflection of the concealed FDWA→C (see t d = 30 ms in Fig. 7). For this reason, the true influence of wave reflection on the BDWdia should be assessed with respect to FDWA→C as the incident wave, rather than FDWdia.
Diastolic forward compression wave (FCWdia)
An aortic FCWdia arises from the transitory increase in pressure and flow associated with the dicrotic notch as the valve closes. This wave is also commonly observed in the coronary arterial wave intensity profile (Davies et al. 2006a; Hadjiloizou et al. 2008; Kyriacou et al. 2012; Lu et al. 2012; Ladwiniec et al. 2016; Raphael et al. 2016) and has been interpreted to arise from transmission of the aortic FCWdia into the coronary arteries (Davies et al. 2006a; Narayan et al. 2015). However, inspection of published figures from these two locations suggest that the FCWdia is generally more prominent in coronary arteries (Davies et al. 2006a; Kyriacou et al. 2012; Lu et al. 2012; Sinclair et al. 2015; Raphael et al. 2016) than in the aorta (Parker & Jones, 1990; Jones et al. 2002; Khir & Parker, 2005; Penny et al. 2008; Lu et al. 2012). Consistent with this impression, our comparison of concurrent aortic and coronary wave intensity profiles indicated that the coronary FCWdia peak intensity was 2–21 times higher than aortic FCWdia, relative to FCWsys (see Fig. 12); in the computational studies, aortic FCWdia was of negligible size but the coronary FCWdia was very prominent (see Fig. 5).
The combination of experimental and computational data suggested that two mechanisms contributed to the FCWdia. As expected, the first was transmission of the aortic FCWdia into coronary arteries. However, the second and perhaps dominant mechanism was negative reflection of the BDWdia at the coronary ostium (FCWref), which was clearly demonstrated with a virtual experiment in which aortic waves were prevented from passing into the coronary arteries (see Fig. 6).
Overcoming the smoke and mirrors effect
A major conclusion of this study is that wave reflection obscures the true (i.e. wave reflection‐independent) influence of upstream and downstream forces on the early diastolic forward and backward waves. However, by making use of information from concurrent aortic wave intensity, we proposed novel techniques for estimating the reflection‐independent waves that pass from the aorta into the coronary arteries (FDWA→C and FCWA→C) and arise from distal suction effects (BCWmr), as well as the reflection‐related contribution to BDWdia (i.e. BDWref). In silico validation with our model suggested that these waves can be estimated with errors of less than 10%.
The proposed wave estimation techniques depend on several assumptions being satisfied. The first is that aorto‐coronary wave transmission does not vary significantly between early systole and early diastole; indeed, based on transmission line theory, we calculated differences in transmission coefficient of less than 5% (Mynard et al. 2017). The second assumption (required for estimation of BDWref and BCWmr) is that the coronary reflection coefficient is similar during early systole and early diastole (when FCWsys and FDWdia occur). When we tested this assumption using our model, we found that model‐predicted reflection index was comparable to that in the experimental studies (0.21 vs. 0.29) and changed by less than 0.01 between early systole and early diastole (Fig. 9 A). Model data also suggested that both intramyocardial resistance and distal reflection index were ∼10% higher during early diastole than early systole (Fig. 9 B–D), which, if anything, would tend to cause the contribution of wave reflection on BDWdia to be underestimated.
Study limitations
This study had a number of limitations. Experimental studies were performed in healthy, young, anaesthetized open‐chest sheep undergoing mechanical ventilation, and thus caution should be applied in extrapolating results to humans and other species with or without pathophysiology undergoing spontaneous respiration. While previous studies suggest that the anaesthetic alpha‐chloralose has (Covert et al. 1989) or does not have (Cox, 1972) a vasoconstrictive effect, systemic vascular resistance at baseline in this study (21.9 ± 4.4 mmHg.L min−1) was only slightly higher than the range 18.8–21.2 mmHg.L min−1 in chronically instrumented conscious sheep (Kemp et al. 1995) and effective circumflex coronary resistance in this study (2.4 ± 1.1 mmHg.min mL−1) was the same as that derived from data in conscious sheep (Bednarik & May, 1995; Parkes et al. 1997). However, we cannot exclude the possibility that some coronary vasoconstriction could have led to a greater influence of wave reflection on the BDWdia, although any such effect is likely to be small.
