

Summary
The interactions between proteins and biological membranes are important for drug development, but remain notoriously refractory to structural investigation. We combine non-denaturing mass spectrometry (MS) with molecular dynamics (MD) simulations to unravel the connections among co-factor, lipid, and inhibitor binding in the peripheral membrane protein dihydroorotate dehydrogenase (DHODH), a key anticancer target. Interrogation of intact DHODH complexes by MS reveals that phospholipids bind via their charged head groups at a limited number of sites, while binding of the inhibitor brequinar involves simultaneous association with detergent molecules. MD simulations show that lipids support flexible segments in the membrane-binding domain and position the inhibitor and electron acceptor-binding site away from the membrane surface, similar to the electron acceptor-binding site in respiratory chain complex I. By complementing MS with MD simulations, we demonstrate how a peripheral membrane protein uses lipids to modulate its structure in a similar manner as integral membrane proteins.
Keywords: non-denaturing mass spectrometry, membrane protein folding, molecular dynamics simulations, protein-lipid interactions
Graphical Abstract
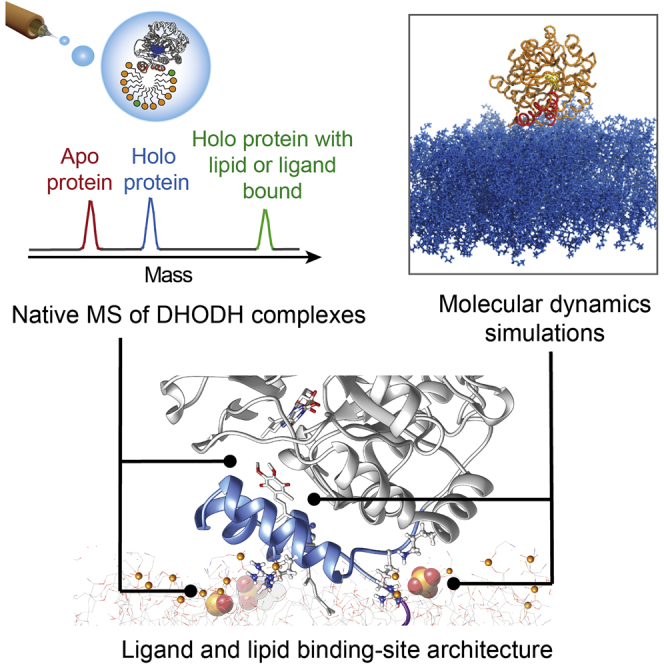
Highlights
-
•
Mass spectrometry captures intact complexes of the peripheral membrane protein DHODH
-
•
Detergent removal in the gas phase reveals lipid and co-factor binding
-
•
DHODH attaches to the membrane by binding charged phospholipids
-
•
Lipids stabilize the flexible substrate- and drug-binding site
The combination of mass spectrometry and molecular dynamics simulations provides insights into the relationship between lipid and substrate binding to the peripheral membrane protein dehydroorotate dehydrogenase, revealing ligand-induced stabilization of the flexible membrane-binding region.
Introduction
Membrane-associated proteins constitute up to 30% of the human proteome and include several important drug targets (Hopkins and Groom, 2002, Krogh et al., 2001). Yet the highly amphipathic nature of these proteins and their dependence on lipid interactions continue to limit structural investigations. It has recently been established that non-denaturing nano-electrospray ionization mass spectrometry (nESI-MS) is well suited to study integral membrane protein complexes (Konijnenberg et al., 2015, Landreh and Robinson, 2015). Here, the protein is ionized while embedded in a protective detergent micelle that is subsequently removed by collisional activation to release the intact complex for MS analysis (Barrera et al., 2008). The approach has been successfully applied to yield pioneering insights into the structural arrangements of membrane protein complexes (Wang et al., 2010, Zhou et al., 2011, Zhou et al., 2014), as well as the effects of lipids on conformational stability, oligomerization, and drug binding in membrane-embedded transporters, receptors, and proteases (Bechara et al., 2015, Gupta et al., 2017, Konijnenberg et al., 2014, Laganowsky et al., 2014, Marcoux et al., 2013). It can furthermore provide constraints for computational methods to yield high-resolution models of complex architectures (Politis and Schmidt, 2017).
The corresponding interactions with lipids are far less well understood in peripheral membrane proteins, which are only partially inserted into the bilayer. Despite being largely hydrophilic, their biological functions require association with lipids, and therefore pose similar challenges for structural investigation as integral membrane proteins (Hurley, 2006). Several methods have been applied successfully to investigate membrane-binding proteins, such as hydrogen/deuterium exchange MS, which can localize association sites and topology, electron spin resonance, which reports on changes in the membrane organization, and molecular dynamics (MD) simulations, which predict molecular determinants of membrane binding (Vadas et al., 2017, Whited and Johs, 2015).
