

Abstract
Crowdsourcing has been used to address computational challenges in systems biology and assess translation of findings across species. Sub-challenge 2 of the sbv IMPROVER Systems Toxicology Challenge was designed to determine whether a common set of genes can be used to identify exposure to cigarette smoke in both human and mouse. Participating teams used a training set of human and mouse blood gene expression data to derive parsimonious models (up to 40 genes) that classify subjects into exposure groups: smokers, former smokers, and never-smokers. Teams were ranked based on two classification performance metrics evaluated on a blinded test dataset. Prediction of current exposure to cigarette smoke in human and mouse by a common prediction model was achieved by the top ranked team (Team 219) with 89% balanced accuracy (BAC), while past exposure was predicted with only 57% BAC. The prediction model of the top ranked team was a random forest classifier trained on sets of genes that appeared best for each species separately with no overlap between species. By contrast, Team 264, ranked second (tied with Team 250), selected genes that were simultaneously predictive in both species and achieved 80% and 59% BAC when predicting current and past exposure, respectively. These performance values were lower than the 96.5% and 61% BAC estimates for current and past exposure, respectively, obtained by Team 264 (top ranked in sub-challenge 1) when using only human data. Unlike past exposure, current exposure to cigarette smoke can be accurately assessed in both human and mouse with a common prediction model based on blood mRNAs. However, requiring a common gene signature to be predictive in both species resulted in a substantial decrease in balanced accuracy for prediction of current exposure to cigarette smoke (from 96.5% to 80%), suggesting species-specific responses exist.
Keywords: Systems toxicology, computational challenge, species-translatable gene signature, smoking biomarker, predictive modeling
Introduction
The assumption at the basis of many omics experiments in animal models is that biological insight is translatable to humans. An important requirement to ensure translatability of findings is the use of robust analytical approaches in the analysis of omics data. Computational challenges in the area of omics data analysis have been addressed using crowdsourcing[1], as a way to both: i) explore existing approaches and identify those that work best and ii) test the robustness of omics-based findings by comparing signatures derived from a training dataset to an analyst-blinded independent test dataset. An example of a crowdsourcing-based initiative to test the rodent to human translatability assumption is the sbv IMPROVER[2] Species Translation Challenge (2013), which aimed at determining to what extent biological processes perturbed by various stimuli in human cells can be inferred from omics data collected in rodents[3]. Similarly, sub-challenge 2 of the sbv IMPROVER Systems Toxicology Challenge (2106), which is the subject of this article, was designed to test whether exposure to cigarette smoke can be detected/predicted in both mouse and human by a parsimonious common set of blood mRNAs. The ability to translate the impact of such toxicants from animals to humans is key in systems toxicology[4-6], which is enabled by the ability to profile tens of thousands of molecules (e.g. mRNAs) in biological samples using omics technologies such as gene expression microarrays [7]. While sub-challenge 1 of the Systems Toxicology Challenge showed that microarray gene expression data generated from human blood samples allowed confident discrimination of current smokers from former and never-smokers, it was not known whether the same genes were implicated in both rodent and human. The challenge organizers provided a training gene expression dataset derived from mouse and human blood samples for current, former, and never cigarette smoke exposed individuals and ranked participating teams based on the performance of their models in predicting the exposure status on a new set of samples (test dataset). The aim of this article is to describe the approaches and results of the top three teams that participated in this sub-challenge, focusing on key aspects of the methodologies that might explain the similarity and differences in the cigarette smoke-exposure classification models that they developed.
Methods
Challenge organization
The human training dataset that was made available to participating teams was based on the Queen Ann Street Medical Center (QASMC) clinical case–control study conducted at the Heart and Lung Centre (London, UK) (see ClinicalTrials.gov, ID: NCT01780298) and included gene expression data from 109 smokers, 57 former smokers, and 58 never-smokers. The test dataset was obtained by expression profiling blood samples from a banked repository (BioServe Biotechnologies Ltd., Beltsville, MD, USA) and included samples from 27 smokers, 26 former smokers, and 28 never-smokers. After total RNA extraction, hybridization was performed on Affymetrix GeneChip® Human Genome U133 Plus 2.0 arrays.
