

Abstract
Influenza viruses are respiratory pathogens that are responsible for seasonal influenza and sporadic influenza pandemic. The therapeutic efficacy of current influenza vaccines and small molecule antiviral drugs is limited due to the emergence of multidrug-resistant influenza viruses. In response to the urgent need for the next generation of influenza antivirals, we utilized a fast-track drug discovery platform by exploring multi-component reaction products for antiviral drug candidates. Specifically, molecular docking was applied to screen a small molecule library derived from the Ugi-azide four-component reaction methodology for inhibitors that target the influenza polymerase PAC-PB1N interactions. One hit compound 5 was confirmed to inhibit PAC-PB1N interactions in an ELISA assay and had potent antiviral activity in an antiviral plaque assay. Subsequent structure-activity relationship studies led to the discovery of compound 12a, which had broad-spectrum antiviral activity and a higher in vitro genetic barrier to drug resistance than oseltamivir. Overall, the discovery of compound 12a as a broad-spectrum influenza antiviral with a high in vitro genetic barrier to drug resistance is significant, as it offers a second line of defense to combat the next influenza epidemics and pandemics if vaccines and oseltamivir fail to confine the disease outbreak.
Introduction
Influenza virus infection is responsible for both seasonal influenza as well as sporadic influenza pandemics1. In the annual influenza season, an estimated 10–20% of the human population is infected with the influenza virus. Despite the availability of influenza vaccines and small molecule antiviral drugs, the death toll of influenza virus-related illness surpasses that of breast cancer, which places the influenza virus among the top ten leading causes of death in the United States2. Moreover, the convenient transmission through airways, coupled with the high mortality rates associated with pandemic influenza viruses and highly pathogenic avian influenza (HPAI) viruses, renders the influenza virus a major public health concern3. For example, the CDC estimated the 2009 H1N1 influenza pandemic led to 284,000 deaths globally in the first 12 months of outbreak4. Over 400 cases of human infection by HPAI H7N9 were reported in the recent 2017 outbreak in China and the mortality rate was ~40%5,6. Therefore, next-generation vaccines and antiviral drugs are clearly needed with improved efficacy and antiviral spectrum to combat influenza virus infection.
Influenza vaccines remain the mainstay for the prophylaxis of influenza infection. They are normally effective in preventing seasonal influenza virus infection with an overall effectiveness of ~60%7. However, there is often a six-month delay from strain identification to batch production, which impedes its use at the beginning of an influenza outbreak8. Thus, small molecule antivirals are highly desired. They are not alternatives, but essential complements of influenza vaccines.
There are currently two classes of FDA-approved small molecule influenza antivirals: M2 channel blockers such as amantadine and rimantadine that inhibit the early stage of viral uncoating9, and neuraminidase inhibitors, such as oseltamivir, peramivir, and zanamivir that inhibit the last stage of viral egress10. The increasing incidences of drug-resistant viruses now call for the development of next generation influenza antivirals11. Indeed, amantadine and rimantadine are no longer recommended, due to the widespread M2-S31N mutant12,13. The 2008–2009 seasonal H1N1 strain circulating in the United States and Japan is completely resistant to the only orally bioavailable drug, oseltamivir, owing to H275Y mutation in the neuraminidase14,15. The emergence of drug-resistant viruses with acquired fitness of transmission is a timely reminder of the urgent need for new antivirals with a novel mechanism of action and a high genetic barrier to drug resistance.
In the search for novel influenza antivirals, we sought to utilize a fast-track drug discovery program by exploring multi-component reaction (MCR) products for inhibitors that target the influenza polymerase subunit PAC-PB1N interactions. As drug discovery involves iterative cycles of design, synthesis, and pharmacological characterization, we envisioned that MCRs would greatly accelerate structure-activity relationship (SAR) studies during the drug discovery process, as final products can be conveniently synthesized in a one-pot one-step reaction, often containing 3 or more point of diversification16–18.
Influenza polymerase consists of three subunits PA, PB1, and PB2 (Fig. 1A)19,20. X-ray crystal structures show that the N-terminal tail of PB1 (PB1N) interacts extensively with the C-terminal domain of PA (PAC) (Fig. 1B), and as such the concave shape of the PB1N-binding site in PAC presents an attractive target for rational drug design20,21. Moreover, the PAC domain is highly conserved among different types and subtypes of influenza viruses, and compounds that inhibit PAC-PB1N interactions have shown to have broad-spectrum antiviral activity22–24. Using the crystal structure of PAC bound to the PB1N peptide (PDB: 3CM8) as a template, we screened two thousand compounds from an in-house library using the Schrödinger Glide standard precision docking program. The in-house library comprises a diverse set of compounds prepared by multicomponent reaction methodologies25. Top hits prioritized by in silico docking were tested in a PAC-PB1N ELISA assay. Compound 5 was found to inhibit PAC-PB1N interaction in a dose-dependent manner with an IC50 of 4.3 ± 0.1 µM. The antiviral activity of compound 5 was confirmed by the plaque assay and it had single to submicromolar EC50 values against several influenza A and B viruses, including both oseltamivir-sensitive and oseltamivir-resistant strains. Subsequent SAR led to the discovery of 12a with an improved selectivity index. Similarly, compound 12a had potent and broad-spectrum antiviral activity against several human clinical isolates of influenza A and B viruses. More importantly, no resistant virus was selected under drug selection pressure of compound 12a. Overall, we view the discovery of compound 12a - a broad-spectrum influenza antiviral - as proof-of-concept for the inherent advantage of our in-house fast-track MCR drug discovery platform, which has been recapitulated throughout the drug discovery arena.
Figure 1.

Structures of the influenza polymerase. (A) X-ray crystal structures of the viral polymerase complexes from the bat influenza A/H17N10 virus (PDB: 4WSB)26. PA: green; PB1: yellow; PB2: magenta. (B) X-ray crystal structure of PAC-PB1N (PDB: 3CM8)27. PA: green; PB1: yellow.
Results and Discussion
Chemistry
Compound 5 was synthesized by the Ugi-azide 4-CR in methanol at room temperature in 72% yield (Fig. 2A). A focused library of compounds 9a-9u with diversity elements amine (R1), aldehyde (R2), and isocyanide (R3) was synthesized according to the general procedure employed to afford compound 5 (Fig. 2B). Compounds 5 and 9a-9u were synthesized and tested as enantiomeric mixtures, whilst compounds 12a and 12b were prepared as diastereomeric mixtures (Fig. 2C), and separated by silica gel flash chromatography. The absolute stereochemistry of compound 12b was determined by X-ray crystallography (Fig. 2D).
Figure 2.

Synthesis routes and structures for compounds 5, 9a-9u and 12a-12b. (A) Synthesis of compound 5 by Ugi-azide 4-CR. (B) Analogs synthesized for the structure-activity relationship studies. (C) Synthesis of diastereomers by the chiral isocyanide strategy. (D) X-ray crystal structure of the diastereomer 12b.
PAC-PB1N inhibition and antiviral activity of the initial hit compound 5
The initial hit 5 was confirmed as a potent inhibitor of the PAC-PB1N polymerase subunit interactions in the ELISA assay with an IC50 of 4.3 ± 0.1 µM (Fig. 3) and its antiviral activity was tested in an antiviral plaque assay. Compound 5 was found to inhibit multiple strains of influenza A and B viruses with single to submicromolar EC50 values (Table 1). Its cellular cytotoxicity in MDCK cells with a 48 h incubation time was 17.4 ± 1.2 µM; therefore, the antiviral activity was not due to its cellular cytotoxicity.
Figure 3.

Inhibition of PAC-PB1N by compound 5 in ELISA assay. (A) Cartoon representation of the PAC-PB1N ELISA assay. (B) IC50 curve of compound 5 in the PAC-PB1N ELISA assay.
Table 1.
Broad-spectrum antiviral activity and cytotoxicity of compound 5.
| Influenza viruses | Drug sensitivity | EC50 (µM) | CC50 (µM) | SIa |
|---|---|---|---|---|
| A/WSN/33 (H1N1) | 1.2 ± 0.6 | 14.5 | ||
| A/California/07/2009 (H1N1) | Amantadine resistant Oseltamivir sensitive | 4.5 ± 0.7 | 3.9 | |
| A/Denmark/524/2009 (H1N1) | 1.8 ± 0.4 | 9.7 | ||
| A/Texas/04/2009 (H1N1) | Amantadine resistant | 3.5 ± 1.1 | 17.4 ± 1.2 | 5.0 |
| A/Washington/29/2009 (H1N1) | Oseltamivir resistant | 1.6 ± 0.5 | 10.9 | |
| B/Wisconsin/1/2010 (Yamagata) | Amantadine resistant | 0.9 ± 0.2 | 19.3 | |
| B/Brisbane/60/2008 (Victoria) | Oseltamivir sensitive | 1.0 ± 0.3 | 17.4 |
The values are the mean ± S.D. from two independent experiments. aSI = selectivity index.