The functions of peripheral membrane proteins are often determined by a complex interplay between lipid and ligand interactions, and structural dynamics. A well-known example is the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH), which catalyses the oxidation of dihydroorotate to orotate during de novo cellular pyrimidine synthesis and is the focus of treatment strategies against malaria, autoimmune diseases, and cancer (Munier-Lehmann et al., 2013, Sykes et al., 2016). Membrane association occurs via the N-terminal region, which contains a mitochondrial signal sequence, a putative transmembrane (TM) helix, and two amphipathic helices. The role of the TM helix is unclear as it does not affect enzymatic activity in vitro and does not appear to be crucial for membrane association in vivo (Liu et al., 2000, Norager et al., 2002, Rawls et al., 2000). The amphipathic region (residues 31–68 of the full-length protein) constitutes the membrane-binding domain that facilitates the transfer of two electrons from the soluble substrate dihydroorotate via the endogenous co-factor flavin mononucleotide (FMN) to the membrane-bound acceptor ubiquinone-10 (Q10). Consequently, targeted occupation of the putative coenzyme Q10 binding site in the membrane-binding domain has emerged as a viable strategy to inhibit DHODH activity (Malmquist et al., 2007, Walse et al., 2008). Although the protein is dependent on membrane interactions in vivo, the role of lipids has so far been elusive (Norager et al., 2002). Here, we show that using the non-denaturing MS strategy developed for integral membrane proteins, we are able to detect intact complexes of DHODH with FMN, phospholipids, and an inhibitor. Integration of the insights from MS with MD simulations and crystallographic evidence reveals that phospholipid binding mediates the stabilization of the Q10 binding channel and the correct positioning of the enzyme on the membrane. This study shows that the combination of non-denaturing MS and MD provides insights into the dynamic nature of peripheral membrane proteins, revealing characteristics of both soluble and membrane protein complexes.
Results and Discussion
Non-denaturing MS Captures Intact DHODH Complexes
Human DHODH in vivo is permanently membrane-associated via its membrane-binding domain and a TM helix. In this study, we used a truncated protein lacking the 30 N-terminal residues that constitute the membrane-binding domain and a short linker region, which exhibits the same enzymatic activity in vitro as the full-length protein and still requires detergent for solubilization, indicating that it retains membrane association (Liu et al., 2000, Norager et al., 2002, Rawls et al., 2000). We therefore hypothesized that the MS strategy employed for integral membrane proteins may be equally suited for DHODH. nESI was performed directly from ammonium acetate-buffered protein solutions containing 2× critical micelle concentration of the detergent lauryldimethylamine N-oxide (LDAO), in order to use detergent micelles to protect the protein during transfer to the gas phase (Barrera et al., 2008).
Using gentle MS conditions commonly employed for soluble protein complexes, we observed a series of low-intensity peaks corresponding in mass to apo-DHODH with a broad charge state distribution ranging from approximately 25+ to 10+, indicative of unfolding during transfer to the gas phase (Figure 1A). However, after isolation of the high mass region and moderate collisional activation (see STAR Methods), we detected a major population of DHODH with an additional mass of 455 Da, in good agreement with binding of one FMN molecule per protein (Figure 1B). A comparison of the average charge states of holo- and apo-DHODH revealed that FMN binding is observed exclusively for low charge states associated with compact conformations, based on empirical charge state predictions for native-like protein ions (Figure S1) (Bush et al., 2010, Hall and Robinson, 2012, Kaltashov and Mohimen, 2005). Circular dichroism (CD) spectroscopy shows that DHODH remains folded under the solvent conditions used for nESI-MS (Figure S1). To elucidate the relationship between FMN binding and the protein's folding state in the gas phase, we measured the collision cross section (CCS) using ion mobility (IM-) MS. The data show that holo-DHODH retains a compact conformation with a CCS of 2,930–3,000 Å2, close the value of 2,800 Å2 expected for the native protein, while apo-DHODH appears to be fully unfolded (Figure S1). We conclude that we are able to preserve compactly folded DHODH with intact co-factor interactions in the gas phase, which may reflect a global stabilizing effect of FMN binding (Beveridge et al., 2016).
Figure 1.

Non-denaturing MS of DHODH Complexes
(A) The nESI-MS spectrum of the full m/z range shows peaks corresponding to apo-DHODH with a broad charge state distribution.
(B) Collisional activation and isolation of the region between m/z 2,700 and 3,300 (shown in gray in A) reveals two series of well-resolved protein signals corresponding to apo-DHODH as well as DHODH bound to the FMN co-factor. The deconvolved zero-charge spectrum is shown as an insert.
(C) The MS strategy for integral membrane proteins facilitates the analysis of intact DHODH complexes. Release of the desolvated protein from detergent by collisional activation preserves interactions with the FMN co-factor as well as exogenous ligands.
See also Figure S1.