The mouse training dataset was based on a 7-month cigarette smoke inhalation study conducted with C57BL/6 mice and included three groups of animals: exposed to smoke for 7 months (equivalent to a human current smoker), exposed to smoke for 2 months followed by exposure to air (equivalent to a human former smoker) and mice continuously exposed to air (equivalent to human never-smoker) [8]. The mouse test dataset was based on an 8-months inhalation study conducted with Apoe−/− mice and involved similar groups as the training dataset. After total RNA extraction, hybridization was performed on GeneChip® Mouse Genome 430 2.0 arrays.
Raw gene expression data (CEL files) were background corrected, normalized, and summarized into one expression value per Entrez gene ID using frozen robust microarray analysis (fRMA)[9]. Mouse genes were mapped to human genes using the NCBI/HCOP search tool [10]. See the technical document from the challenge organizers [11] as well as Belcastro et al. in this issue for more details.
Participating teams were requested to develop a prediction rule that classifies human and rodent expression profiles into exposure groups using training data and then apply it on the test data. For each test sample, the teams provided a confidence value (probability ranging from 0 to 1) that a sample was taken from a smoker (pS) as opposed to a non-current smoker (former smoker or never-smoker (1−ps). For samples assigned to the non-current smoker group (ps ≤0.5) a second classification was requested to determine whether the sample came from a former smoker (pfs) or from a never-smoker (1−pfs). Of note, teams that wrongly classified non-current smokers (former smokers or never-smokers) as smokers were penalized twice by imputing the missing confidence values for those subjects with the worst-case scenario confidence values (e.g., pfs for a former smoker misclassified as smoker was set to 0.0 instead of the ideal 1.0). To enable a direct comparison of the prediction performance between the two classification tasks (smoker vs non-current smoker and former smoker vs never-smoker), predictions for all test samples obtained with former smoker vs never-smoker prediction models were requested post-challenge for top three teams discussed in this article.
The submissions from the different teams were then ranked by computing the area under the precision recall curve (AUPR)[12] and the Matthew’s correlation coefficient (MCC)[13] for both classification tasks (smoker vs non-current smoker and former smoker vs never-smoker) on the test dataset. The sum of ranks of each team based on four statistics (two metrics × two classification tasks) was used to rank the teams. To determine whether or not the performance metric (AUPR or MCC) of a given team was better than expected by chance, the empirical distribution of AUPR and MCC statistics were determined as described in Belcastro et al. in this issue. The top three teams were invited to contribute to this manuscript.
Approach of first-ranked team (Team 219)
The team ranked first in this challenge (OSS, ATB, ISB, Team ID 219) relied on a classifier development pipeline based on the selection of a set of features/genes for each classification task, followed by fitting a classification model. All modeling decisions were based solely on the prediction performance assessed on the training data via 10-fold cross-validation. Performance metrics that were considered were accuracy and area under the ROC curve. LASSO regression[14] and random forest[15] algorithms implemented in the corresponding R packages glmnet[16] and randomForest[17] were tested. Both these methods can be used for feature selection and classification, and therefore can be seen as embedded feature selection methods. LASSO is a sparse linear estimator that selects only the most relevant features among all features used as input via a regularization term in the optimization function. Random forest classifiers rely on multiple binary classification trees and can be used to evaluate the importance of features by determining their frequency of selection among 1,000 decision trees built with different bootstrap samples of the training set starting with a random set of features among all available.
After testing LASSO regression, random forests, and random forests with preselected genes by LASSO regression, the last option was determined to be best (87%, 81%, and 91% accuracy, respectively) for the smoker vs non-current smoker classification. The same approach was then used in the former smoker vs never-smoker classification. The second modeling decision was whether to select features using both human and mouse data at the same time or select features separately for human and mouse datasets and then use the union of these features in the classifier. Selecting predictors for each species separately resulted in 95% accuracy compared with 91% accuracy obtained when feature selection was performed on combined sets at the same time.