Structure-activity relationship studies of compound 5
Encouraged by the broad-spectrum antiviral activity of 5, we were thus interested in pursuing SAR studies to further optimize antiviral potency and its selectivity index. To this end, we synthesized a focused library of 21 compounds (9a-9u) using the expeditious Ugi-azide 4-CR. The library includes compounds that have points of diversity at the amine component R1 (9a-9c), the aldehyde component R2 (9d-9o), and the aldehyde and isocyanide components combined (R2 & R3) (9p-9u, and 12a-12b). All compounds were initially tested at 5 µM against the A/WSN/33 (H1N1) virus in the plaque assay to rule out compounds that have no cellular antiviral activity (Fig. 4). Nucleozin and oseltamivir carboxylate, two known influenza antivirals, were included as positive controls. It was found that the amine component, 4-(2-keto-1-benzimidazolinyl)piperidine, is essential for the antiviral activity, as compounds with amine modifications 9a-9c had no antiviral activity (Fig. 4 and Table 2). Amongst compounds with modifications of the aldehyde input R2 (9d-9o), compounds 9i and 9k-9o had significantly improved antiviral activity compared with compound 5, and infected cells had less than 30% plaque formation when treated with 5 µM of compound. Compounds 9d, 9e, 9 g, and 9 h had moderate antiviral activity, while compounds 9 f and 9j were not active. These results suggest that hydrophobic aromatic groups such as benzene (9 g, 9i-9l), thiophene (9 m, 9o), and furan (9n) are preferred at the R2 position. When R2 substitution was benzene, a methoxyl group was tolerated at the ortho-, meta-, and para-positions (9k and 9 l). A small alkyl group such as ethyl (9 g) was also tolerated at the para- position; however, branched alkyl group such as isopropyl (9j) and a bulky substitution such as benzene (9 f) were not tolerated. For compounds with simultaneous modifications at both the aldehyde component R2 and the isocyanide component R3 (9p-9u), it was found that benzyl is preferred at R3 (9p, 9 u versus 9q, 9r, 9 s and 9t), and compound 9p had similar antiviral activity as 9o (12.3% versus 19.5% plaque formation at 5 µM). Compound 9t was not active (73.0% plaque formation at 5 µM), possibly due to the presence of the bulky naphthalene substitution at the R2 position. Compound 9 u had antiviral activity similar to compound 5 (22.7% vs 16.5% plaque formation at 5 µM) as it combines favorable substitutions from both the aldehyde component ((o-trifluoromethoxy)phenyl) and the isocyanide component (benzyl).
Figure 4.
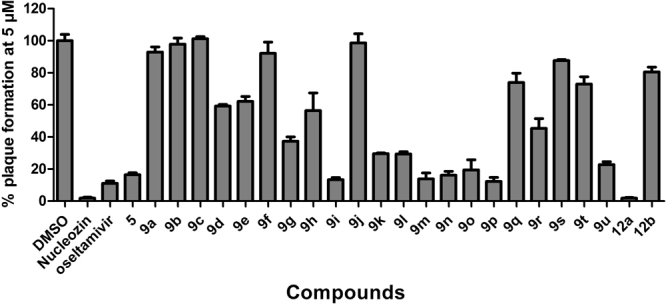
Structure-activity relationship studies of compound 5. Oseltamivir carboxylate and nucleozin were tested at 200 nM and 1 µM, respectively. All other compounds were tested at 5 µM. The results are the mean ± S.D. from two independent experiments.
Table 2.
Antiviral activity and PA-PB1 inhibition of tetrazole analogs.
| Compounds | % plaque formation at 5 µM | PAC-PB1N ELISA IC50 (µM) |
|---|---|---|
| 5 | 16.5 ± 1.2 | 4.3 ± 0.1 |
| 9a | 92.9 ± 4.5 | N.D. |
| 9b | 97.9 ± 5.3 | N.D. |
| 9c | 101.3 ± 1.8 | 26.9 ± 4.1 |
| 9d | 59.3 ± 0.8 | N.D. |
| 9e | 62.2 ± 4.3 | N.D. |
| 9 f | 92.2 ± 9.8 | >30 |
| 9 g | 37.4 ± 3.7 | N.D. |
| 9 h | 56.5 ± 12.3 | N.D. |
| 9i | 13.4 ± 0.8 | 13.6 ± 0.9 |
| 9j | 98.7 ± 8.0 | N.D. |
| 9k | 29.5 ± 1.0 | 7.0 ± 0.6 |
| 9 l | 29.3 ± 1.3 | N.D. |
| 9 m | 13.8 ± 1.8 | 15.1 ± 1.2 |
| 9n | 16.1 ± 1.2 | N.D. |
| 9o | 19.5 ± 8.8 | 9.9 ± 0.7 |
| 9p | 12.3 ± 2.1 | N.D. |
| 9q | 73.9 ± 8.2 | N.D. |
| 9r | 45.4 ± 8.6 | N.D. |
| 9 s | 87.7 ± 0.9 | N.D. |
| 9t | 73.0 ± 6.5 | N.D. |
| 9 u | 22.7 ± 2.2 | N.D. |
| 12a | 1.9 ± 0.3 | 7.6 ± 1.4 |
| 12b | 80.6 ± 4.5 | 37.0 ± 7.0 |
| Nucleozin | 0 | >30 |
| Oseltamivir | 11.1 ± 2.1 | >30 |
The results are the mean ± S.D. from two independent experiments. N.D. = not determined.
One feature of Ugi-azide 4-CR is that it produces a new chiral center during the reaction, and hence the product is a mixture of enantiomers. Although the enantiomers could be separated by chiral HPLC or other methods, chiral separation is generally time consuming and expensive, which presents a challenge for future development. Indeed, this somewhat compromises the advantages of exploring these specific chemotypes in the drug discovery arena. With grams of material required for possible downstream pharmacokinetic and in vivo animal studies, we therefore sought to develop a convenient synthesis and separation strategy to bypass chiral separation. Hence, we employed a chiral isocyanide, (S)-(−)-α-methylbenzyl isocyanide in Ugi-azide 4-CR, which we felt would be well-tolerated based on prior SAR results of 9a-9u. With this chiral isocyanide strategy, a mixture of diastereomers was thus produced (Fig. 2C) that were conveniently separated by silica gel flash column chromatography. The absolute stereochemistry of compound 12b was determined by X-ray crystallography as (R, S). It was found that the (S, S) diastereomer 12a had potent antiviral activity (1.9% plaque formation at 5 µM), while the (R, S) diastereomer 12b was not active (80.6% plaque formation at 5 µM) (Fig. 4).
Selected compounds were also tested for their inhibition of PAC-PB1N interactions in the ELISA assay (Table 2). For compounds 9i, 9k, 9 m, 9o, and 12a, which had potent antiviral activity (less than 30% plaque formation at 5 µM), the IC50 values in the ELISA assay ranged from 7.6 µM to 15.1 µM. For compounds 9c, 9 f, and 12b which had no antiviral activity (greater than 90% plaque formation at 5 µM), the IC50 values were above 26.9 µM. Overall, there is in general a positive correlation between the compounds’ PAC-PB1N inhibition and their antiviral efficacy. The lack of a straight linear correlation was expected as the antiviral efficacy is a combined effect of PAC-PB1N inhibition, cellular permeability, and potential off-target interactions.
Subsequently, the cellular cytotoxicity and broad-spectrum antiviral activity of one of the most potent compounds 12a was further profiled (Table 3). Compound 12a was not cytotoxic to either MDCK or A549 cells and the CC50 values were greater than 150 µM and 98.1 ± 2.5 µM, respectively. When tested against a panel of human clinical isolates of influenza A and B viruses, compound 12a showed broad-spectrum antiviral activity, with EC50 values ranging from 0.6 µM to 2.7 µM. It is noteworthy that compound 12a had no cross-resistance with the FDA-approved influenza antivirals amantadine and oseltamivir, as shown by the fact that compound 12a had potent antiviral activity against viruses that are resistant to amantadine, oseltamivir, or both (Table 3).
Table 3.
Broad-spectrum antiviral activity and cytotoxicity of compound 12a.
| Influenza viruses | Drug sensitivity | EC50 (µM)a | MDCK CC50 (µM)a | A549 CC50 (µM)a | SIb |
|---|---|---|---|---|---|
| A/WSN/33 (H1N1) | 0.7 ± 0.1 | >214.3/140.1 | |||
| A/California/07/2009 (H1N1) | Amantadine resistant | 1.3 ± 0.4 | >115.4/75.5 | ||
| A/Denmark/524/2009 (H1N1) | Oseltamivir sensitive | 0.8 ± 0.2 | >187.5/122.6 | ||
| A/Switzerland/9715293/2013 (H3N2) | 1.4 ± 0.6 | >107.1/70.1 | |||
| A/Texas/04/2009 (H1N1) | 1.1 ± 0.4 | >136.4/89.2 | |||
| A/Denmark/528/2009 (H1N1) | Amantadine resistant Oseltamivir resistant | 0.9 ± 0.3 | >166.7/109.0 | ||
| A/Washington/29/2009 (H1N1) | 0.9 ± 0.3 | >150 | 98.1 ± 2.5 | >166.7/109.0 | |
| B/Wisconsin/1/2010 (Yamagata) | 0.8 ± 0.3 | >187.5/122.6 | |||
| B/Memphis/20/1996 (Yamagata) | 2.7 ± 0.2 | >55.6/36.3 | |||
| B/Utah/09/2014 (Yamagata) | Amantadine resistant Oseltamivir sensitive | 1.5 ± 0.3 | >100/65.4 | ||
| B/Phuket/3073/2013 (Yamagata) | 1.2 ± 0.2 | >125/81.8 | |||
| B/Brisbane/60/2008 (Victoria) | 0.6 ± 0.3 | >250/163.5 |
aThe values are the mean ± S.D. from two independent experiments. bSelectivity index is expressed as MDCK/A549.