Importantly, we find that the intact DHODH-FMN complexes are released from detergent clusters in a similar manner as integral membrane proteins (Figure 1C). However, the low activation energy required and the presence of detergent-free, unfolded protein suggest that the association with detergent micelles is less stable for DHODH than for integral membrane proteins, in line with its predicted peripheral membrane association (Hanson et al., 2003, Ilag et al., 2004, Landreh et al., 2017). Taken together, our findings show that non-denaturing MS is a suitable tool to probe non-covalent interactions of peripheral membrane proteins. We are therefore able to study the broad range of dynamic interactions previously elucidated for integral membrane proteins and soluble protein complexes (Lössl et al., 2016).
DHODH Binds Charged Phospholipid Head Groups
Since DHODH in vivo is permanently membrane-associated, we speculated that mitochondrial lipids may be of importance for its biological function. We performed DHODH activity assays either in detergent alone or supplemented with any of the three major human mitochondrial lipids phosphatidyl ethanolamine (PE), phosphatidyl choline (PC), and cardiolipin (CDL). Monitoring the reduction of the soluble electron acceptor benzoquinone showed that all three lipids increased the reaction rate (Vmax) by 30%–60% relative to the detergent-solubilized protein. Since all three lipids caused a similar increase in activity, the effect is unlikely to be mediated by specific lipid recognition in DHODH (Figure S2).
Having established that we can detect intact DHODH complexes in the gas phase, we used MS to investigate the interactions between DHODH and phospholipids in detail (Figure 2A). nESI-MS spectra recorded in the presence of any of the three lipids show additional peaks indicating the formation of protein-lipid complexes. For CDL and PE, these were readily detected at a protein to lipid ratio of 1:4. Interactions with PC, on the other hand, resulted in fewer lipid adducts that could only be detected at higher lipid concentrations (Figure 2B). Since the lipids chosen for analysis have identical acyl chains, the different extent to which their association is preserved after desolvation can be attributed to differences in head group interactions. For PC, for which only minor adducts are detected, hindrance from the tertiary amine may negatively affect favorable electrostatic head group interactions. CDL, for which strong adduct peaks are observed, contains two negatively charged phosphates that may easily form hydrogen-bonding interactions with DHODH. The observed preference for anionic head groups suggests that the protein-lipid complexes are held together by charge interactions in the gas phase. To further characterize the interaction, we recorded mass spectra of the protein in the presence of increasing concentrations of PE, the most abundant of the three mitochondrial lipids. Interestingly, a maximum of three lipid adducts could be observed even at a 20-fold excess of PE over DHODH, and the protein-lipid complex peaks remained at lower intensities than the peaks corresponding to the lipid-free protein (Figure 2C). The limited lipid binding indicates that the protein makes little contact with the detergent-solubilized lipids. As a result, only a few lipid molecules remain attached to the protein after detergent release, in contrast to integral membrane proteins, which bind and retain a large number of lipid adducts (Hanson et al., 2003, Laganowsky et al., 2014, Landreh et al., 2017). We conclude that DHODH associates only superficially with membrane lipids, consistent with binding to the membrane-associated region. To test this hypothesis, we measured the effect of lipids on the thermal stability of detergent-solubilized DHODH. All three lipids induced only a very minor shift in melting temperature (Figure S2). The fact that lipids do not provide stabilization against global unfolding of the protein is compatible with a peripheral association.
Figure 2.

DHODH Exhibits Limited Binding of Mitochondrial Phospholipids
(A) Structures of the detergent LDAO and the three major phospholipid species in the human mitochondrial membrane.
(B) nESI-MS shows that binding of PC, PE, and CDL, can be preserved in the gas phase. CDL exhibits the most stable association with DHODH, while PC interactions are not well retained.
(C) Mass spectra of DHODH in the presence of increasing amounts of PE reveal a maximum number of lipid adducts for both apo- and holo-DHODH. Above a 20-fold excess (180 μM) of PE over DHODH, a maximum of three lipids are bound per protein.
(D) The DHODH-lipid complexes are less sensitive to DMSO-induced dissociation than the DHODH-FMN complexes (indicated by blue arrows). Spectra recorded without and with DMSO are normalized to the 14+ charge state of holo- and apo-DHODH, respectively.
See also Figure S2.
Next we asked whether ligand binding to DHODH can be disrupted by chemical destabilization. This was achieved by adding small amounts of dimethyl sulfoxide (DMSO), which is enriched 3–5 times in the electrospray droplets during desolvation due to its high boiling point (189°C), distorting protein structure and ligand interactions (Cubrilovic and Zenobi, 2013, Sterling et al., 2011). Accordingly, low DMSO concentrations (<6%) showed no significant effect on the integrity of the native complex in solution, as judged by FMN fluorescence (Figure S2). When subjected to nESI-MS in the presence of >1% DMSO, however, DHODH exhibited progressive loss of FMN (Figure S2). Complexes between DHODH and PE, on the other hand, could still be detected in the presence of 6% DMSO, suggesting that they are less sensitive to destabilization (Figure 2D). In summary, we find that all major mitochondrial lipid species bind to peripheral regions of DHODH. The stability of the resulting complexes in MS is governed predominantly by head group interactions with only a few additional contacts with the protein.