Approach of (tied) second-ranked team (Team 250)
One of the teams ranked second (in a tie) in this challenge (RK, SKD Team ID 250) used a pipeline that included feature selection followed by classification by multiple algorithms. For feature selection, the WEKA (version 3.6.13) platform[18] was used to apply the BestFirst algorithm in the forward direction to build a combination of features predictive of the outcome. The BestFirst algorithm searches the space of attribute subsets by forward inclusion of the features that maximize the classification performance followed by backward deletion of the feature that decreases the performance by the smallest amount. The same algorithm was applied for both classification tasks selecting the maximum allowed number of predictor (40) genes that showed best correlation with the outcome and minimum inter-correlation with other predictor genes.
Several machine-learning techniques including support vector machines (SVM), artificial neural networks (ANN), K-nearest neighbor (KNN), Naïve Bayes classifier, and random forests were considered (see[19] for a review). The different approaches were compared in terms of their accuracy and mean absolute error estimated by 10-fold cross-validation on the training dataset. The random forest classifiers were selected as the best alternative for both classification tasks. Random forests provide a probability that a given sample belongs to the positive class (e.g. smoker class in the smoker vs non-current smoker classification task) based on the votes of individual trees in the random forest.
Approach of (tied) second-ranked team (Team 264)
The approach of the other second ranked team (ALT and RR, Team ID 264) was similar to the one used in previous sbv IMPROVER Challenges[20,21] but was modified to use a common gene signature for mouse and human. The approach was applied in the same manner to both classification tasks (i.e., smoker vs non-current smoker, and former smoker vs never-smoker and involved the following steps:
Fit a linear model to gene expression data including the exposure indicator variable (e.g., smoker vs non-current smoker) and the species (mouse vs human) as covariate. The goal was to find genes that changed with the exposure status regardless of the species and hence relevant for both human and mouse simultaneously.
Rank genes by moderated t-test[22] p-values of the exposure group coefficient and select the first NF candidate genes as those with p <0.05 and fold change in expression between groups greater than a given fold change threshold (FCT). If there were no such NF genes, use (or complement with) top genes ranked solely by p-values.
Implement a 3-fold cross-validation repeated 10 times to estimate the cross-validated performance (PCV) of a linear discriminant analysis (LDA) model using d genes as predictors, d=1,2, …, NF. The optimal value of d will be the value that maximizes the average performance over the 3×10 cross-validation test sets.
In the steps above, the prediction performance was the average over five different metrics used in previous sbv IMPROVER challenges: belief confusion metric, correct class enrichment metric, area under the ROC curve, AUPR, and balanced accuracy described in detail elsewhere [23]. The optimal values for FCT and NF (leading to highest PCV) were chosen by trial and error over a set of pre-specified FCT values (1.25, 1.5, 2.0) and NF values (5, 6, …, 25). Let p denote the optimal number of predictor genes determined using the optimal value of FCT and NF as described in steps 1 and 2 above. A p-genes final model was fitted using the entire training dataset and then applied to the test data to compute the posterior probability that a given sample belongs to a class of interest (e.g., to the smoker group for smoker vs non-current smoker, or to the former smoker group for former smoker vs never-smoker). All analysis was performed using the R statistical language[24] (version 3.2.2) as well as specialized packages limma (version 3.24.15) (for linear model fitting), and MASS (version 7.3-45) (for LDA model fitting).