The computational model is based on findings of many physiological studies from other research groups (Fisher et al. 1982; Spaan, 1985; Bruinsma et al. 1988; Rabbany et al. 1989; Dodge et al. 1992; Spaan et al. 2000; Algranati et al. 2010), has been validated in sheep (Mynard et al. 2014) and produced wave intensity profiles that were very consistent with published in vivo waveforms (Davies et al. 2006a). However, a number of factors were not accounted for in the model, such as the effects of cardiac motion (Ramaswamy et al. 2004), wave propagation effects in the microcirculation (Guiot et al. 1990) and the effects of external constraint on conduit arteries (Liu et al. 2008). The coronary microcirculation was modelled using lumped elements and simplifying assumptions were made about intramyocardial pressure. More detailed microvascular models, such as the poromechanical model described by Lee et al. (2016), may be useful for further investigating some of the issues raised in this paper.
While this study highlighted the complexity of coronary arterial wave dynamics, we did not study the effect of multiple re‐reflections. In the sheep under baseline conditions, the magnitude of second‐ and third‐generation waves would be expected to be in the order of ∼6% and ∼1.5% that of initial incident wave, not accounting for wave dispersion and dissipation. However, in cases where wave reflection is high (e.g. vasoconstriction or stenosis), these re‐reflections may be more influential. Use of the theoretical framework described in this paper could be used in future to investigate these wave dynamics in even greater detail, although the limited fidelity of experimental data may make this challenging. We also did not investigate issues related to the ‘windkesselness’ of the coronary arterial network, which can lead to errors when calculating wave speed and separating wave intensity into forward and backward components (Kolyva et al. 2008; Siebes et al. 2009).
Conclusions
This study revealed a ‘smoke and mirrors’ effect caused by wave reflection that obscures the true upstream and downstream contributions to early diastolic coronary waves. Experimental and computational data supported our hypothesis that the FDWdia, FCWdia and BDWdia are all affected by wave reflection and, hence, both upstream and downstream forces. In particular, the dominant BDWdia appears to arise from a combination of distal suction and wave reflection, with non‐linear addition of the two component waves being a significant factor underlying the prominence of this wave. The FDWdia and FCWdia are attenuated and augmented respectively by negative reflection of the BDWdia at the coronary ostium. These findings have significant implications for the prevailing interpretation of the coronary wave intensity profile. We also proposed techniques for recovering the waves arising from non‐reflection‐related upstream forces (for forward waves) and downstream forces (for backward waves). While the precise contribution of wave reflection and active mechanisms in humans under various conditions must be established in future studies, the principles and techniques described in this study are likely to have an important impact on the interpretation of coronary wave intensity profiles in health and disease.
Additional information
Conflicts of interest
None.
Author contributions
JPM: conception and design, computational studies, data analysis, preparation of figures and manuscript; DJP and JJS: design and conduct of laboratory experiments. All authors edited and revised the manuscript, approved the final version and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
JPM was supported by a CJ Martin Early Career Research Fellowship from the National Health and Medical Research Council of Australia. This work was supported by the Victorian Government's Operational Infrastructure Support Program.
Translational perspective
Wave intensity analysis is an emerging technique that is being used to unravel the upstream (aortic) and downstream (intramyocardial) forces that determine coronary blood flow. A dominant backward decompression wave (BDWdia) accompanies the early‐diastolic surge in coronary flow and, based on a number of published studies, has been shown to have potential clinical value. To date, it has been widely held that the BDWdia is generated in the coronary microvasculature via a suction effect caused by myocardial relaxation. Our study provides evidence that another mechanism contributes to the BDWdia, namely passive wave reflection of a forward wave entering the coronary arteries from the aorta. We also show that summation of the wave reflection and myocardial relaxation forces is non‐linear, i.e. the resultant combined wave is substantially larger than the simple sum of waves from each mechanism acting in isolation. This finding may impact on the interpretation of clinical studies assessing BDWdia magnitude under healthy or disease conditions and how it changes with intervention. With an additional measurement of ascending aortic wave intensity, the separate contributions of wave reflection and myocardial relaxation to BDWdia may be estimated, thus providing more detailed and accurate insights into the upstream and downstream forces that determine coronary flow.
Acknowledgements
The current address for D.J. Penny is Department of Pediatrics, Baylor College of Medicine and Department of Cardiology, Texas Children's Hospital, 19th Floor West Tower, Texas Children's Hospital, MC19345‐C Houston, TX 77030, USA. The authors thank Mubin Yousuf for contributing to data analysis.
Appendix 1.