Ligands Stabilize the Membrane-Binding Domain
Next, we used MS to probe the interactions between DHODH and the potent inhibitor brequinar, a promising agent for the treatment of acute myeloid leukemia (Rawls et al., 2000). Direct addition of equimolar amounts of brequinar to the protein led to the appearance of peaks that could be assigned to protein-inhibitor complexes (Figure 3A). Interestingly, these complexes additionally retained up to three LDAO molecules. For apo-DHODH, which is likely unfolded according to the IMMS data (Figure S1), only minor peaks corresponding to protein-ligand complexes could be observed. Brequinar adducts could easily be dissociated by low levels of collisional activation, suggesting that the underlying interactions are much more easily disrupted in the gas phase than complexes with lipids, which require higher collisional activation for detection.
Figure 3.

Effects of Ligand Interactions on the Flexible Membrane-Binding Domain
(A) nESI-MS of DHODH in the presence of brequinar shows a series of adduct peaks for holo-DHODH corresponding to one bound brequinar molecule, as well as one brequinar and one, two, or three LDAO molecules. The deconvolved zero-charge spectrum is shown above the corresponding mass spectrum. One and two asterisks indicate peaks corresponding to apo-DHODH with LDAO and brequinar, respectively.
(B) The crystal structure of human DHODH with a brequinar analogue (PDB: 1D3G) shows binding of the inhibitor and an LDAO molecule in the hydrophobic channel leading to the FMN co-factor.
(C) Comparison with ligand-free DHODH (PDB: 2PRM) and in complex with brequinar and LDAO (PDB: 1D3G) rendered according to the average B factor show that binding of brequinar and LDAO significantly reduces the conformational flexibility of the membrane-binding domain.
(D) MD simulations of ligand-free truncated DHODH in solution confirm the conformational flexibility of the membrane-binding domain. The protein is rendered according to the RMSDs between the equilibrated starting structure and the end structure after 200 ns all-atom MD simulations.
See also Figure S3.
High-resolution crystal structures of DHODH with a brequinar analogue show that the inhibitor inserts between the two helices that form the membrane-binding domain (Liu et al., 2000). Interestingly, the resulting complexes additionally contain an LDAO molecule co-inserted into the membrane-binding domain, acting as a “plug” for the inhibitor-binding site (Figure 3B). In the absence of bound inhibitors, this region is marked by high crystallographic B factors, indicating considerable structural heterogeneity that is reduced in the presence of inhibitors and LDAO (Figure 3C) (Liu et al., 2000, Walse et al., 2008). We therefore asked whether the structural heterogeneity apparent in the crystal structure translates to conformational flexibility in solution. To address this question, we performed all-atom MD simulations of solvated ligand-free DHODH in the absence of lipids and detergents. Mapping the structural deviations of the protein backbone between the equilibrated starting structure and the simulation endpoint shows that the membrane-binding domain retains its helical fold but exhibits a considerable degree of conformational freedom that is reflective of the crystallographic B factors (Figure 3D).
Interestingly, we also found a second highly flexible region in the ligand-free protein, the loop between residues 214 and 226, which in the crystal structure is well-structured in the presence of orotate (Figure S3). It appears likely that this segment can move aside to provide access to the dihydroorotate binding site. Since the rest of the protein exhibited remarkably little conformational change, the MD data demonstrate that the protein contains a membrane- and a substrate-binding site that is flexible in solution and can undergo ligand-mediated stabilization. Although we can only speculate whether LDAO and brequinar occupy the same sites in the desolvated DHODH complex as in the static high-resolution structure, the insights from crystallography and MD simulations are in agreement with the MS results, which suggest concomitant retention of the inhibitor and detergent molecules in the gas phase.
MD Simulations Reveal Molecular Determinants of Lipid Binding
Since DHODH in vivo is associated with the mitochondrial membrane, we asked whether the conformational flexibility of the membrane-binding domain may be specifically adapted to its native lipid environment. To better understand the impact of lipid binding on the protein structure, we performed all-atom MD simulations of the truncated as well as the full-length protein associated with a model PE bilayer. First, we determined the preferred positioning of membrane-associated DHODH. To achieve this, we performed MD simulations in which the protein was pulled toward the center of the bilayer while monitoring the opposing force. Interestingly, once the N-terminal amphipathic helices (residues 35–51 and 52–67) that constitute the membrane-binding domain (Figure 4A) were inserted up to the lipid head groups, the force was found to increase in a nearly linear fashion. The finding that even shallow insertion is energetically unfavorable is in good agreement with the superficial detergent and lipid association inferred from MS (Figure S4). We then performed MD simulations of the protein with and without the N-terminal transmembrane helix attached (see STAR Methods). In all cases, we found that the soluble domain forms additional contacts with the membrane by extending the side chains of the surface residue K167, and intermittently also R162, toward the lipid head groups (Figure 4B). This is surprising, as the resulting orientation partially lifts the amphipathic helices off the membrane surface. All three trajectories of the full-length protein, as well as two trajectories of the truncated protein, converged toward similar orientations (Figure S4). In the equilibrated system, the structures of the TM helix and the soluble domain were independently oriented, suggesting that the TM helix does not have a pronounced effect on the fold of the soluble domain (Figure S4). In one trajectory, the truncated protein formed much more extensive contacts with the membrane, leading to the complete detachment of the membrane-binding domain from the bilayer (Figure S4). Therefore, the TM helix may help to safeguard the enzyme from getting trapped in such unfavorable orientations by providing extra anchorage to the membrane bilayer.