Results
Prediction performance in the sbv IMPROVER Systems Toxicology sub-challenge 2
Of the 15 international teams that provided submissions in sub-challenge 2, only six met all the criteria to be included in the final ranking. Participating teams submitted confidence levels (ranging from 0 to 1) for classifying human samples from 27 smokers, 26 former smokers, and 28 never-smokers, and mouse samples from 12, 8, and 13 animals from corresponding exposure groups. Samples were classified first as smoker vs non-current smoker (task 1) and then as former smoker vs never-smoker (task 2). After ranking teams by AUPR and MCC for each classification task, the sum of ranks was computed for all teams (see Table 1 and Figure 1). Based on principal components analysis using the genes selected as predictors by each of the top three teams, the main source of variability in the combined dataset was determined to be the species/microarray platform, captured by the first principal component, as depicted in Figure S1. The variability in gene expression due to exposure status was captured by the second principal component.
Table 1. Performance metrics for participating teams in sub-challenge 2 of the sbv IMPROVER Systems Toxicology Challenge.
Teams were ranked by area under the precision recall curve (AUPR) and Matthew’s correlation coefficient (MCC) in each of the two classification tasks. The ranks of teams (the smaller the better) on each individual metric and task were summed to determine the final rank. The AUPR and MCC values shown in parentheses for the former smoker vs never-smoker classification were determined from all actual confidence values, as opposed to the values outside the parentheses (used in the official ranking) in which missing predictions for some non-current smokers (wrongly classified as smokers) were imputed with the worst case scenario value (see methods section). This explains why even apparently worse than chance performance values (e.g., AUPR <0.5 or MCC <0.0 for Teams 250 and 264) appear as significantly better than chance (p <0.05) based on a simulation of the null distribution of these statistics.
| Smoker vs Non-Current Smoker | Former Smoker vs Never-smoker | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Team | Final rank | Sum Ranks | AUPR | MCC | p AUPR | p MCC | AUPR | MCC | p AUPR | p MCC |
| 219 | 1 | 4 | 0.93 | 0.78 | 0.000 | 0.000 | 0.45(0.537) | 0.04(0.13) | 0.000 | 0.000 |
| 250 | 2 | 10 | 0.793 | 0.65 | 0.000 | 0.000 | 0.36(0.57) | −0.17(0.05) | 0.000 | 0.001 |
| 264 | 2 | 10 | 0.789 | 0.59 | 0.000 | 0.000 | 0.41(0.541) | −0.01(0.20) | 0.000 | 0.000 |
| 221 | 4 | 19 | 0.55 | 0.54 | 0.001 | 0.000 | 0.34 | −0.39 | 0.000 | 0.141 |
| 225 | 5 | 19.5 | 0.75 | 0.31 | 0.000 | 0.108 | 0.30 | −0.45 | 0.000 | 0.305 |
| 247 | 6 | 21.5 | 0.63 | 0.20 | 0.000 | 0.218 | 0.29 | −0.52 | 0.016 | 0.567 |
Figure 1. Classification performance of the six teams with valid submissions in the sbv IMPROVER Systems Toxicology sub-challenge 2.
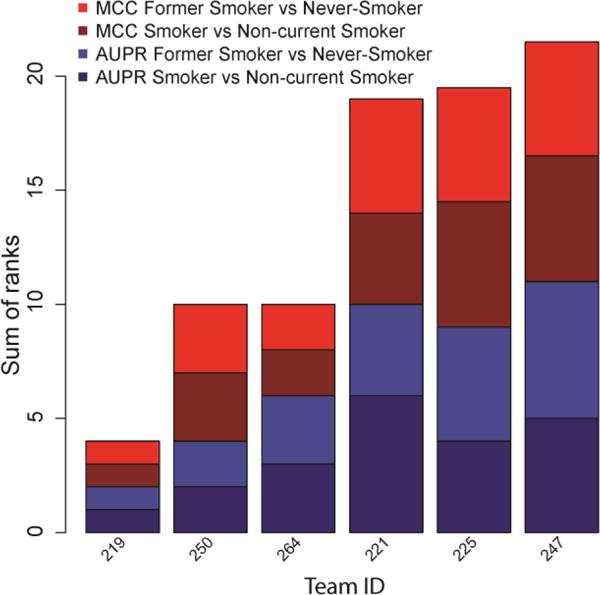
Data shown represent the ranks (1–6, the smaller the better) for two prediction performance metrics (area under the precision-recall curve, AUPR, and Mathew’s correlation coefficient, MCC) in two classification tasks (smoker vs non-current smoker and former smoker vs never-smoker). The final team ranking is based on the sum of the four individual ranks.