Cellular antiviral mechanism of compound 12a
To gain insights into the cellular antiviral mechanism of 12a, we performed time-of-addition and RT-qPCR. As 12a was confirmed to inhibit influenza polymerase PA-PB1 interactions, it is expected to inhibit the intermediate stage of viral replication post viral fusion in the time-of-addition experiment. Furthermore, when the viral polymerase is inhibited, viral RNA transcription and replication should also be reduced. To confirm which stage of viral replication was affected by 12a, we performed a time-of-addition experiment by adding 12a at different time points during viral replication (Fig. 5A and B). Thus, it was found that the efficacy of 12a gradually decreased when it was added at later stages of viral replication (Fig. 5B). Pre-treatment of cells with 12a has little to no effect on viral replication, indicating that 12a might not target the host factors (Fig. 5B). As a control, oseltamivir carboxylate retained potent antiviral activity even when it was added 8 h post viral infection (Fig. 5A), which is consistent with its known mechanism of inhibiting viral egress. Next, to test whether 12a inhibits viral RNA transcription and replication, we performed RT-qPCR assay. Quantification of the viral nucleoprotein (NP) RNA expression levels by RT-qPCR revealed that the levels of vRNA, cRNA, and mRNA were all inhibited in a dose-dependent manner by 12a (Fig. 5C). The IC50 values are consistent with the antiviral efficacy EC50 values of compound 12a. Collectively, the results from the time-of-addition experiment and RT-qPCR are consistent with the antiviral mechanism of 12a by inhibiting the viral polymerase PA-PB1 interactions.
Figure 5.

Cellular antiviral mechanism of compound 12a. (A) Time-of-addition experiments with oseltamivir carboxylate. (B) Time-of-addition experiments with compound 12a. For the time-of-addition experiments, MDCK cells were infected with the A/WSN/33 (H1N1) at MOI of 0.01 at −2 h time point; viruses were first incubated at 4 °C for 1 h for attachment followed by 37 °C for 1 h for viral entry. At time point 0 h, cells were washed with PBS buffer and viruses were harvested at 12 h p.i. The titer of harvested virus was determined by plaque assay. Arrows indicate the period in which (A) 1 µM oseltamivir carboxylate or (B) 10 µM 12a was present. (C) RT-qPCR quantification of the NP mRNA, cRNA and vRNA levels upon compound 12a treatment. Asterisks indicate statistically significant difference in comparison with the DMSO control (student’s t-test, **p < 0.01, ***p < 0.001). The results represent the average of two repeats ± standard deviation.
Compound 12a has a higher in vitro genetic barrier to drug resistance than oseltamivir
Drug-induced resistance is one of the major obstacles facing antiviral drugs10. We therefore designed serial viral passage experiments to characterize the genetic barrier to drug resistance of compound 12a (Fig. 6)28–31. In this experiment, the A/WSN/33 (H1N1) virus was amplified in the presence of increasing concentrations of compound 12a and the drug sensitivity of the resulting viruses at different passages was assayed against compound 12a using a plaque assay. Oseltamivir carboxylate was included as a control. Gratifyingly, viruses at passage 10 remained sensitive to compound 12a, and no increase in EC50 value was observed (Fig. 6B and Table 4). In contrast, the EC50 for oseltamivir carboxylate increased 10-fold at passage six and onwards, which is consistent with previous reports32,33. These results suggest that compound 12a targets a vital viral replication component such as the viral polymerase that is less prone to mutate. Overall, compound 12a demonstrated a high in vitro genetic barrier to drug resistance, rendering it a desirable drug candidate for further development.
Figure 6.

Compound 12a has a high in vitro genetic barrier to drug resistance as shown by the serial viral pasage experiment. (A) Cartoon representation of the serial viral passage experiment. (B) Comparison of the in vitro genetic barrier of drug resistance between oseltamivir carboxylate and compound 12a.
Table 4.
In vitro serial drug passage experiment with compound 12a.
| Passage # | Oseltamivir carboxyalte applied (nM) | EC50 (nM) | Compound 12a applied (µM) | EC50 (µM) |
|---|---|---|---|---|
| 0 | 6.7 ± 1.8 | 0.7 ± 0.1 | ||
| 1 | 10 | N.D. | 1 | N.D. |
| 2 | 20 | N.D. | 2 | N.D. |
| 3 | 40 | 7.9 ± 3.1 | 4 | 0.7 ± 0.5 |
| 4 | 80 | N.D. | 8 | N.D. |
| 5 | 160 | N.D. | 16 | N.D. |
| 6 | 320 | 60.1 ± 14.7 | 32 | 0.5 ± 0.3 |
| 7 | 640 | N.D. | 64 | N.D. |
| 8 | 640 | N.D. | 64 | N.D. |
| 9 | 640 | N.D. | 64 | N.D. |
| 10 | 640 | 70.2 ± 18.0 | 64 | 0.7 ± 0.4 |
Passage was performed using the A/WSN/33 (H1N1) virus by following our reported procedure29–31. Oseltamivir carboxylate was included as a control. The EC50 values at selected passages were determined by plaque assay. The values are the mean ± S.D. from two independent experiments. N.D. = not determined.
Docking model of compound 12a in PAc
To gain insights on how compound 12a binds to the PB1N-binding pocket in PAC, we performed molecular docking using Schrödinger Glide software. In the docking model of 12a in PA, 12a snuggly fits in the PB1-binding pocket in PA (Fig. 7A), forming extensive hydrophobic interactions and multiple π–π interactions (Fig. 7B). For example, the phenyl ring from the isocyanide input (R3) in 12a interacts with F707 and K643 through π–π and cation–π interactions, respectively. The thiophene ring from 12a forms π–π interactions with F710. In addition, the carbonyl from the benzimidazol-2-one in 12a forms a hydrogen bond with the E623 backbone amide NH, while the benzene ring of the benzimidazol-2-one fits in the hydrophobic pocket formed by F411 and I621.
Figure 7.

Docking model of compound 12a in the PB1N-binding pocket in PAC. (A) Surface view of the docking model of compound 12a in PAC. (B) Ligand interaction diagram of compound 12a with residues in the binding site.
Conclusions
Drug discovery is an expensive and time-consuming endeavor. On average, it costs billions of dollars and 10–15 years to advance a drug to the market34. Development of antiviral drugs comes with an even greater challenge because of the intrinsic issue of drug resistance, which could render years of research efforts futile14. To shorten the length of antiviral drug development, we are interested in further building our fast-track drug discovery platform by exploiting MCR products to identify hits with broad-spectrum antiviral activity and a high genetic barrier to drug resistance. Exploring MCRs for antiviral drug discovery offers the key advantage of expeditious synthesis that could feasibly enable timely development of newer generations of antivirals if resistance to earlier generations were to emerge. Toward this goal, we launched an in silico virtual screening campaign of an in-house, MCR-derived small-molecule library25,35,36 against the highly conserved PAC subunit of the influenza polymerase. The rationale for choosing this library were: (1) compounds in this library were synthesized by one-pot MCR reactions, which will ensure expeditious turnover of subsequent structure–activity relationship (SAR) studies; (2) the synthesis methodologies for compounds in this library are established and optimized, which ensures expeditious re-synthesis of interesting hits in gram quantities for subsequent mechanistic studies and in vivo animal studies and (3) the tetrazole ring embedded in all the molecules are cis-amide bond isosteres and as such are attractive constrained peptidomimetics37. As a proof-of-concept, our efforts have already yielded several hits. For example, compound 5 (Table 1) is active against multiple influenza A and B strains. Subsequent SAR by the expeditious Ugi-azide 4-CR led to the discovery of compound 12a that: (1) inhibits PAC-PB1N interactions in the ELISA assay, (2) has broad-spectrum antiviral activity against a panel of human clinical isolates of influenza A and B viruses, (3) has high selectivity index, and (4) has a high in vitro genetic barrier to drug resistance. The cellular antiviral mechanism of 12a was further confirmed by time-of-addition and RT-qPCR experiments. Importantly, by introducing a chiral isocyanide as the building block in the Ugi-azide 4-CR, we circumvent the problem of chiral separation and the diastereomeric product mixture was conveniently separated by silica gel flash chromatography. In summary, the promising in vitro antiviral efficacy of compound 12a warrants its further development as a next-generation influenza antiviral.
Experimental Section
Molecular Docking
The X-ray crystal structure of PAc-PB1N (PDB: 3CM8) was used as the protein template for the molecular docking. Docking was performed using Schrödinger Glide. In house small molecule library was processed in Ligprep and all possible enantiomers and diasteromers were generated and docked individually. The grid box center was set as the centroid of workspace ligand (PB1N). The grid box size was set to dock ligands similar in size to the workspace ligand (PB1N). All compounds were initially docked using standard precision algorithm, and top 10% of the hits were further docking using extra precision algorithm. Hits were ranked by Glide score.