Figure 4.

Membrane Interactions Shape the Q10 Binding Site
(A) The N-terminal segment of truncated DHODH contains two amphipathic helices. Hydrophobic residues are shown in green, basic residues in contact with the membrane are labeled in blue.
(B) All-atom MD of full-length DHODH on a PE bilayer shows association of charged residues with the lipid head groups, partially lifting the membrane-binding domain (blue) off the membrane surface. Here, a partial Q10 molecule has been modeled into the binding site.
(C) The close clustering of the charged residues limits the number of simultaneously bound PE molecules. Phosphate head groups within 3 Å of the protein surface after 200 ns simulations are rendered as spheres and basic residues as sticks.
(D) Membrane association reduces conformational fluctuations in the membrane-binding domain. The protein is rendered according to the RMSDs between the equilibrated starting structure and the end structure after 200 ns.
(E) In the membrane-bound protein, the Q10 binding site forms a narrow reaction chamber that extends away from the membrane and is sealed by the hydrophobic tail (shown as sticks). The dashed line indicates the membrane surface.
(F) In this model, Q10 is coordinated by the strictly conserved residues R136 and Y356.
See also Figure S4.
The MD trajectories also provide detailed insights into the structural impact of DHODH-lipid interactions. Examination of membrane-bound DHODH over the course of the simulations (200 ns) reveals that the protein associates with only 3–6 lipid molecules at a time (Figure 4C). The interaction sites correlate with a patch of basic residues on the N-terminal end of helix 1 and at the C-terminal end of helix 2, as well as K167 on the soluble domain. Lipids are bound via salt bridges between their negatively charged phosphate moieties and the positively charged side chains without further protein-lipid contacts, in line with what we inferred from the MS experiments. Considering that these interactions suffice to position the protein, changes in local membrane composition may therefore alter the orientation of the enzyme, potentially providing a regulatory mechanism as described for other peripheral membrane proteins (Li and Buck, 2017). We also note that the closely clustered charged residues push apart neighboring head groups, giving rise to minor defects in the membrane surface. Together, these observations potentially provide the molecular basis for a lipid-operated “gate” in the membrane-binding region (Basso et al., 2016). We also compared the structural changes of the membrane-binding domain by calculating the backbone root-mean-square deviation (RMSD) values between the start and the end configuration of the simulations and found that the protein exhibited reduced conformational flexibility around the lipid-binding sites compared with the lipid-free protein (Figures 3C and 3D). Therefore, membrane interactions likely stabilize the membrane-binding domain, comparable with the effect of brequinar and detergent binding.
Membrane Association Shapes the Q10 Binding Site
Since the membrane-binding domain also constitutes the binding sites for inhibitors and the electron acceptor coenzyme Q10, we speculated that lipid interactions may be of importance for shaping the binding site configuration of DHODH in vivo. However, no high-resolution structure providing insights into the Q10-DHODH interaction has been reported to date. We therefore modeled the benzoquinol head group and the first four isoprenoid units of Q10 into the structure of the membrane-bound full-length protein based on the coordination observed in the crystal structure of DHODH with 3-amido-5-biphenyl-benzoic acid (PDB: 2B0M) (Figures 4E and S6). In this orientation, the Q10 head group is coordinated by hydrogen bonding with the strictly conserved residues R136 and Y356 (Figure 4F). Notably, mutation of R136 to cysteine gives rise to familial DHODH deficiency in Miller Syndrome (Fang et al., 2012). The hydrophobic tail of Q10 folds into the channel occupied by the LDAO molecule in the crystal structure and reaches through the lipid-binding site down into the membrane (Figures 4D, 4E, and S6). Since the entire Q10 molecule is too long to be accommodated inside the protein, the associated membrane lipids should be considered part of the substrate-binding site. Therefore, the development of inhibitors that additionally engage in lipid interactions may represent a promising approach to target DHODH in vivo.
Our model of the membrane-bound full-length DHODH complex reveals some surprising features of its likely architecture. In the preferred orientation of the lipid-bound protein, the binding channel extends away from the membrane, placing the Q10 head group between 10 and 20 Å above its surface (Figures 4B and S6). Strikingly, a similar Q10 binding site architecture is conserved from bacteria to humans in respiratory complex I, with an active site tyrosine located approximately 15–25 Å above the membrane (Angerer et al., 2012, Baradaran et al., 2013, Zhu et al., 2016). Here, Q10 also binds in an extended conformation so that its hydrophobic tail acts as a seal to prevent water and oxygen molecules from entering the active site and interfering with Q10 reduction. We speculate that the comparable layout of the Q10 binding site in DHODH induced by lipid interactions enables the protein to utilize a similar mechanism, ensuring specificity for Q10 as electron acceptor in a closed reaction chamber. Considering the spatial association of DHODH with the respiratory chain complexes, it may be possible that this architecture also aids the transfer of Q10 to other enzymes associated with DHODH (Fang et al., 2013).