The classification of subjects (mouse and human) into smoker vs non-current smoker (or equivalent for mouse) was performed with high accuracy. The top team achieved an AUPR of 0.93, MCC of 0.78 (Table 1), and sensitivity of 85% at 93% specificity (Table 2). Notably, the prediction performance was, in general, higher for the mouse samples than for the human samples for all teams (mouse/human MCC and AUPR: 0.99/0.93 and 0.87/0.78 (Team 219); 0.75/0.79 and 0.96/0.65 (Team 250); 0.81/0.79 and 0.96/0.6 (Team 264)).
Table 2. Additional performance metrics for the top three teams in sub-challenge 2 of the sbv IMPROVER Systems Toxicology Challenge.
The metrics were computed using a cut-off of 0.5 on the confidence values that a sample was taken from a current smoker (left) or former smoker (right). For the former smoker vs never-smoker comparison (right) performance metrics were obtained from all samples and hence correspond to values in parentheses in Table 1.
| Smoker vs Non-Current Smoker | Former Smoker vs Never-smoker | |||||
|---|---|---|---|---|---|---|
| Team | Sensitivity (%) | Specificity (%) | Balanced accuracy (%) | Sensitivity (%) | Specificity (%) | Balanced accuracy (%) |
| 219 | 85 | 93 | 89 | 74 | 39 | 56 |
| 250 | 90 | 79 | 84 | 59 | 46 | 53 |
| 264 | 74 | 85 | 80 | 85 | 32 | 59 |
Additional information about the quality of the classifications not directly captured by the AUPR and MCC statistics is presented in Figure 2 as the distribution of confidence values for the predictions of the top three teams. The first-ranked team (Team 219) classified both actual smokers and non-current smokers correctly with higher confidence (paired Wilcoxon test p <0.00001) than Team 250 but with lower confidence (p <0.00001) than Team 264 (Figure 2A). This is because neither the MCC nor AUPR statistics that were used to rank the teams, reward this qualitative aspect of predictions, which was considered important in previous sbv IMPROVER Challenges.
Figure 2. Classification confidence values for the top three teams in the sbv IMPROVER Systems Toxicology sub-challenge 2.
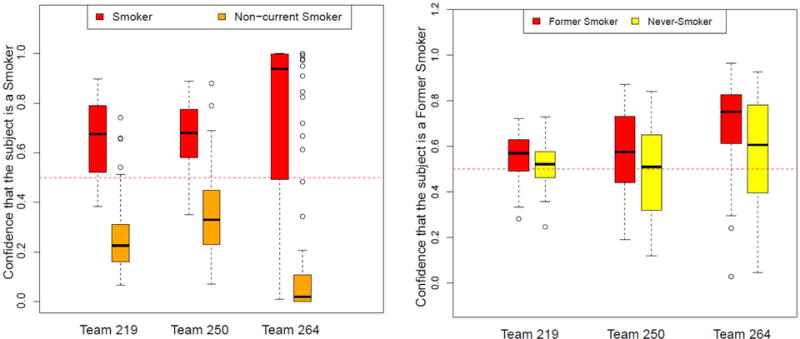
Data shown represent the confidence (0.0–1.0) that blood gene expression profiles belonged to a smoker (left) or former smoker (right). Distribution boxplots are shown by actual smoking status. Thick horizontal lines in the boxes represent median values, while the boxes encompass the first and third quartile.