Model parameters
Cardiovascular model
Parameters for the closed‐loop cardiovascular model were taken from Mynard et al. (2012) and adjusted to represent an older adult arterial system with target haemodynamic values described in the Methods. Parameters governing the heart chambers, valves, 1D segments and vascular beds shown in Fig. 4 are given in A1, A2, A3, A4.
Table A1.
Model parameters for heart chambers
| LV | RV | LA | RA | |
|---|---|---|---|---|
| E max (mmHg mL−1) | 2.2 | 0.35 | 0.13 | 0.09 |
| E min (mmHg mL−1) | 0.07 | 0.035 | 0.09 | 0.045 |
| V 0 (mL) | 10 | 60 | 3 | 7 |
| Vt = 0 (mL) | 150 | 155 | 80 | 60 |
| K S (10−3 s mL−1) | 1.00 | 1.00 | 0.25 | 0.50 |
| τ1 (s) | 0.215 | 0.215 | 0.042 | 0.042 |
| τ2 (s) | 0.362 | 0.362 | 0.138 | 0.138 |
| m 1 (–) | 1.32 | 1.32 | 1.99 | 1.99 |
| m 2 (–) | 21.9 | 21.9 | 11.2 | 11.2 |
| κ (–) | 6 | 6 | 2 | 2 |
| μAV (g cm−7 s−1) | 0 | 0 | 0.033 | 0.05 |
| t onset (s) | 0 | 0 | 0.65 | 0.65 |
E max, maximum free wall elastance; E min, minimum free wall elastance; V 0, pressure‐axis intercept of the pressure–volume relation; Vt = 0, initial chamber volume; K S, source resistance coefficient; τ1/τ2, contraction/relaxation time constants; m 1/m 2, contraction/relaxation rate constants; κ, septal elastance constant (ventricular interaction via the septum); μAV, atrioventricular plane piston constant (atrioventricular interaction); t onset, onset time of contraction. Note: no pericardial constraint was applied. See Mynard and Smolich (2015) for an explanation of these parameters.
Table A2.
Model parameters for heart valves
| Av | Pv | Mv | Tv | Vv | |
|---|---|---|---|---|---|
| A eff,max (cm2) | 4.9 | 5.7 | 5.1 | 6 | 6 |
| A eff,min (cm2) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| l eff (cm) | 1.5 | 1.5 | 2 | 2 | 1.5 |
| K vo (cm2 s2 g−1) | 0.02 | 0.02 | 0.02 | 0.03 | 0.03 |
| K vc (cm2 s2 g−1) | 0.02 | 0.02 | 0.04 | 0.04 | 0.03 |
A eff,max, maximum effective valve area (when fully open); A eff,max, minimum effective valve area (when closed); l eff, effective length; K vo, valve opening rate coefficient; K vc, valve closure rate coefficient. Valve abbreviations: Av, aortic valve; Pv, pulmonary valve; Mv, mitral valve; Tv, tricuspid valve; Vv, venous valve. See Mynard et al. (2012) and Mynard and Smolich (2015) for an explanation of these parameters.
Table A3.
Model parameters for one‐dimensional segments of the cardiovascular model (see Fig. 4 B)
| L (cm) | A 0 (cm2) | c 0 (cm/s) | P 0 (mmHg) | |
|---|---|---|---|---|
| LVot | 1 | 7.2 | 900 | 90 |
| AoRt | 1 | 7.2 | 900 | 90 |
| SA | 40 | 7.2 → 5.0 | 900 → 1286 | 90 |
| SV1 | 10 | 6 | 150 | 4.5 |
| SV2 | 5 | 6 | 150 | 4.5 |
| RVot | 1 | 7.1 | 250 | 14 |
| PA | 5 | 7.1 → 10.6 | 250 | 14 |
| PV | 5 | 10 | 150 | 8.5 |
L is segment length. A 0, c 0 and P 0 are reference cross‐sectional area, wave speed and pressure. Abbreviations: LVot, left ventricular outflow tract; AoRt, aortic root proximal to the coronary ostia; SA, systemic artery; SV, systemic vein (two segments separated by a venous valve); RVot, right ventricular outflow tract; PA, pulmonary artery; PV, pulmonary vein. Arrows indicate linear tapering from proximal to distal values.
Table A4.