Significance
Lipid interactions are a key feature of membrane proteins; however, the structural and functional impact remains challenging to study. Here, we demonstrate that MS strategies developed for integral membrane proteins can be applied to study the interactions of a human peripheral membrane protein and in this manner outline the molecular determinants of lipid binding. Using low-resolution structural data from MS to complement high-resolution models from MD simulations, we are able to identify a distinctive electron acceptor-binding site architecture where lipid binding anchors the opening of the Q10 binding channel to the bilayer, ensuring that the catalytic center of the enzyme, which is located outside the membrane, remains accessible from the membrane core. The surprising similarities to the Q10 binding site in respiratory chain complex I are not grounded in structural homology but probably reflect similar functional requirements. The design of DHODH inhibitors as therapeutics should therefore take into consideration the impact of membrane association on the inhibitor-binding site. The combination of MS and MD employed here is therefore well suited for detailed investigations of the elusive interactions between peripheral membrane proteins and lipids.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| ArcticExpress (DE3) competent E.coli | Agilent | Cat#230192 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Brequinar sodium salt hydrate | Sigma Aldrich | Cat#SML0113 |
| n-Dodecyl-N,N-Dimethylamine-N-Oxide (LDAO) | Anatrace | Cat#D360 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) | Avanti Polar Lipids | Cat#850757P |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | Cat#850457P |
| 1',3'-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (CDL) | Avanti Polar Lipids | Cat#710335P |
| Dimethylsulfoxide (DMSO) | Sigma Aldrich | Cat#276855 |
| Ammonium acetate | Sigma Aldrich | Cat#5.43834 |
| H. sapiens DHODHNΔ30 | Ullrich et al., 2001 | N/A |
| 3,4-Dimethoxy-5-Methyl-p-Benzoquinone | Sigma Aldrich | Cat#D9150 |
| Deposited Data | ||
| Crystal structure of hDHODH with 3-amido-5-biphenyl-benzoic acid | Hurt et al., 2006 | PDB: 2B0M https://www.rcsb.org/pdb/explore/explore.do?structureId=2B0M |
| Crystal structure of hDHODH with brequinar analogue | Liu et al., 2000 | PDB: 1D3G https://www.rcsb.org/pdb/explore/explore.do?structureId=1d3g |
| Crystal structure of ligand-free DHODH | Walse et al., 2008 | PDB: 2PRM https://www.rcsb.org/pdb/explore/explore.do?structureId=2prm |
| Recombinant DNA | ||
| pNIC28-Bsa4 | Savitsky et al., 2010 | Addgene plasmid # 26103 |
| H. sapiens DHODHNΔ30 coding sequence (Liu et al., 2000) | GeneArt | N/A |
| Software and Algorithms | ||
| PULSAR | Allison et al., 2015 | http://www.pulsar.chem.ox.ac.uk |
| UniDec | Marty et al., 2015 | http://www.unidec.chem.ox.ac.uk |
| IMPACT | Marklund et al., 2015 | http://www.impact.chem.ox.ac.uk |
| mMass | Strohalm et al., 2010 | http://www.mmass.org |
| Xcalibur | Thermo Scientific | N/A |
| UCSF Chimera v1.11.02 | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| PyMol | Schrödinger, LLC Version 1.8 | www.sourceforge.net/projects/pymol |
| GROMACS 2016.1 | Abraham et al., 2015 | http://www.gromacs.org |
| MemGen | Knight and Hub, 2015 | http://memgen.uni-goettingen.de |
| ModLoop | Fiser and Sali, 2003 | https://modbase.compbio.ucsf.edu/modloop/ |
| AMBER99SB-ILDN force field | Lindorff-Larsen et al., 2010 | http://ambermd.org/#ff |
| FMN force field parameters | Schneider and Sühnel, 1999 | http://research.bmh.manchester.ac.uk/bryce/amber |
| POPE force field parameters and coordinates | Jämbeck and Lyubartsev, 2012b | http://www.fos.su.se/∼sasha/SLipids/Downloads.html |
| Other | ||
| Medium nanoES capillaries | Thermo Scientific | Cat#ES380 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Landreh (Michael.landreh@ki.se).