The classification of subjects into former smoker vs never-smoker was more difficult. The top team for this classification task, Team 264, achieved an AUPR of 0.54, MCC of 0.20 (Table 1), and sensitivity of 85% at 32% specificity (74% sensitivity at 40% specificity for Team 219, which was ranked best overall, Table 2). The interquartile range of confidence values that a given subject was a former smoker overlapped between the actual former smokers and never-smokers for all teams (Figure 2, right panel).
Analysis of the predictor genes identified by top three ranked teams
For the classification of subjects into smoker vs non-current smoker, the number of predictor genes used in the models of Teams 264, 219, and 250 were 25, 34, and 40, respectively. AHRR, COX6B2, DSC2, LRRN3, P2RY6, and SASH1 were among the genes chosen as predictors by two of the three teams. A post-challenge analysis revealed that of the 89 genes selected by at least one team in this comparison, F2R was the only one that reached a false discovery rate adjusted p-value (q-value) of <0.1 significance based on moderated t-tests[22] in both the training and test datasets of both human and mouse. Another eight genes, AHRR, KLRG1, PGRMC1, TBX21, WDFY1, GUCY1B3, PF4, and SYTL4, met this significance criterion in three of the four datasets. All these nine genes were down-regulated in the blood samples from current smokers compared with non-current smokers.
Gene Ontology [25] functional profiling of the set of 89 predictor genes used by at least one of the top three teams revealed 11 biological processes (such as response to external biotic stimulus, and thrombin receptor signaling pathway) and three molecular functions (enzyme inhibitor activity, peptide receptor activity, and receptor activity) associated with current smoking (q-values <0.05, odds ratios 2.3–93.0). The neuroactive ligand-receptor interaction, Parkinson’s disease, and oxidative phosphorylation pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG)[26] were also associated with current smoking (q-values <0.1, odds ratios 3.9–4.7).
For the comparison former smoker vs never-smoker, Teams 219 and 250 each selected 40 predictor genes, while Team 264, which achieved the highest performance in this comparison, selected only eight predictor genes. Among the 85 unique genes chosen by at least one of the teams, CLDN19 was selected by all three teams, while CNTROB was selected by two of the teams, suggesting there was little overlap. None of these genes met the q-value of <0.1 significance level on any of the four datasets.
Discussion
The expectation that biological processes and pathways in human and animal models are perturbed in a similar manner by external stimuli is the assumption that underpins many animal model studies, with controversies occurring when lack of translatability of findings are reported [27]. After demonstrating that changes in protein phosphorylation status and gene set activation induced by cellular responses to 52 stimuli in human cells can be predicted to some extent given responses generated in rat cells[3,21,28], the crowdsourcing-based initiative sbv IMPROVER, addressed the question of whether a common set of blood mRNAs can identify exposure to cigarette smoke in both mouse and human. According to the results of sub-challenge 1 of the sbv IMPROVER Systems Toxicology Challenge (see Tarca et al. in this issue), current smokers were distinguished from non-current smokers with 96.5% balanced accuracy (BAC) (100% sensitivity at 93% specificity) (task 1), while former smokers were discriminated from never-smokers with only 61% BAC (65% sensitivity at 57% specificity) (task 2). When a common set of genes was required, and the same analytical approach was used (Team 246) to predict both human and mouse exposure to cigarette smoke (sub-challenge 2), the BAC dropped to 80% (74% sensitivity at 85% specificity) for task 1 and 59% (85% sensitivity at 32% specificity) for task 2. Therefore, we concluded that although good prediction performances for current exposure to smoking can be achieved with a common gene signature for both species, the performance is lower than when a separate model is used for each species because species-specific changes also occur in blood mRNAs.
From a computation perspective, sub-challenge 2 can be seen as a classification problem involving a strata, in this case the species (mouse or human), where the goal was to design a model to predict accurately the outcome/class (exposed vs non-exposed) in a new dataset containing samples (observations) from both strata using a common set of genes. Although the test datasets on which the different models were ranked were not balanced for the two strata (2.5 times more human than mouse samples), all three top teams trained their final model without weighting the samples to favor equally both species. Hence, by default, all models of the top three teams are expected to have favored the most dominant strata (human). However, there are fundamental differences in the way the predictor genes were selected and used by the prediction models of the three teams that can explain, at least in part, the higher accuracy of the Team 219 model compared with the models of Teams 250 and 264.