Chemistry
All commercially available chemicals were used without further purification. All final compounds were purified by flash column chromatography. Compound 5 and 9a-9u were synthesized and tested as a mixture of enantiomers. Compounds 12a and 12b were separated as pure diastereomers and tested individually. 1H and 13C NMR spectra were recorded on a Bruker-400 NMR spectrometer. Chemical shifts are reported in parts per million referenced with respect to residual solvent (DMSO-d6) 2.50 ppm and (Chloroform-d) 7.26 ppm or from internal standard tetramethylsilane (TMS) 0.00 ppm. The following abbreviations were used in reporting spectra: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets; ddd, doublet of doublet of doublets. HPLC-grade solvents were used for all reactions. Flash column chromatography was performed using silica gel (230–400 mesh, Merck). Low-resolution mass spectra were obtained using an ESI technique on a 3200 Q Trap LC/MS/MS system (Applied Biosystems). High-resolution mass spectra were obtained using the positive ESI method for all the compounds, obtained in an Ion Cyclotron Resonance (ICR) spectrometer. The purity was assessed by using a Shimadzu LC-MS with a Waters XTerra MS C-18 column (part #186000538), 50 × 2.1 mm, at a flow rate of 0.3 mL/min; λ = 250 and 220 nm; mobile phase A, 0.1% formic acid in H2O, and mobile phase B’, 0.1% formic in 60% isopropanol, 30% CH3CN and 9.9% H2O. All compounds submitted for testing in the ELISA and plaque assays were confirmed to be >95.0% purity by LC-MS traces. All compounds were characterized by proton and carbon NMR and MS.
Synthesis Procedures
Amine (1.0 equiv) and aldehyde (1.0 equiv) were mixed in methanol (5 ml) and stirred at room temperature for 15 minutes. Then TMS-azide (1.0 equiv) and isocyanide (1.0 equiv) were added sequentially and the resulting mixture was stirred at room temperature overnight. After that, the solvent was removed under reduced pressure and the crude product was purified with flash silica gel chromatography (Ethyl acetate in hexane 20–70%).
1‐(1‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1,2,3,4‐tetrazol‐5‐yl]‐3‐phenylpropyl}piperidin‐4‐ yl)‐3 H‐1,3‐benzodiazol‐2‐one (5). White solid. Yield: 65%. 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1 H), 7.27–6.89 (m, 12 H), 4.24–4.22 (m, 5 H), 3.95–3.85 (m, 1 H), 2.97–2.95 (m, 1 H), 2.86–2.19 (m, 9 H), 1.84–1.70 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 144.8, 143.5, 134.8, 133.6, 130.2, 118.6, 118.1, 117.9, 117.7, 116.7, 115.8, 110.9, 110.6, 107.8, 107.6, 104.3, 99.5, 99.0, 54.0, 53.9, 46.7, 40.3, 38.6, 37.5, 22.1, 19.2, 19.1, 18.6; C30H31N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 538.25611, found 538.25612.
1‐(1‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]‐3‐phenylpropyl}piperidin‐4‐yl)‐3‐methyl‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9a). Beige oil. Yield: 89%. 1H NMR (400 MHz, DMSO-d6) δ 7.28–7.24 (m, 3 H), 7.18–7.10 (m, 5 H), 7.07–7.06 (m, 2 H), 7.04–7.02 (m, 2 H), 4.33 (2, 4 H), 4.14–3.96 (m, 2 H), 3.28 (s, 3 H), 2.87 (d, J = 12.1 Hz, 1 H), 2.66–2.59 (m, 2 H), 2.46 (d, J = 9.4 Hz, 1 H), 2.31–2.05 (m, 5 H), 1.63 (d, J = 13.8 Hz, 1 H), 1.52 (d, J = 11.5 Hz, 1 H), 1.33–1.17 (m, 1 H); 13C NMR (101 MHz, DMSO-d6) δ 154.1, 152.9, 144.9, 143.6, 141.1, 129.7, 128.3, 128.2, 127.8, 126.7, 125.9, 120.7, 120.6, 118.5, 117.6, 114.7, 108.6, 107.8, 64.22, 64.15, 56.3, 50.5, 47.9, 47.7, 31.8, 29.03, 28.97, 26.7; C31H33N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 552.27245, found 552.27176.
8‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]‐3‐phenylpropyl}‐1‐phenyl‐ 1,3,8‐triazaspiro[4.5]decan‐4‐one (9b). White solid. Yield: 66%. 1H NMR (400 MHz, DMSO-d6) δ 8.62 (s, 1 H), 7.31 (d, J = 2.3 Hz, 1 H), 7.28–7.13 (m, 7 H), 7.12–7.01 (m, 2 H), 6.81–6.68 (m, 3 H), 4.53 (s, 2 H), 4.33–4.30(m, 4 H), 3.95–3.92 (m, 1 H), 3.08–3.02 (m, 1 H), 2.89–2.78 (m, 2 H), 2.62–2.57 (m, 3 H), 2.49–2.40 (m, 1 H), 2.38–2.22 (m, 3 H), 1.56 (d, J = 13.2 Hz, 1 H), 1.50–1.41 (m, 1 H); 13C NMR (101 MHz, DMSO-d6) δ 176.0, 154.1, 144.9, 143.7, 143.2, 141.1, 128.9, 128.3, 128.2, 126.7, 125.9, 118.3, 117.6, 117.5, 114.4, 113.9, 64.2, 64.1, 58.6, 58.1, 56.4, 45.4, 44.2, 31.8, 28.7, 28.6; C31H33N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 552.27176, found 552.27210.
3‐(1‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]‐3‐phenylpropyl}piperidin‐4‐yl)‐1 H‐indole (9c). White solid. Yield: 75%. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1 H), 7.66–7.57 (m, 1 H), 7.41–7.34 (m, 1 H), 7.30–7.23 (m, 3 H), 7.23–7.08 (m, 6 H), 7.00–6.92 (m, 3 H), 4.46–4.21 (m, 4 H), 3.92 (dd, J = 8.6, 5.4, 1 H), 2.97 (d, J = 11.4, 1 H), 2.84–2.72 (m, 2 H), 2.74–2.62 (m, 2 H), 2.60–2.46 (m, 2 H), 2.45–2.26 (m, 2 H), 2.11 (d, J = 13.1, 1 H), 1.97 (d, J = 12.8, 1 H), 1.83–1.56 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 154.10, 145.11, 143.93, 140.88, 136.41, 128.47, 128.37, 127.18, 126.55, 126.09, 121.94, 121.23, 119.64, 119.14, 119.11, 118.35, 117.87, 114.81, 111.21, 64.40, 64.29, 57.41, 49.81, 49.09, 33.58, 33.42, 33.06, 32.61, 29.12; C31H32N6O2 HRMS (ESI) m/z calculated for [M + H]+ = 521.25867, found 521.26690.
1‐(1‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]propyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9d). Beige solid. Yield: 34%. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1 H), 7.35–7.34 (m, 1 H), 7.17–7.11 (m, 2 H), 7.08–7.05 (m, 1 H), 6.99–6.94 (m, 3 H), 4.34 (s, 4 H), 4.13–3.93 (m, 2 H), 2.90 (d, J = 10.4 Hz, 1 H), 2.51–2.46 (m, 3 H), 2.23–1.99 (m, 4 H), 1.62 (d, J = 13.7 Hz, 1 H), 1.51 (d, J = 11.8 Hz, 1 H), 0.89 (t, J = 7.3 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 154.3, 153.6, 144.9, 143.7, 129.1, 128.3, 127.0, 120.5, 120.3, 118.6, 117.7, 114.7, 108.8, 108.6, 64.2, 58.9, 49.9, 48.4, 47.5, 29.0, 28.9, 20.7, 11.2; C24H27N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 462.22481, found 462.22522.
1‐(1‐{1‐[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]‐2‐phenylethyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9e). White solid. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1 H), 7.30–7.13 (m, 5 H), 7.08–6.92 (m, 5 H), 6.83 (d, J = 2.5 Hz, 1 H), 6.71 (dd, J = 8.6, 2.5 Hz, 1 H), 4.35–4.23 (m, 5 H), 4.07–3.96 (m, 1 H), 3.44–3.27 (m, 2 H), 3.07 (d, J = 11.4 Hz, 1 H), 2.65 (d, J = 11.3 Hz, 1 H), 2.50–2.42 (m, 1 H), 2.30 (dd, J = 12.2, 9.8 Hz, 1 H), 2.22–2.01 (m, 2 H), 1.64 (d, J = 11.9 Hz, 1 H), 1.54 (d, J = 11.7 Hz, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 154.36, 154.05, 145.46, 144.08, 138.50, 129.77, 129.53, 128.80, 128.76, 126.96, 126.88, 120.97, 120.76, 118.85, 118.10, 114.93, 114.26, 109.28, 109.07, 64.67, 64.61, 60.14, 50.27, 49.07, 48.45, 35.35, 29.42, 29.30; C29H29N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 524.23319, found 524.24136.