Method Details
Protein Preparation
N-terminally His6-tagged human DHODH with a 30-residue N-terminal deletion to remove the mitochondrial localization sequence and a putative transmembrane helix was expressed and purified as described (Liu et al., 2000, Ullrich et al., 2001) with the following modifications: The coding sequence was synthesized by GeneArt and cloned into pNIC28-Bsa4 (a gift from Opher Gileadi, Addgene plasmid # 26103). Expression was carried out using E. coli Arctic Express, DE3 (Agilent Technologies). Tunair shake flasks containing 750 mL LB media (Sigma Aldrich) were inoculated with 50 mL of an overnight culture and incubated at 30°C. At an OD of 0.6 the temperature was lowered to 13°C and expression was induced by the addition of IPTG (Thermo Scientific) to a final concentration of 1 mM. FMN (SigmaAldrich) was added to a final concentration of 100 μM, cultures were incubated overnight and harvested by centrifugation. Attempts to purify a C-terminally His6-tagged variant of the full-length protein resulted in significant aggregation and degradation. Protein samples were stored at a concentration of approximately 10 μM in 20 mM phosphate buffer, pH 8.0, containing 10 mM LDAO (Generon, Berkshire, UK). Prior to MS analysis, protein samples were exchanged into MS buffer (100 mM ammonium acetate, pH 7.5, containing 4 mM LDAO) using BioSpin microcentrifuge columns (BioRad Laboratories, Waltford, UK). 1.4 mM stock solutions of PE, PC, or CDL (Avanti Polar lipids, Inc, AL) in H2O were prepared as described (Laganowsky et al., 2014). Brequinar (Sigma-Aldrich, Darmstadt, Germany) was dissolved in DMSO at a concentration of 40 mM. For binding studies, the brequinar stock solution was diluted 1:40 in MS buffer and added to the protein to a final inhibitor concentration of 20 μM. All chemicals were purchased from Sigma (Sigma-Aldrich, Darmstadt, Germany).
Mass Spectrometry
Mass spectra were recorded on an Orbitrap Fusion (Thermo Fisher Scientific, Waltham, MA) equipped with an offline nanoelectrospray source. Samples were introduced using gold-coated Proxeon borosilicate capillaries (Thermo Scientific, Waltham, MA). The instrument was operated in intact protein mode. The capillary voltage was 1.2-1.8 kV, the transfer tube temperature was maintained at 40°C and the pressure in the ion-routing multipole was 0.011 torr. Precursor ion selection was performed in the ion trap with a selection window width of 600 m/z. Detergent was removed by increasing the in-source fragmentation to 100%. Additional collisional activation was performed by increasing the HCD energy (Gault et al., 2016). While protein peaks could already be detected at 0% HCD energy, an HCD energy of 10% increased spectral quality and was employed for lipid binding studies. High-purity nitrogen was used as collision gas. Spectra were recorded using the Orbitrap mass analyser at a resolution of 60 000 at m/z 200 with a high mass mode acquisition window of 1000-6000 m/z and an AGC target of 1 000 000. Data were analysed using the Xcalibur 3.0 (Thermo Scientific, Waltham, MA), UniDec (www.unidec.chem.ox.ac.uk) and mMass software packages (http://www.mmass.org) (Marty et al., 2015, Strohalm et al., 2010). The appearance of DHODH in the mass spectra was invariant between different protein preparations and could be reproduced by using the same instrument parameters.
IM-MS measurements were performed on a Waters Synapt G1 ion mobility mass spectrometer equipped with a linear field drift tube to facilitate direct CCS determination, and an offline nanoelectrospray source. Samples were introduced using gold-coated borosilicate capillaries produced in-house. Detergent was removed by increasing the cone voltage to 100 V. The pressure in the source region was maintained at 4.0 mbar. Mass spectra were recorded at drift voltages between 40 and 120 V at a ion trap voltage of 20 V for CCS determination. The trap voltage was ramped from 20 to 100 V in 10 V increments for collisional unfolding experiments. The drift gas was He with a flow of 2.2 L/h. Data were analysed using the PULSAR software package (www.pulsar.chem.ox.ac.uk) (Allison et al., 2015).
Fluorescence Spectroscopy
FMN fluorescence was measured using 100 μL of 10 μM DHODH in MS buffer, with step-wise addition of DMSO. Measurements were performed on a Tecan Spark 20M multimode reader (Tecan Instruments, Männedorf, Switzerland) using Corning COSTAR flat-bottom 96-well plates (Sigma-Aldrich, Darmstadt, Germany). Buffer containing 6% DMSO was used for blank measurements. Excitation wavelength was 450 nm, emission was recoded between 495-600 nm in 2 nm steps with 10 nm bandwidth and an integration time of 40 μs.
CD Spectroscopy
Prior to analysis, DHODH was exchanged into MS buffer to a final protein concentration of 10 μM. Experiments were performed on a 410-model CD spectrometer (Aviv Biomedical Inc., Lakewood, NJ) using 300 μL quartz cuvettes with a 1 mm path length. Spectra were recorded from 260 to 195 nm at 25°C. The data is shown as an average of four scans.