The least computationally restrictive model, and hence the model that allowed for the best fit of the data, was developed by Team 219, which was deemed best overall in sub-challenge 2. The Team 219 model selected genes by LASSO regression based on human and mouse datasets separately with virtually no overlap between the optimal species-specific gene lists. Then, a random forest model was trained using a combined list of the predictor genes on the combined human and mouse datasets. The second least restrictive model was developed by Team 250 (ranked second) in which genes predictive of the outcome were selected based on the combined human and mouse training datasets. The selected genes were then used in a random forest classifier. The random forest classifiers used by Teams 219 and 250 could easily have leveraged systematic biases between the human and mouse data to determine internally the predictor genes to use for each species separately in the decision trees; hence, defying the goal of the sub-challenge of finding a common (consistent) gene expression signature (genes that were up- or down-regulated in both species at the same time). The accurate identification of the species was easily achievable even based on genes that were initially intended to be predictive for cigarette smoke exposure. Indeed, 68% and 38% of the predictor genes selected by Teams 219 and 250, respectively, for classification task 1 showed systematic differences between the two species (q-value <0.1 and fold change >1.5, consistent direction of change in both training and test datasets). The strong effect of species on overall expression data is depicted in Figure S1, because the first principal component captures differences between species and not between exposure groups.
The most restrictive approach (from a computational perspective), yet aligned most with the main aim of the challenge, was developed by Team 264, in which genes that appeared to be changing consistently with exposure in both strata were selected by adjusting for the species variable in a linear model fitting expression data as a function of the exposure status. Indeed, as can be seen in Figure S1, the gene predictors identified by Team 264 for the smoker vs non-current smoker comparison led to consistent separation of exposure groups in both the human and mouse training and test datasets based on the second principal component (which was not the case when predictor genes of Teams 219 and 250 were used in the principal components analysis). Therefore, a likely explanation for the high performance of the Team 219 prediction model was more freedom (less restriction) in designing the model to fit the available expression data.
The main biological finding of sub-challenge 2 of the sbv IMPROVER Systems Toxicology Challenge was that exposure to cigarette smoke had a significant effect on the blood transcriptome of both human and mouse and that this effect attenuated after one or more years (or equivalently two months for mouse) of exposure cessation. Exposure to cigarette smoke is known to have a broad and long-term impact on genome-wide methylation [29], and this epigenetic mechanism allows exposed species to respond to the environment through changes in gene expression [30]. Interestingly, AHRR, KLRG1, PGRMC1, TBX21, WDFY1, GUCY1B3, PF4, and SYTL4 mRNAs were all down-regulated with smoking in three or four of the human and mouse datasets used in this study. Moreover, the gene expression changes with current exposure to smoking reflected the perturbation of several biological processes and pathways.
Supplementary Material
Highlights.