1‐[1‐({[1,1′‐biphenyl]‐4‐yl}[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl]methyl)piperidin‐4‐yl]‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9 f). White solid. Yield: 62%. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1 H), 7.74–7.65 (m, 4 H), 7.56–7.43 (m, 4 H), 7.42–7.33 (m, 1 H), 7.24 (d, J = 2.5 Hz, 1 H), 7.17–6.92 (m, 6 H), 5.27 (s, 1 H), 4.41–4.22 (m, 4 H), 4.06–3.84 (m, 1 H), 2.89 (dd, J = 30.1, 10.5 Hz, 2 H), 2.35–2.03 (m, 4 H), 1.66–1.53 (m, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 154.93, 154.07, 145.64, 144.15, 140.44, 140.06, 134.07, 130.30, 129.62, 129.43, 128.74, 128.07, 127.17, 126.98, 120.98, 120.77, 119.35, 118.15, 115.46, 109.25, 109.01, 64.74, 64.63, 62.55, 50.28, 50.16, 49.28, 29.21, 29.11; C34H31N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 586.24884, found 586.25730.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](4‐ethylphenyl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9 g). White solid. Yield: 69%. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1 H), 7.32 (d, J = 8.2 Hz, 2 H), 7.29–7.15 (m, 3 H), 7.15–7.06 (m, 2 H), 7.04–6.92 (m, 4 H), 5.14 (s, 1 H), 4.44–3.78 (m, 5 H), 2.84 (dd, J = 26.0, 10.1 Hz, 2 H), 2.62 (q, J = 7.6 Hz, 2 H), 2.31–2.13 (m, 3 H), 2.02 (t, J = 11.4 Hz, 1 H), 1.57 (t, J = 14.4 Hz, 2 H), 1.20 (t, J = 7.6 Hz, 3 H). 13C NMR (101 MHz, DMSO-d6) δ 155.13, 154.07, 145.62, 144.19, 144.14, 132.29, 129.60, 128.73, 128.10, 127.12, 120.97, 120.77, 119.32, 118.14, 115.43, 114.26, 109.24, 109.00, 64.73, 64.62, 62.69, 50.31, 50.13, 49.34, 29.16, 29.07, 28.28, 15.88; C30H31N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 538.24884, found 538.25708.
1‐(1‐{[1‐(2,3‐dihydro-1,4-benzodioxin-6-yl)-1 H-1,2,3,4-tetrazol-5-yl](pyridin-4-yl)methyl}piperidin-4-yl)-2,3-dihydro-1 H-1,3-benzodiazol-2-one (9 h). Whites solid. Yield: 43%. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1 H), 8.60 (d, J = 6.0 Hz, 2 H), 7.61–7.37 (d, J = 6.0 Hz, 2 H), 7.30 (d, J = 2.1 Hz, 1 H), 7.19–7.03 (m, 3 H), 6.97 (m, 3 H), 5.39 (s, 1 H), 4.34 (t, J = 3.3 Hz, 4 H), 4.18–3.81 (m, 1 H), 2.88 (d, J = 9.3 Hz, 1 H), 2.74 (d, J = 10.0 Hz, 1 H), 2.35–1.91 (m, 4 H), 1.72–1.40 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 153.58, 153.23, 149.56, 145.17, 143.66, 142.55, 129.09, 128.27, 126.75, 124.25, 120.52, 120.28, 118.83, 117.65, 114.93, 108.80, 108.55, 79.16, 64.28, 64.16, 61.14, 49.60, 49.32, 48.45, 28.73, 28.66; C27H26N8O3 HRMS (ESI) m/z calculated for [M + H]+ = 511.21279, found 511.22126.
1-(1-{[1-(2,3-dihydro‐1,4‐benzodioxin-6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](phenyl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9i). White solid. Yield: 88%. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1 H), 7.41–7.33 (m, 5 H), 7.19–7.18 (m, 1 H), 7.11–7.07 (m, 2 H), 7.01–6.94 (m, 4 H), 5.19 (s, 1 H), 4.36–4.33 (m, 4 H), 4.02–3.94 (m, 1 H), 2.87 (d, J = 9.7 Hz, 1 H), 2.80 (d, J = 11.9 Hz, 1 H), 2.51–2.50 (m, 1 H), 2.28–2.12 (m, 2 H), 1.61–1.52 (m, 1 H); 13C NMR (101 MHz, DMSO-d6) δ 154.5, 153.6, 145.1, 143.7, 134.5, 129.2, 129.1, 128.3, 128.2, 126.7, 120.5, 120.3, 118.8, 117.7, 115.0, 108.8, 108.5, 64.3, 64.1, 62.4, 59.7, 49.8, 49.6, 48.8, 28.6, 20.7, 14.1, 10.8; C28H27N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 510.22481, found 510.22513.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl][4‐(propan‐2‐yl)phenyl]methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9j). White solid. Yield: 65%. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1 H), 7.33 (d, J = 8.3 Hz, 2 H), 7.26 (d, J = 8.3 Hz, 2 H), 7.21–7.07 (m, 3 H), 7.05–6.92 (m, 4 H), 5.14 (s, 1 H), 4.48–4.24 (m, 4 H), 4.07–3.89 (m, 1 H), 2.94–2.77 (m, 3 H), 2.30–2.10 (m, 3 H), 2.03 (t, J = 11.5 Hz, 1 H), 1.64–1.52 (m, 2 H), 1.22 (d, J = 6.9 Hz, 6 H). 13C NMR (101 MHz, DMSO-d6) δ 155.15, 154.07, 148.77, 145.62, 144.13, 132.43, 129.60, 128.73, 127.13, 126.63, 120.97, 120.77, 119.35, 118.13, 115.46, 109.24, 109.01, 64.73, 64.62, 62.67, 50.30, 50.16, 49.35, 33.58, 29.16, 29.06, 24.29, 24.24; C31H33N7O3 HRMS (ESI) m/z calculated for [M + H]+ = 552.26449, found 552.27210.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](2,3‐dimethoxyphenyl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9k). White solid. Yield: 56%. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1 H), 7.32–7.29 (m, 1 H), 7.18–7.16 (m, 1 H), 7.10–7.06 (m, 2 H), 7.04–6.98 (m, 4 H), 6.92–6.85 (m, 2 H), 5.56 (s, 1 H), 4.35–4.30 (m, 4 H), 4.26–4.21 (m, 1 H), 3.87 (s, 3 H), 3.64 (s, 3 H), 3.10–2.98 (m, 2 H), 2.54–2.34 (m, 3 H), 2.32–2.16 (m, 1 H), 1.84–1.71 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 155.2, 154.8, 152.7, 147.3, 145.5, 144.1, 129.2, 129.0, 128.2, 127.0, 123.9, 122.1, 121.3, 121.1, 119.1, 118.1, 115.4, 112.7, 109.8, 109.7, 64.5, 64.4, 60.7, 56.0, 55.9, 50.8, 50.5, 50.1, 29.6; C30H31N7O5 HRMS (ESI) m/z calculated for [M + H]+ = 570.24594, found 570.24716.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](4‐methoxyphenyl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9l). White solid. Yield: 76%. 1H NMR (400 MHz, CDCl3) δ 10.16 (s, 1 H), 7.35 (d, J = 8.3 Hz, 2 H), 7.26–7.15 (m, 1 H), 7.15–6.96 (m, 4 H), 6.96–6.84 (m, 3 H), 6.77 (dd, J = 8.6, 2.5 Hz, 1 H), 4.89 (s, 1 H), 4.34 (t, J = 3.8 Hz, 4 H), 4.31 (s, 1 H), 3.81 (s, 3 H), 3.07 (d, J = 11.1 Hz, 1 H), 2.96 (d, J = 7.0 Hz, 1 H), 2.58–2.30 (m, 3 H), 2.11 (m, 1 H), 1.75 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 159.85, 155.25, 154.95, 145.63, 144.16, 130.59, 129.15, 128.19, 126.89, 126.79, 121.30, 121.11, 118.95, 118.15, 115.27, 114.10, 109.88, 109.73, 64.53, 64.43, 63.19, 55.44, 50.74, 50.50, 49.99, 29.47, 29.35; C29H29N7O4 HRMS (ESI) m/z calculated for [M + H]+ = 540.22810, found 540.23656.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](thiophen‐2‐yl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9 m). White solid. Yield: 61%. 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1 H), 7.37–7.35 (m, 1 H), 7.18–6.92 (m, 8 H), 5.35 (s, 1 H), 4.35–4.21 (m, 5 H), 3.13–3.02 (m, 2 H), 2.64–2.56 (m, 1 H), 2.49–2.37 (m, 2 H), 2.26–2.19 (m, 1 H), 1.84–1.76 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 155.3, 153.6, 145.7, 144.3, 136.8, 129.1, 128.6, 128.2, 126.9, 126.8, 126.7, 121.4, 121.2, 118.6, 118.3, 115.0, 110.0, 109.6, 64.5, 64.4, 60.5, 58.4, 50.6, 50.0, 48.8, 29.5, 29.3, 21.2, 14.3, 13.8; C26H25N7O3S HRMS (ESI) m/z calculated for [M + H]+ = 516.18123, found 516.18154.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](furan‐3‐yl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9n). White solid. Yield: 68%. 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1 H), 7.66–7.39 (m, 2 H), 7.25–7.15 (m, 2 H), 7.14–7.00 (m, 5 H), 6.67 (dd, J = 1.9, 0.9 Hz, 1 H), 5.05 (s, 1 H), 4.41–4.30 (m, 4 H), 4.30–4.18 (m, 1 H), 3.04 (dd, J = 40.0, 11.3 Hz, 2 H), 2.71–2.04 (m, 4 H), 1.92–1.72 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 154.90, 153.74, 145.45, 144.08, 143.33, 142.36, 129.11, 127.97, 127.00, 121.21, 121.05, 118.44, 118.12, 118.10, 114.81, 111.53, 109.68, 109.47, 64.44, 64.37, 54.67, 50.54, 50.33, 47.81, 29.50, 29.16; C26H25N7O4 HRMS (ESI) m/z calculated for [M + H]+ = 500.19680, found 500.20514.