Model parameters for the vascular beds in the cardiovascular model (see Fig. 4 B)
| R 0 (mmHg.s mL−1) | C art (mL mmHg−1) | C ven (mL mmHg−1) | |
|---|---|---|---|
| SVB | 1.8 | 0.5 | 11 |
| PVB | 0.05 | 5.0 | 15 |
R 0, reference resistance; C art, arterial compliance; C ven, venous compliance. Vascular bed abbreviations: SVB, systemic vascular bed; PVB, pulmonary vascular bed. Note that arterial and venous characteristic impedances (Z 0,art, Z 0,ven in Fig. 4 B) were calculated from the connecting 1D segment as ρc 0/A 0, where ρ = 1.06 g cm−3 is blood density (see A3).
One‐dimensional conduit coronary arteries
The geometry of the right‐dominant 1D coronary conduit arterial model (i.e. reference cross‐sectional area, length and connectivity of all 1D segments) was identical to that in Mynard & Smolich (2016a) and is therefore not reproduced here. The only difference was that, compared with the model in Mynard & Smolich (2016a), the wave speed of all segments was increased by 250% compared with those representing a healthy young circulation, resulting in a proximal LAD wave speed of 20.8 m s−1, similar to reported values in older adult patients (Davies et al. 2006a; Rolandi et al. 2012).
Coronary microcirculation model
Terminal 1D segments perfuse parts of the LV free wall (LVfw), right ventricular free wall (RVfw) and septum (Sep). As in previous work (Mynard et al. 2014; Mynard & Smolich, 2016a), total microcirculatory resistance (i.e. R 1 + R m + R 2 in Fig. 4 C) was determined iteratively to achieve a target mean flow (2.54%, 0.66% and 1.35% of cardiac output, for the LVfw, RVfw and septum respectively). This flow was distributed amongst instances of the 0D model in proportion to their myocardial weights, which in turn were distributed according to the inverse cube of penetrating artery radii (i.e. Murray's law). Total weights for the adult human LVfw, RVfw and septum of 104, 46 and 54 g respectively were taken from Lorenz et al. (1999). Subendocardial‐to‐subepicardial flow ratios (1.24 and 1.18 for LVfw and RVfw) and left‐to‐right septal flow ratios (1.37) were taken from Fisher et al. (1982). We assumed R 1 = 1.2R m and R 2 = 0.5R m in all myocardial regions and that 75% of R m is dependent on the volume of chamber 1 and the remainder is dependent on chamber 2 (Spaan et al. 2000). Compliances were set to C 1 = 0.013 and C 2 = 0.254 mL mmHg−1/100 g, and reference volumes V 0,1 = 2.5 and V 0,2 = 8.0 mL/100 g, both with a subendocardial‐to‐subepicardial (or left‐to‐right septal) ratio of 1.14 (Weiss & Winbury, 1974; Bruinsma et al. 1988). CEP was assumed to decline linearly from ventricular cavity pressure at the endocardium to pericardial pressure (assumed to be zero) at the epicardium, while SIP was assumed to be the same in each transmural layer, with a peak value equal to 20% of cavity pressure and a waveform shape identical to the ventricular chamber elastance curve (Mynard et al. 2014).
Edited by: Don Bers & Michael Shattock
References
- Algranati D, Kassab GS & Lanir Y (2010). Mechanisms of myocardium‐coronary vessel interaction. Am J Physiol Heart Circ Physiol 298, H861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts T, Kruger RT, van Gerven W, Lambregts JA & Reneman RS (1979). Propagation velocity and reflection of pressure waves in the canine coronary artery. Am J Physiol Heart Circ Physiol 237, H469–474. [DOI] [PubMed] [Google Scholar]
- Bednarik JA & May CN (1995). Evaluation of a transit‐time system for the chronic measurement of blood flow in conscious sheep. J Appl Physiol 78, 524–530. [DOI] [PubMed] [Google Scholar]
- Bessems D, Rutten M & Van de Vosse FN (2007). A wave propagation model of blood flow in large vessels using an approximate velocity profile function. J Fluid Mech 580, 145–168. [Google Scholar]