Common blood mRNAs predict current exposure to cigarette smoke in mouse and human
Organism specific signature is more accurate for prediction of current exposure
Best predictive methods found in previous challenges are proven again to be robust
Abreviations
- IMPROVER
Industrial Methodology for PROcess VErification in Research
- LDA
Linear Discriminant Analysis
- LASSO
Least Absolute Shrinkage and Selection Operator
- sbv
systems biology verification
References
- 1.Saez-Rodriguez J, Costello JC, Friend SH, Kellen MR, Mangravite L, Meyer P, et al. Crowdsourcing biomedical research: leveraging communities as innovation engines. Nat Rev Genet. 2016;17:470–486. doi: 10.1038/nrg.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer P, Alexopoulos LG, Bonk T, Califano A, Cho CR, de la Fuente A, et al. Verification of systems biology research in the age of collaborative competition. Nat Biotechnol. 2011;29:811–815. doi: 10.1038/nbt.1968. [DOI] [PubMed] [Google Scholar]
- 3.Rhrissorrakrai K, Belcastro V, Bilal E, Norel R, Poussin C, Mathis C, et al. Understanding the limits of animal models as predictors of human biology: lessons learned from the sbv IMPROVER Species Translation Challenge. Bioinformatics under review. 2014 doi: 10.1093/bioinformatics/btu611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahuja V, Sharma S. Drug safety testing paradigm, current progress and future challenges: an overview. J Appl Toxicol. 2014;34:576–594. doi: 10.1002/jat.2935. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Xuan J, Couch L, Iyer A, Wu Y, Li QZ, et al. Sertraline induces endoplasmic reticulum stress in hepatic cells. Toxicology. 2014;322C:78–88. doi: 10.1016/j.tox.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sturla SJ, Boobis AR, FitzGerald RE, Hoeng J, Kavlock RJ, Schirmer K, et al. Systems toxicology: from basic research to risk assessment. Chem Res Toxicol. 2014;27:314–329. doi: 10.1021/tx400410s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 8.Phillips B, Veljkovic E, Peck MJ, Buettner A, Elamin A, Guedj E, et al. A 7-month cigarette smoke inhalation study in C57BL/6 mice demonstrates reduced lung inflammation and emphysema following smoking cessation or aerosol exposure from a prototypic modified risk tobacco product. Food Chem Toxicol. 2015;80:328–345. doi: 10.1016/j.fct.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 9.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eyre TA, Wright MW, Lush MJ, Bruford EA. HCOP: a searchable database of human orthology predictions. Brief Bioinform. 2007;8:2–5. doi: 10.1093/bib/bbl030. [DOI] [PubMed] [Google Scholar]
- 11.SBV IMPROVER P. The Systems Toxicology Challenge 2015 [Google Scholar]
- 12.Schein AI, Popescul A, Ungar LH, Pennock DM. Proceedings of the 25th annual international ACM SIGIR conference on Research and development in information retrieval. Tampere, Finland: ACM; 2002. Methods and metrics for cold-start recommendations; pp. 253–260. [Google Scholar]
- 13.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. BiochimBiophysActa. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 14.Tibshirani R. Regression shrinkage and selection via the lasso. JRoyStatistSocSerB. 1996;58:267–288. [Google Scholar]
- 15.Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 16.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. JStatSoftw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 17.Liaw A, Wiener M. Classification and Regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 18.Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 19.Tarca AL, Carey VJ, Chen XW, Romero R, Draghici S. Machine learning and its applications to biology. PLoS Comput Biol. 2007;3:e116. doi: 10.1371/journal.pcbi.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarca AL, Than NG, Romero R. Methodological approach from the Best Overall Team in the sbv IMPROVER Diagnostic Signature Challenge. Systems Biomedicine. 2013;1 [Google Scholar]
- 21.Dayarian A, Romero R, Wang Z, Biehl M, Bilal E, Hormoz S, et al. Predicting protein phosphorylation from gene expression: top methods from the IMPROVER Species Translation Challenge. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; 2012. pp. 397–420. [Google Scholar]
- 23.Tarca AL, Lauria M, Unger M, Bilal E, Boue S, Kumar Dey K, et al. Strengths and limitations of microarray-based phenotype prediction: lessons learned from the IMPROVER Diagnostic Signature Challenge. Bioinformatics. 2013;29:2892–2899. doi: 10.1093/bioinformatics/btt492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Team RDC. R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- 25.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. NatGenet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hafemeister C, Romero R, Bilal E, Meyer P, Norel R, Rhrissorrakrai K, et al. Inter-species pathway perturbation prediction via data-driven detection of functional homology. Bioinformatics. 2015;31:501–508. doi: 10.1093/bioinformatics/btu570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet. 2016 doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.