1‐(1‐{[1‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](thiophen‐3‐yl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9o). White solid. Yield: 76%. 1H NMR (400 MHz, CDCl3) δ 9.82 (s, 1 H), 7.42–7.26 (m, 3 H), 7.25–7.15 (m, 1 H), 7.16–7.00 (m, 5 H), 6.93 (dd, J = 8.6, 2.5 Hz, 1 H), 5.17 (d, J = 0.5 Hz, 1 H), 4.42–4.32 (m, 4 H), 4.25 (tt, J = 12.2, 4.2 Hz, 1 H), 3.52 (d, J = 4.9 Hz, 1 H), 3.18–3.10 (m, 1 H), 3.04–2.96 (m, 1 H), 2.62–2.34 (m, 3 H), 2.23–2.12 (m, 1 H), 1.86–1.78 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 154.97, 154.08, 145.49, 144.08, 134.78, 129.09, 128.44, 128.00, 126.83, 126.10, 125.46, 121.21, 121.04, 118.60, 118.10, 114.95, 109.70, 109.52, 64.43, 64.35, 58.57, 50.57, 50.34, 48.79, 29.48, 29.23; C26H25N7O3S HRMS (ESI) m/z calculated for [M + H]+ = 516.17396, found 516.18240.
1‐{1‐[(1‐benzyl‐1 H‐1,2,3,4‐tetrazol‐5‐yl)(thiophen‐3‐yl)methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9p). White solid. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1 H), 7.55 (d, J = 4.5 Hz, 2 H), 7.46–7.16 (m, 6 H), 7.16–6.79 (m, 4 H), 6.01–5.76 (m, 2 H), 5.63 (s, 1 H), 3.97 (m, 1 H), 3.00 (d, J = 6.9 Hz, 1 H), 2.89 (d, J = 10.8 Hz, 1 H), 2.37–1.93 (m, 4 H), 1.68–1.45 (m, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 154.22, 153.64, 134.86, 134.37, 129.10, 128.80, 128.74, 128.25, 128.15, 127.72, 125.88, 125.66, 120.48, 120.25, 108.76, 108.62, 79.16, 57.27, 54.88, 50.29, 49.84, 48.62, 47.89, 28.79, 28.52; C25H25N7OS HRMS (ESI) m/z calculated for [M + H]+ = 472.18413, found 472.19237.
1‐{1‐[(1‐cyclohexyl‐1 H‐1,2,3,4‐tetrazol‐5‐yl)(thiophen‐3‐yl)methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9q). White solid. Yield: 65%. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1 H), 7.71–7.44 (m, 2 H), 7.27 (dd, J = 4.8, 1.4 Hz, 1 H), 7.22–7.06 (m, 1 H), 7.06–6.79 (m, 3 H), 5.66 (s, 1 H), 4.03 (m, 1 H), 3.04 (d, J = 10.8 Hz, 1 H), 2.87 (d, J = 10.8 Hz, 1 H), 2.46–2.15 (m, 3 H), 2.16–2.00 (m, 2 H), 2.00–1.77 (m, 5 H), 1.77–1.38 (m, 5 H), 1.38–1.20 (m, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 153.63, 153.24, 134.77, 129.21, 128.67, 128.32, 125.92, 125.41, 120.48, 120.20, 108.81, 108.34, 79.16, 57.51, 57.12, 50.29, 49.76, 47.93, 32.87, 32.60, 28.91, 28.63, 24.85, 24.82, 24.63; C24H29N7OS HRMS (ESI) m/z calculated for [M + H]+ = 464.21543, found 464.22364.
1‐[1‐({1‐[(4‐methylbenzenesulfonyl)methyl]‐1 H‐1,2,3,4‐tetrazol‐5‐yl}(thiophen‐3‐yl)methyl)piperidin‐4‐yl]‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9r). White solid. Yield: 53%. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1 H), 7.73–7.62 (m, 2 H), 7.58 (dd, J = 5.0, 2.9 Hz, 1 H), 7.55–7.42 (m, 3 H), 7.25–7.14 (m, 2 H), 7.04–6.86 (m, 3 H), 6.72–6.50 (m, 2 H), 5.47 (s, 1 H), 4.04–3.87 (m, 1 H), 2.92 (dd, J = 53.6, 11.2 Hz, 2 H), 2.42 (s, 3 H), 2.39–2.14 (m, 3 H), 2.03–1.84 (m, 1 H), 1.62 (dd, J = 31.7, 11.8 Hz, 2 H). 13C NMR (101 MHz, DMSO) δ 154.84, 153.64, 146.08, 133.76, 133.12, 130.24, 129.19, 128.72, 128.27, 125.80, 125.77, 120.50, 120.28, 108.74, 108.61, 64.73, 57.23, 49.82, 49.71, 47.81, 28.86, 28.56, 21.22; C26H27N7O3S2 HRMS (ESI) m/z calculated for [M + H]+ = 550.16168, found 550.17011.
1‐(1‐{[1‐(2,6‐dimethylphenyl)‐1 H‐1,2,3,4‐tetrazol‐5‐yl](thiophen‐3‐yl)methyl}piperidin‐4‐yl)‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9 s). White solid. Yield: 72%. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1 H), 7.55 (dd, J = 5.0, 3.0 Hz, 1 H), 7.50 (t, J = 7.6 Hz, 1 H), 7.44 (dd, J = 3.0, 1.3 Hz, 1 H), 7.41 (m, 1 H), 7.29 (m, 1 H), 7.21 (dd, J = 5.0, 1.3 Hz, 1 H), 7.08 (m, 1 H), 7.02– 6.92 (m, 3 H), 4.87 (s, 1 H), 3.98 (m, 1 H), 3.10–2.72 (m, 2 H), 2.39–2.11 (m, 3 H), 2.06 (s, 3 H), 2.04–1.90 (m, 1 H), 1.70–1.53 (m, 2 H), 1.48 (s, 3 H). 13C NMR (101 MHz, DMSO) δ 155.34, 153.61, 135.41, 135.35, 134.17, 131.42, 131.05, 129.23, 128.90, 128.79, 128.52, 128.25, 126.45, 126.10, 120.46, 120.28, 108.76, 108.34, 79.16, 57.98, 51.00, 49.84, 48.14, 28.67, 28.41, 17.15, 16.28; C26H27N7OS HRMS (ESI) m/z calculated for [M + H]+ = 486.19978, found 486.20824.
1‐{1‐[(1‐cyclopentyl‐1 H‐1,2,3,4‐tetrazol‐5‐yl)(naphthalen‐1‐yl)methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9t). White solid. Yield: 62%. 1H NMR (400 MHz, CDCl3) δ 9.66 (s, 1 H), 8.53–8.50 (m, 1 H), 7.91–7.85 (m, 2 H), 7.65–7.61 (m, 1 H), 7.58–7.53 (m, 2 H), 7.49–7.45 (m, 1 H), 7.22–7.20 (m, 1 H), 7.12–7.05 (m, 3 H), 5.93 (s, 1 H), 4.91–4.83 (m, 1 H), 4.40–4.33 (m, 1 H), 3.27–3.23 (m, 1 H), 2.92–2.88 (m, 1 H), 2.81–2.76 (m, 1 H), 2.72–2.59 (m,1 H), 2.53–2.44 (m, 1 H), 1.92–1.78 (m, 8 H), 1.64–1.59 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 155.1, 153.5, 134.3, 131.7, 131.5, 129.7, 129.4, 129.1, 128.1, 127.0, 126.6, 126.3, 125.1, 123.7, 121.4, 121.2, 109.8, 109.4, 61.3, 59.3, 52.0, 51.2, 50.0, 33.5, 33.3, 29.6, 24.9, 24.7; C29H31N7O HRMS (ESI) m/z calculated for [M + H] + = 494.26629, found 494.26652.
1‐{1‐[(1‐benzyl‐1 H‐1,2,3,4‐tetrazol‐5‐yl)[2‐(trifluoromethoxy)phenyl]methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (9u). White solid. Yield: 79%. 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1 H), 7.89–7.86 (m, 1 H), 7.40–7.31 (m, 5 H), 7.28–7.26 (m, 1 H), 7.22–7.19 (m, 2 H), 7.12–7.01 (m, 4 H), 5.78 (d, J = 15.3 Hz, 1 H), 5.56 (d, J = 15.3 Hz, 1 H), 5.36 (s, 1 H), 4.19–4.12 (m, 1 H), 2.88–2.85 (m, 1 H), 2.76–2.75 (m, 1 H), 2.46–2.29 (m, 2 H), 2.19–2.12 (m, 2 H), 1.73–1.67 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 155.3, 154.0, 147.6, 133.3, 131.9, 130.2, 129.3, 129.2, 129.1, 128.2, 127.6, 126.9, 126.2, 121.8, 121.4, 121.2, 119.8, 109.9, 109.5, 55.8, 51.5, 50.6, 50.5, 49.3, 29.4, 29.3; C28H26F3N7O2 HRMS (ESI) m/z calculated for [M + H]+ = 550.21728, found 550.21728.