- Brosh D, Higano ST, Slepian MJ, Miller HI, Kern MJ, Lennon RJ, Holmes DR Jr & Lerman A (2002). Pulse transmission coefficient: a novel nonhyperemic parameter for assessing the physiological significance of coronary artery stenoses. J Am Coll Cardiol 39, 1012–1019. [DOI] [PubMed] [Google Scholar]
- Broyd CJ, Nijjer S, Sen S, Petraco R, Jones S, Al‐Lamee R, Foin N, Al‐Bustami M, Sethi A, Kaprielian R, Ramrakha P, Khan M, Malik I, Francis D, Parker KH, Hughes AD, Mikhail G, Mayet J & Davies JE (2016). Estimation of coronary wave intensity analysis using non‐invasive techniques and its application to exercise physiology. Am J Physiol Heart Circ Physiol 210, H619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma P, Arts T, Dankelman J & Spaan JAE (1988). Model of the coronary circulation based on pressure dependence of coronary resistance and compliance. Basic Res Cardiol 83, 510–524. [DOI] [PubMed] [Google Scholar]
- Claridge S, Chen Z, Jackson T, De Silva K, Behar J, Sohal M, Webb J, Hyde E, Lumley M, Asrress K, Williams R, Bostock J, Ali M, Gill J, O'Neill M, Razavi R, Niederer S, Perera D & Rinaldi CA (2015). Effects of epicardial and endocardial cardiac resynchronization therapy on coronary flow: insights from wave intensity analysis. J Am Heart Assoc 4, e002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert RF, Drummond WH, Gimotty PA & Carter RL (1989). Chloralose alters both basal hemodynamics and cardiovascular responses to alveolar hypoxia in chronically instrumented, spontaneously breathing lambs. Pediatr Res 25, 389–395. [DOI] [PubMed] [Google Scholar]
- Cox RH ( 1972). Influence of chloralose anesthesia on cardiovascular function in trained dogs. Am J Physiol 223, 660–667. [DOI] [PubMed] [Google Scholar]
- Davies JE, Sen S, Broyd C, Hadjiloizou N, Baksi J, Francis DP, Foale RA, Parker KH, Hughes AD, Chukwuemeka A, Casula R, Malik IS, Mikhail GW & Mayet J (2011). Arterial pulse wave dynamics after percutaneous aortic valve replacement. Circulation 124, 1565–1572. [DOI] [PubMed] [Google Scholar]
- Davies JE, Whinnett ZI, Francis DP, Manisty CH, Aguado‐Sierra J, Willson K, Foale RA, Malik IS, Hughes AD, Parker KH & Mayet J (2006a). Evidence of a dominant backward‐propagating “suction” wave responsible for diastolic coronary filling in humans, attenuated in left ventricular hypertrophy. Circulation 113, 1768–1778. [DOI] [PubMed] [Google Scholar]
- Davies JE, Whinnett ZI, Francis DP, Willson K, Foale RA, Malik IS, Hughes AD, Parker KH & Mayet J (2006b). Use of simultaneous pressure and velocity measurements to estimate arterial wave speed at a single site in humans. Am J Physiol Heart Circ Physiol 290, 878–885. [DOI] [PubMed] [Google Scholar]
- De Silva K, Foster P, Guilcher A, Bandara A, Jogiya R, Lockie T, Chowiencyzk P, Nagel E, Marber M, Redwood S, Plein S & Perera D (2013). Coronary wave energy: a novel predictor of functional recovery after myocardial infarction. Circ Cardiovasc Interv 6, 166–175. [DOI] [PubMed] [Google Scholar]
- De Silva K, Lumley M, Kailey B, Alastruey J, Guilcher A, Asrress KN, Plein S, Marber M, Redwood S & Perera D (2014). Coronary and microvascular physiology during intra‐aortic balloon counterpulsation. JACC Cardiovasc Interv 7, 631–640. [DOI] [PubMed] [Google Scholar]
- Dodge JT Jr, Brown BG, Bolson EL & Dodge HT (1992). Lumen diameter of normal human coronary arteries. Influence of age, sex, anatomic variation, and left ventricular hypertrophy or dilation. Circulation 86, 232–246. [DOI] [PubMed] [Google Scholar]
- Fisher DJ, Heymann MA & Rudolph AM (1982). Regional myocardial blood flow and oxygen delivery in fetal, newborn, and adult sheep. Am J Physiol Heart Circ Physiol 243, H729–731. [DOI] [PubMed] [Google Scholar]
- Guiot C, Pianta G, Cancelli C & Pedley TJ (1990). Prediction of coronary blood flow with a numerical model based on collapsible tube dynamics. Am J Physiol Heart Circ Physiol 258, H1606–1614. [DOI] [PubMed] [Google Scholar]
- Hadjiloizou N, Davies JE, Malik IS, Aguado‐Sierra J, Willson K, Foale RA, Parker KH, Hughes AD, Francis DP & Mayet J (2008). Differences in cardiac microcirculatory wave patterns between the proximal left mainstem and proximal right coronary artery. Am J Physiol Heart Circ Physiol 295, H1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineman FW & Grayson J (1985). Transmural distribution of intramyocardial pressure measured by micropipette technique. Am J Physiol Heart Circ Physiol 249, H1216–1223. [DOI] [PubMed] [Google Scholar]
- Hollander EH, Wang JJ, Dobson GM, Parker KH & Tyberg JV (2001). Negative wave reflections in pulmonary arteries. Am J Physiol Heart Circ Physiol 281, H895–902. [DOI] [PubMed] [Google Scholar]
- Hughes AD, Parker KH & Davies JE (2008). Waves in arteries: a review of wave intensity analysis in the systemic and coronary circulations. Artery Res 2, 51–59. [Google Scholar]
- Jones CJ, Sugawara M, Kondoh Y, Uchida K & Parker KH (2002). Compression and expansion wavefront travel in canine ascending aortic flow: wave intensity analysis. Heart Vessels 16, 91–98. [DOI] [PubMed] [Google Scholar]
- Kemp BK, Smolich JJ, Ritchie BC & Cocks TM (1995). Endothelium‐dependent relaxations in sheep pulmonary arteries and veins: resistance to block by N G‐nitro‐l‐arginine in pulmonary hypertension. Br J Pharmacol 116, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khir AW, O'Brien A, Gibbs JS & Parker KH (2001). Determination of wave speed and wave separation in the arteries. J Biomech 34, 1145–1155. [DOI] [PubMed] [Google Scholar]
- Khir AW & Parker KH (2005). Wave intensity in the ascending aorta: effects of arterial occlusion. J Biomech 38, 647–655. [DOI] [PubMed] [Google Scholar]
- Kolyva C, Spaan JAE, Piek JJ & Siebes M (2008). Windkesselness of coronary arteries hampers assessment of human coronary wave speed by single‐point technique. Am J Physiol Heart Circ Physiol 295, H482–490. [DOI] [PubMed] [Google Scholar]
- Kyriacou A, Whinnett ZI, Sen S, Pabari PA, Wright I, Cornelussen R, Lefroy D, Davies DW, Peters NS, Kanagaratnam P, Mayet J, Hughes AD, Francis DP & Davies JE (2012). Improvement in coronary blood flow velocity with acute biventricular pacing is predominantly due to an increase in a diastolic backward‐travelling decompression (suction) wave. Circulation 126, 1334–1344. [DOI] [PubMed] [Google Scholar]
- Ladwiniec A, White PA, Nijjer SS, O'Sullivan M, West NEJ, Davies JE & Hoole SP (2016). Diastolic backward‐traveling decompression (suction) wave correlates with simultaneously acquired indices of diastolic function and is reduced in left ventricular stunning. Circ Cardiovasc Interv 9, e003779. [DOI] [PubMed] [Google Scholar]
- Lee J, Nordsletten D, Cookson A, Rivolo S & Smith N (2016). In silico coronary wave intensity analysis: application of an integrated one‐dimensional and poromechanical model of cardiac perfusion. Biomech Model Mechanobiol 15, 1535–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang W & Kassab GS (2008). Effects of myocardial constraint on the passive mechanical behaviors of the coronary vessel wall. Am J Physiol Heart Circ Physiol 294, H514–523. [DOI] [PubMed] [Google Scholar]
- Lockie TPE, Rolandi MC, Guilcher A, Perera D, De Silva K, Williams R, Asrress KN, Patel K, Plein S, Chowienczyk P, Siebes M, Redwood SR & Marber MS (2012). Synergistic adaptations to exercise in the systemic and coronary circulations that underlie the warm‐up angina phenomenon. Circulation 126, 2565–2574. [DOI] [PubMed] [Google Scholar]
- Lorenz CH, Walker ES, Morgan VL, Klein SS & Graham TP (1999). Normal human right and left ventricular mass, systolic function, and gender differences by cine magnetic resonance imaging. J Cardiovasc Magn Reson 1, 7–21. [DOI] [PubMed] [Google Scholar]
- Lu P‐J, Yang C‐FJ, Wu M‐Y, Hung C‐H, Chan M‐Y & Hsu T‐C (2011). Wave energy patterns of counterpulsation: a novel approach with wave intensity analysis. J Thorac Cardiovasc Surg 142, 1205–1213. [DOI] [PubMed] [Google Scholar]