1‐{1‐[(S)‐{1‐[(1 S)‐1‐phenylethyl]‐1 H‐1,2,3,4‐tetrazol‐5‐yl}(thiophen‐3‐yl)methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (12a). White solid. Yield: 35%. 1H NMR (400 MHz, CDCl3) δ 10.53 (s, 1 H), 7.46–7.34 (m, 4 H), 7.34–7.25 (m, 3 H), 7.22 (dd, J = 5.0, 1.3 Hz, 1 H), 7.17–7.08 (m, 1 H), 7.07–7.01 (m, 3 H), 5.73 (q, J = 7.0 Hz, 1 H), 5.15 (s, 1 H), 4.26–4.14 (m, 1 H), 3.06–2.73 (m, 2 H), 5.53–2.32 (m, 2 H), 2.28–2.07 (m, 2 H), 2.00 (d, J = 7.0 Hz, 3 H), 1.74 (d, J = 11.6 Hz, 1 H), 1.70–1.52 (d, J = 11.6 Hz, 1 H). 13C NMR (101 MHz, CDCl3) δ 155.30, 139.51, 129.20, 128.99, 128.70, 128.49, 128.17, 126.40, 126.31, 121.29, 120.97, 109.91, 109.61, 59.39, 58.55, 50.84, 50.52, 48.91, 29.39, 28.98, 22.69; C26H27N7OS HRMS (ESI) m/z calculated for [M + H]+ = 486.19978, found 486.20677.
1‐{1‐[(R)‐{1‐[(1 S)‐1‐phenylethyl]‐1 H‐1,2,3,4‐tetrazol‐5‐yl}(thiophen‐3‐yl)methyl]piperidin‐4‐yl}‐2,3‐dihydro‐1 H‐1,3‐benzodiazol‐2‐one (12b). White solid. Yield: 31%. 1H NMR (400 MHz, CDCl3) δ 10.57 (s, 1 H), 7.39–7.24 (m, 6 H), 7.22 (dd, J = 2.9, 1.3 Hz, 1 H), 7.20–7.10 (m, 3 H), 7.10–7.00 (m, 2 H), 6.15 (q, J = 6.8 Hz, 1 H), 5.31–5.01 (m, 1 H), 4.32–4.13 (m, 1 H), 3.25–3.01 (m, 1 H), 2.95–2.74 (m, 1 H), 2.63–2.35 (m, 3 H), 2.31–2.16 (m, 1 H), 2.02 (d, J = 7.0 Hz, 3 H), 1.80 (dd, J = 23.7, 10.9 Hz, 2 H). 13C NMR (101 MHz, CDCl3) δ 155.32, 139.26, 129.15, 129.11, 128.69, 128.26, 128.22, 128.18, 126.42, 126.14, 121.42, 121.07, 110.00, 109.24, 59.81, 58.91, 51.48, 50.48, 49.32, 29.14, 22.62; C26H27N7OS HRMS (ESI) m/z calculated for [M + H]+ = 486.19978, found 486.20677. Compound 12b was crystalized by solvent evaporation in ethyl acetate and chloroform. The X-ray crystal structure of 12b was deposited in Cambridge Crystallographic Data Centre and the deposition number is CCDC 1573497.
Plaque assay
The plaque reduction assay was performed as previously reported38, except MDCK cells expressing ST6Gal I were used instead of regular MDCK cells39. Briefly, the confluent monolayers of ST6Gal MDCK cells were incubated with ~100 pfu virus samples in DMEM with 0.5% BSA for 1 h at 4 °C, then 37 °C for 1 h. The inoculums were removed, and the cells were washed with phosphate buffered saline (PBS). The cells were then overlaid with DMEM containing 1.2% Avicel microcrystalline cellulose (FMC BioPolymer, Philadelphia, PA) and NAT (2.0 µg/mL). To examine the effect of the compounds on plaque formation, the overlay media was supplemented with compounds at testing concentrations. At two days after infection, the monolayers were fixed and stained with crystal violet dye solution (0.2% crystal violet, 20% methanol). Influenza A virus A/WSN/33 (H1N1) was obtained from Dr. Robert Lamb at the Northwestern University. The influenza viruses A/Texas/04/2009 (H1N1), B/Wisconsin/1/2010, and B/Brisbane/60/2008 were obtained from Dr. James Noah at the Southern Research Institute. Influenza A and B viruses A/Switzerland/9715293/2013 × −247 (H3N2), FR-1366, A/Washington/29/2009 (H1N1), FR-460, A/California/07/2009 (H1N1), FR-201, A/Washington/29/2009 (H1N1), FR-460, B/Memphis/20/1996, FR-486, B/Utah/9/2014, FR-1372, and B/Phuket/3073/2013, FR-1364, were obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA. The influenza viruses A/Denmark/524/2009 (H1N1) and A/Denmark/528/2009 (H1N1) was obtained from Dr. Elena Govorkova at St. Jude Children’s Research Hospital. All influenza strains used in this study are biosafety level 2 (BSL-2) pathogens and all experiments using influenza viruses were performed in BSL-2 certified laboratory.
Cytotoxicity assay
Evaluation of the cytotoxicity of compounds was carried out using the neutral red uptake assay40. Briefly, 80,000 cells/mL of MDCK or A549 cells in DMEM medium supplemented with 10% FBS and 100 U/mL Penicillin-Streptomycin were dispensed into 96-well cell culture plates at 100 µL/well. Twenty-four hours later, the growth medium was removed and washed with 100 µL PBS buffer; then for the cytotoxicity assay, 200 µL fresh DMEM (no FBS) medium containing serial diluted compounds was added to each well. After incubating for 48 h at 37 °C with 5% CO2 in a CO2 incubator, the medium was removed and replaced with 100 µL DMEM medium containing 40 µg/mL neutral red for four hours at 37 °C. The amount of neutral red uptake was determined at absorbance 540 nm using a Multiskan FC Microplate Photometer (Fisher Scientific). The CC50 values were calculated from best-fit dose response curves with variable slope in GraphPad Prism version 5.
ELISA assay
To test the inhibitory activity of compound on PAC–PB1N interaction, ELISA was performed22. Briefly, microtiter plates were coated with 400 ng of His-tagged PA239-716 (PAC) for 3 h at 37 °C, followed by blocking with 2% (wt/vol) BSA in phosphate buffer saline (PBS) for 1 h. After washing with PBS containing 0.3% Tween 20, plates were incubated with 200 ng of GST-tagged PB11-25 (PB1N) protein and compounds overnight at room temperature. Then the PAC–PB1N interaction was detected using a horseradish peroxidase (HRP)-conjugated anti-GST monoclonal antibody and its chromogenic substrate TMB. The absorbance at 450 nm was read on a plate reader. The IC50 values were calculated from best-fit dose response curves with variable slope in GraphPad Prism version 5.
Time-of-Addition Experiment
A time-of-addition experiment was performed according to the procedure described earlier29,41,42. Briefly, MDCK cells were seeded at 6 cm2 dishes at a 2 × 105 cells/dish cell density. After incubating for 24 h to allow cells to attach, the cells were infected with A/WSN/33 (H1N1) virus at a MOI of 0.01 at −2 h time point. Antiviral compounds, such as oseltamivir carboxylate (1 µM) or 12a (10 µM) was added at different time points before, during, or after viral infection as illustrated in Fig. 4. Viruses were harvested from the cell culture supernatant at 12 h post infection. The virus titers were quantified by plaque assay.
RNA Extraction and Real-Time PCR
Total RNA was extracted from influenza A/WSN/33 (H1N1) infected cells using Trizol reagents (Thermo Fisher Scientific). After removing genomic DNA by RQ1 RNase-Free DNase (Promega), the first strand of cDNA was synthesized using 1.2 μg of total RNA and AMV Reverse Transcriptase (Promega). vRNA specific primer (5′-AGCAAAAGCAGG-3′), cRNA specific primer (5′-AGTAGAAACAAGG-3′) or oligo (dT)18 was used for detecting influenza vRNA, cRNA or mRNA, respectively. Real-time PCR was performed on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) using FastStart Universal SYBR Green Master (Rox) (Roche, Basel, Switzerland) and following influenza NP-specific primers: NP-F: 5′-AGGGTCAGTTGCTCACAAGTCC-3′; NP-R: 5′-TTTGAAGCAGTCTGAAAGGGTCTA-3′. GAPDH was also amplified to serve as a control using human GAPDH-specific primers (GAPDH-F: 5′-ACACCCACTCCTCCACCTTTG-3′ and GAPDH-R: 5′-CACCACCCTGTTGCTGTAGCC-3′). The amplification conditions were: 95 °C for 10 min; 40 cycles of 15 s at 95 °C and 60 s at 60 °C. Melting curve analysis was performed to verify the specificity of each amplification. All experiments were repeated three times independently.
Serial viral passage experiments
Serial drug passage experiments were performed accordingly to previously published protocol29,30,42. Briefly, MDCK cells were infected with the A/WSN/33 (H1N1) virus at MOI 0.001 for 1 h. Then the inoculum was removed and MDCK cells were incubated with 1 µM compound 12a in the first passage and the concentration of 12a was gradually increased 2-fold in passages 2–7 and kept constant at 64 µM in passages 7–10. In each passage, the viruses were harvested when a significant cytopathic effect was observed, which usually takes 2-3 days after virus infection. The titers of harvested viruses were determined by plaque assay. The drug sensitivity after passages 3, 6, and 10 was determined via plaque assay as described previously43. Oseltamivir carboxylate was included as a control and similar fold of drug selection pressure was applied. The drug sensitivity of oseltamivir at passages 3, 6, and 10 was determined via plaque assay.
Electronic supplementary material
Acknowledgements
This research was supported by start-up funding from the University of Arizona and NIH grant AI119187 to J.W. R.M. was supported by the NIH training grant T32 GM008804. We thank Dr. David Bishop for proof-reading and editing the manuscript.