- Lu P‐J, Yang C‐FJ, Wu M‐Y, Hung C‐H, Chan M‐Y & Hsu T‐C (2012). Wave intensity analysis of para‐aortic counterpulsation. Am J Physiol Heart Circ Physiol 302, H1481–1491. [DOI] [PubMed] [Google Scholar]
- Mirsky I ( 1984). Assessment of diastolic function: suggested methods and future considerations. Circulation 69, 836–841. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Lacourciere Y, Ouellet J‐P, Izzo JL Jr, Neutel J, Kerwin LJ, Block AJ & Pfeffer MA (2003). Determinants of elevated pulse pressure in middle‐aged and older subjects with uncomplicated systolic hypertension: the role of proximal aortic diameter and the aortic pressure‐flow relationship. Circulation 108, 1592–1598. [DOI] [PubMed] [Google Scholar]
- Mynard JP, Davidson MR, Penny DJ & Smolich JJ (2012). A simple, versatile valve model for use in lumped parameter and one‐dimensional cardiovascular models. Int J Num Meth Biomed Eng 28, 626–641. [DOI] [PubMed] [Google Scholar]
- Mynard JP, Kowalski R, Cheung MMH & Smolich JJ (2017). Beyond the aorta: partial transmission of reflected waves from aortic coarctation into supra‐aortic branches modulates cerebral hemodynamics and left ventricular load. Biomech Model Mechanobiol 16, 635–650. [DOI] [PubMed] [Google Scholar]
- Mynard JP & Nithiarasu P (2008). A 1D arterial blood flow model incorporating ventricular pressure, aortic valve and regional coronary flow using the locally conservative Galerkin (LCG) method. Comm Num Meth Eng 24, 367–417. [Google Scholar]
- Mynard JP, Penny DJ & Smolich JJ (2008). Accurate automatic detection of end‐diastole from left ventricular pressure using peak curvature. IEEE Trans Biomed Eng 55, 2651–2657. [DOI] [PubMed] [Google Scholar]
- Mynard JP, Penny DJ & Smolich JJ (2014). Scalability and in vivo validation of a multiscale numerical model of the left coronary circulation. Am J Physiol Heart Circ Physiol 306, H517–528. [DOI] [PubMed] [Google Scholar]
- Mynard JP & Smolich JJ (2015). One‐dimensional haemodynamic modeling and wave dynamics in the entire adult circulation. Ann Biomed Eng 43, 1443–1460. [DOI] [PubMed] [Google Scholar]
- Mynard JP & Smolich JJ (2016a). Influence of anatomical dominance and hypertension on coronary conduit arterial and microcirculatory flow patterns: a multi‐scale modeling study. Am J Physiol Heart Circ Physiol 311, H11–23. [DOI] [PubMed] [Google Scholar]
- Mynard JP & Smolich JJ (2016b). Novel wave power analysis linking pressure‐flow waves, wave potential and the forward and backward components of hydraulic power. Am J Physiol Heart Circ Physiol 310, H1026–1038. [DOI] [PubMed] [Google Scholar]
- Narayan O, Leung MCH, Wong DTL, Meredith IT & Cameron JD (2015). Percutaneous coronary intervention enhances accelerative wave intensity in coronary arteries. PLoS ONE 10, e0142998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KH & Jones CJ (1990). Forward and backward running waves in the arteries: analysis using the method of characteristics. J Biomech Eng 112, 322–326. [DOI] [PubMed] [Google Scholar]
- Parker KH, Jones CJ, Dawson JR & Gibson DG (1988). What stops the flow of blood from the heart? Heart Vessels 4, 241–245. [DOI] [PubMed] [Google Scholar]
- Parkes DG, Vaughan J, Rivier J, Vale W & May CN (1997). Cardiac inotropic actions of urocortin in conscious sheep. Am J Physiol Heart Circ Physiol 272, H2115–2122. [DOI] [PubMed] [Google Scholar]
- Penny DJ, Mynard JP & Smolich JJ (2008). Aortic wave intensity analysis of ventricular‐vascular interaction during incremental dobutamine infusion in adult sheep. Am J Physiol Heart Circ Physiol 294, H481–489. [DOI] [PubMed] [Google Scholar]
- Penny DJ & Smolich JJ (2002). Divergent effects of NO synthase inhibition on systemic and myocardial O2 delivery and consumption during dobutamine infusion in sheep. Pflügers Arch Eur J Physiol 443, 601–608. [DOI] [PubMed] [Google Scholar]
- Pijls NHJ, Kern MJ, Yock PG & De Bruyne B (2000). Practice and potential pitfalls of coronary pressure measurement. Catheter Cardiovasc Interv 49, 1–16. [DOI] [PubMed] [Google Scholar]
- Rabbany SY, Kresh JY & Noordergraaf A (1989). Intramyocardial pressure: interaction of myocardial fluid pressure and fiber stress. Am J Physiol Heart Circ Physiol 257, H357–364. [DOI] [PubMed] [Google Scholar]
- Ramaswamy SD, Vigmostad SC, Wahle A, Lai YG, Olszewski ME, Braddy KC, Brennan TMH, Rossen JD, Sonka M & Chandran KB (2004). Fluid dynamic analysis in a human left anterior descending coronary artery with arterial motion. Ann Biomed Eng 32, 1628–1641. [DOI] [PubMed] [Google Scholar]
- Ramsey MW & Sugawara M (1997). Arterial wave intensity and ventriculoarterial interaction. Heart Vessels (Suppl 12), 128–134. [PubMed] [Google Scholar]
- Raphael CE, Cooper R, Parker KH, Collinson J, Vassiliou V, Pennell DJ, de Silva R, Hsu LY, Greve AM, Nijjer S, Broyd C, Ali A, Keegan J, Francis DP, Davies JE, Hughes AD, Arai A, Frenneaux M, Stables RH, Di Mario C & Prasad SK (2016). Mechanisms of myocardial ischemia in hypertrophic cardiomyopathy: insights from wave intensity analysis and magnetic resonance. J Am Coll Cardiol 68, 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivolo S, Hadjilucas L, Sinclair M, van Horssen P, van den Wijngaard J, Wesolowski R, Chiribiri A, Siebes M, Smith NP & Lee J (2016). Impact of coronary bifurcation morphology on wave propagation. Am J Physiol Heart Circ Physiol 311, H855–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolandi MC, Nolte F, van de Hoef TP, Remmelink M, Baan J, Piek JJ, Spaan JAE & Siebes M (2012). Coronary wave intensity during the valsalva manoeuvre in humans reflects altered intramural vessel compression responsible for extravascular resistance. J Physiol 590, 4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumberger JA Jr, Nerem RM & Muir WW III (1979). Coronary artery pressure development and wave transmission characteristics in the horse. Cardiovasc Res 13, 413–419. [DOI] [PubMed] [Google Scholar]
- Siebes M, Kolyva C, Verhoeff B‐J, Piek J & Spaan J (2009). Potential and limitations of wave intensity analysis in coronary arteries. Med Biol Eng Comput 47, 233–239. [DOI] [PubMed] [Google Scholar]
- Sinclair MD, Lee J, Cookson AN, Rivolo S, Hyde ER & Smith NP (2015). Measurement and modeling of coronary blood flow. Wiley Interdiscip Rev Syst Biol Med 7, 335–356. [DOI] [PubMed] [Google Scholar]
- Smolich JJ & Mynard JP (2016). A step towards clinically applicable non‐invasive coronary wave intensity analysis. Am J Physiol Heart Circ Physiol 310, H525–527. [DOI] [PubMed] [Google Scholar]
- Spaan JA ( 1985). Coronary diastolic pressure‐flow relation and zero flow pressure explained on the basis of intramyocardial compliance. Circ Res 56, 293–309. [DOI] [PubMed] [Google Scholar]
- Spaan JAE, Cornelissen AJM, Chan C, Dankelman J & Yin FCP (2000). Dynamics of flow, resistance, and intramural vascular volume in canine coronary circulation. Am J Physiol Heart Circ Physiol 278, H383–403. [DOI] [PubMed] [Google Scholar]
- Stein PD, Sabbah HN & Marzilli M (1985). Intramyocardial pressure and coronary extravascular resistance. J Biomech Eng 107, 46–50. [DOI] [PubMed] [Google Scholar]
- Sun YH, Anderson TJ, Parker KH & Tyberg JV (2000). Wave‐intensity analysis: a new approach to coronary hemodynamics. J Appl Physiol 89, 1636–1644. [DOI] [PubMed] [Google Scholar]
- Sun YH, Anderson TJ, Parker KH & Tyberg JV (2004). Effects of left ventricular contractility and coronary vascular resistance on coronary dynamics. Am J Physiol Heart Circ Physiol 286, H1590–1595. [DOI] [PubMed] [Google Scholar]
- Weiss HR & Winbury MM (1974). Nitroglycerin and chromonar on small‐vessel blood content of the ventricular walls. Am J Physiol 226, 838–843. [DOI] [PubMed] [Google Scholar]