Author Contributions
Jiantao Zhang, Yanmei Hu, and Jun Wang designed and organized this research. Jun Wang performed the docking experiments. Christopher Foley, Yuanxiang Wang, Shuting Xu, and Yongtao Zhang performed synthesis, purification, and compound characterizations. Jiantao Zhang performed the ELISA, minigenome assay, and RT- qPCR experiments. Yanmei Hu, Rami Musharrafieh, and Chunlong Ma performed the antiviral assay, cytotoxicity assay, time-of-addition experiment, and the passage experiments. Christopher Hulme contributed the in-house small molecule library for the initial docking experiments. Jiantao Zhang, Yanmei Hu, and Jun Wang analyzed the obtained data, interpreted the results, and wrote the manuscript with the input from other co-authors. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Jiantao Zhang and Yanmei Hu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22875-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Palese, P. & Shaw, M. L. Orthomyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, eds Fields Virology. 5th ed. Philadelphia: Lippincott Williams & Wilkins. pp 1647–1690 (2007).
- 2.Leading causes of death. https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm. Accessed on Feburary 6th, 2018.
- 3.Zhang WQ, Webster RG. Can we beat influenza? Science. 2017;357:111–111. doi: 10.1126/science.aan7961. [DOI] [PubMed] [Google Scholar]
- 4.Dawood FS, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhu H, Lam TT, Smith DK, Guan Y. Emergence and development of H7N9 influenza viruses in China. Curr Opin Virol. 2016;16:106–113. doi: 10.1016/j.coviro.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Kile JC, et al. Update: Increase in Human Infections with Novel Asian Lineage Avian Influenza A(H7N9) Viruses During the Fifth Epidemic - China, October 1, 2016-August 7, 2017. MMWR Morb Mortal Wkly Rep. 2017;66:928–932. doi: 10.15585/mmwr.mm6635a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson ML, et al. Influenza Vaccine Effectiveness in the United States during the 2015–2016 Season. N Engl J Med. 2017;377:534–543. doi: 10.1056/NEJMoa1700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koszalka P, Tilmanis D, Hurt AC. Influenza antivirals currently in late-phase clinical trial. Influenza Other Respir Viruses. 2017;11:240–246. doi: 10.1111/irv.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Li F, Ma C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers. 2015;104:291–309. doi: 10.1002/bip.22623. [DOI] [PubMed] [Google Scholar]
- 10.Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palu G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci. 2014;71:3659–3683. doi: 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webster RG, Govorkova EA. Continuing challenges in influenza. Ann N Y Acad Sci. 2014;1323:115–139. doi: 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Hu Y, Wang Y, Ma C, Wang J. Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki-Miyaura Cross-Coupling Reaction. J Med Chem. 2017;60:1580–1590. doi: 10.1021/acs.jmedchem.6b01852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Ma C, Hu Y, Wang Y, Wang J. Discovery of Potent Antivirals against Amantadine-Resistant Influenza A Viruses by Targeting the M2-S31N Proton Channel. ACS Infect Dis. 2016;2:726–733. doi: 10.1021/acsinfecdis.6b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurt AC. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol. 2014;8:22–29. doi: 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Matsuzaki Y, et al. A two-year survey of the oseltamivir-resistant influenza A(H1N1) virus in Yamagata, Japan and the clinical effectiveness of oseltamivir and zanamivir. Virol J. 2010;7:53. doi: 10.1186/1743-422X-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dömling A, Wang W, Wang K. Chemistry and Biology Of Multicomponent Reactions. Chem Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Kaim L, Grimaud L, Le Goff XF, Menes-Arzate M, Miranda LD. Straightforward four-component access to spiroindolines. Chem Commun. 2011;47:8145–8147. doi: 10.1039/c1cc12236c. [DOI] [PubMed] [Google Scholar]
- 18.Brauch S, van Berkel SS, Westermann B. Higher-order multicomponent reactions: beyond four reactants. Chem Soc Rev. 2013;42:4948–4962. doi: 10.1039/c3cs35505e. [DOI] [PubMed] [Google Scholar]
- 19.Pflug A, Lukarska M, Resa-Infante P, Reich S, Cusack S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017;234:103–117. doi: 10.1016/j.virusres.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Stevaert A, Naesens L. The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design. Med Res Rev. 2016;36:1127–1173. doi: 10.1002/med.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massari S, Goracci L, Desantis J, Tabarrini O. Polymerase Acidic Protein-Basic Protein 1 (PA-PB1) Protein-Protein Interaction as a Target for Next-Generation Anti-influenza Therapeutics. J Med Chem. 2016;59:7699–7718. doi: 10.1021/acs.jmedchem.5b01474. [DOI] [PubMed] [Google Scholar]
- 22.Yuan S, et al. Identification of a small-molecule inhibitor of influenza virus via disrupting the subunits interaction of the viral polymerase. Antiviral Res. 2016;125:34–42. doi: 10.1016/j.antiviral.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Massari S, et al. A Broad Anti-influenza Hybrid Small Molecule That Potently Disrupts the Interaction of Polymerase Acidic Protein-Basic Protein 1 (PA-PB1) Subunits. J Med Chem. 2015;58:3830–3842. doi: 10.1021/acs.jmedchem.5b00012. [DOI] [PubMed] [Google Scholar]
- 24.Muratore G, et al. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc Natl Acad Sci USA. 2012;109:6247–6252. doi: 10.1073/pnas.1119817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hulme, C., Ayaz, M., Martinez-Ariza, G., Medda, F. & Shaw, A. In Small Molecule Medicinal Chemistry 145–187 (John Wiley & Sons, Inc, 2015).
- 26.Reich S, et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature. 2014;516:361–366. doi: 10.1038/nature14009. [DOI] [PubMed] [Google Scholar]
- 27.He X, et al. Crystal structure of the polymerase PAC-PB1N complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 28.White KM, et al. A Potent Anti-influenza Compound Blocks Fusion through Stabilization of the Prefusion Conformation of the Hemagglutinin Protein. ACS Infect Dis. 2015;1:98–109. doi: 10.1021/id500022h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma C, Li F, Musharrafieh RG, Wang J. Discovery of cyclosporine A and its analogs as broad-spectrum anti-influenza drugs with a high in vitro genetic barrier of drug resistance. Antiviral Res. 2016;133:62–72. doi: 10.1016/j.antiviral.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma C, Zhang J, Wang J. Pharmacological Characterization of the Spectrum of Antiviral Activity and Genetic Barrier to Drug Resistance of M2-S31N Channel Blockers. Mol Pharmacol. 2016;90:188–198. doi: 10.1124/mol.116.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y, et al. An M2-V27A channel blocker demonstrates potent in vitro and in vivo antiviral activities against amantadine-sensitive and -resistant influenza A viruses. Antiviral Res. 2017;140:45–54. doi: 10.1016/j.antiviral.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehrhardt C, et al. The NF-kappaB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol. 2013;15:1198–1211. doi: 10.1111/cmi.12108. [DOI] [PubMed] [Google Scholar]
- 33.Shih SR, et al. BPR2-D2 targeting viral ribonucleoprotein complex-associated function inhibits oseltamivir-resistant influenza viruses. J Antimicrob Chemother. 2010;65:63–71. doi: 10.1093/jac/dkp393. [DOI] [PubMed] [Google Scholar]
- 34.Khanna I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov Today. 2012;17:1088–1102. doi: 10.1016/j.drudis.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Hulme C, Lee YS. Emerging approaches for the syntheses of bicyclic imidazo[1,2-x]-heterocycles. Mol Divers. 2008;12:1–15. doi: 10.1007/s11030-008-9072-1. [DOI] [PubMed] [Google Scholar]
- 36.Hulme, C. Applications of Multicomponent Reactions in Drug Discovery - Lead Generation to Process Development. Multicomponent Reactions, 311–341, 10.1002/3527605118.ch11 (2005).
- 37.Zabrocki J, Smith GD, Dunbar JB, Iijima H, Marshall GR. Conformational Mimicry .1. 1,5-Disubstituted Tetrazole Ring as a Surrogate for the Cis Amide Bond. J Am Chem Soc. 1988;110:5875–5880. doi: 10.1021/ja00225a045. [DOI] [Google Scholar]
- 38.Wang J, et al. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci USA. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatakeyama S, et al. Enhanced Expression of an α2,6-Linked Sialic Acid on MDCK Cells Improves Isolation of Human Influenza Viruses and Evaluation of Their Sensitivity to a Neuraminidase Inhibitor. J Clin Microbiol. 2005;43:4139–4146. doi: 10.1128/JCM.43.8.4139-4146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 41.Hu, Y. et al. Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral. Int J Mol Sci18, 10.3390/ijms18091929 (2017). [DOI] [PMC free article] [PubMed]
- 42.Hu Y, et al. Discovery of dapivirine, a nonnucleoside HIV-1 reverse transcriptase inhibitor, as a broad-spectrum antiviral against both influenza A and B viruses. Antiviral Res. 2017;145:103–113. doi: 10.1016/j.antiviral.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Wang Y, Li F, Ma C, Wang J. Design and expeditious synthesis of organosilanes as potent antivirals targeting multidrug-resistant influenza A viruses. Eur J Med Chem. 2017;135:70–76. doi: 10.1016/j.ejmech.2017.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.