Enzymatic Activity Assay
Kinetic DHODH activity assays were carried out using 6 nM recombinant truncated human DHODH. The reaction mixture contained 1 mM DL-dihydroorotic acid, 100 μM 3,4-dimethoxy-5-methyl-p-benzoquinone (Sigma-Aldrich, Darmstadt, Germany), and 100 μM DCIP in buffer. 20 mM DCIP stock was prepared in enzyme buffer (50 mM Tris-HCl pH 8.0, 0.1 % Triton X-100, 150 mM KCl) and filtered through 25 μm pore size filter paper immediately before use. Loss in DCIP absorbance was measured at 595 nm in a stepped time course (8 × 2 min, 8 × 3 min, 6 × 5 min) carried out at room temperature. The decrease in absorbance over time was linear between 3 and 10 min. Therefore, for each concentration of inhibitor tested, a value for DHODH’s Vmax was estimated by linear regression within this time frame. Three independent measurements (n=3) were carried out with 4 technical replicates each time.
Thermal Unfolding Measurements
For thermal stability measurements, 1 μL of 1.4 mM PC, PE, or CDL stock in 3 × CMC LDAO, or LDAO only, were added to 9 μL of 20 μM DHODH in 50 mM tris-HCl buffer, pH 8.0, and loaded into a Prometheus NT.Plex nanoDLS system (NanoTemper, Munich, Germany). Melting was carried out by ramping the temperature from 20 and 80°C, and the folded-to-unfolded transition was monitored via the 1st derivative of the fluorescence emission at 330 nm.
MD of Truncated DHODH in Saline Solution
A structure of truncated DHODH, missing the C-terminal TM-helix (residues 1-30), and containing FMN, brequinar, and orotate (pdb code 2B0M) was used as a starting point. Brequinar and orotate were removed, and the missing residues (70, 71 and 72) were added with MODLOOP (Fiser and Sali, 2003, Fiser et al., 2000). The protein-FMN complex was solvated in TIP3P water (Mahoney and Jorgensen, 2000) inside a dodecahedron with 10.055 nm box vectors to which NaCl was added, yielding a holo-protein immersed in a 154 mM saline solution. The MD simulations and pre-processing were performed with GROMACS software (Abraham et al., 2015) using amber99sb-ildn forcefield (Lindorff-Larsen et al., 2010) modified with parameters for FMN (Schneider and Sühnel, 1999), using periodic boundary conditions. The velocity-rescaling thermostat (Bussi et al., 2007) was used for all condensed-phase simulations. The equations of motion were integrated with leap-frog algorithm (Hockney et al., 1974). All bonds were constrained with the LINCS algorithm (Hess et al., 1997), non-bonded interactions were calculated within 1 nm cut-off and PME (Essmann et al., 1995) was used for long-range electrostatics. Following steepest-descent energy minimization, the system was equilibrated in the NVE ensemble at 300 K, first with position restraints applied to all heavy atoms for 0.1 ns, then without position restraints for 1 ns. The system was then equilibrated in the NPT ensemble for 1 ns at 300 K and 1 bar using Berendsen pressure coupling (Berendsen et al., 1984). After equilibration, the system was simulated for 200 ns in the NVT ensemble using the Parrinello-Rahman barostat (Nosé and Klein, 1983, Parrinello, 1981). All these procedures were carried out in triplicate, yielding three separate sets of simulations. Theoretical CCS values were calculated from the all MD runs of truncated DHODH in saline solution and membrane-bound truncated DHODH, using IMPACT (www.impact.chem.ox.ac.uk) (Marklund et al., 2015) projection approximation multiplied by an empirical factor 1.14 (Benesch and Ruotolo, 2011).
MD of Membrane-Bound Truncated DHODH
The system was prepared by combining the DHODH starting structure with a hexagonal PE bilayer structure created with MemGen (Knight and Hub, 2015) using 140 lipids per monolayer and the default area per lipid. PE parameters were taken from the Stockholm lipids force field (Jämbeck and Lyubartsev, 2012a, Jämbeck and Lyubartsev, 2012b, Klauda et al., 2010). After following the same equilibration procedure as for the water-solvated DHODH, the protein was associated with the membrane by first aligning the membrane-binding helices with the membrane in a 10-ns simulation, then pulling the protein towards and into the membrane for another 10 ns. For both steps, GROMACS’s pulling code was used, employing distance-based harmonic potentials (using a force constant of 1000 kJ mol-1 nm-2) to manipulate the helices relative to the membrane. The potentials were only applied perpendicular to the membrane, and when pulling the protein into the membrane, a cylinder geometry with a 2.42 nm radius centered on the protein was used to define the part of the membrane used for the pulling. The pulling rate was chosen such that the helices were aligned by the end of the first run, and that the protein touched the middle of the membrane in the other. Since the pull-force gradually increased during the second step, indicating that the protein is on the surface at equilibrium, starting structures for production MD was taken from snapshots at 3 ns from these trajectories, before the protein entered the membrane. The production MD was performed the same way as for DHODH in saline solution. All steps were made in triplicate (n=3) and the results for each run shown in Figure S4.
MD of Membrane-Bound Full-Length DHODH
The TM-helix of DHODH was first simulated in a PE membrane by building an ideal α-helix using the TM-helix sequence and inserting that inside of the same membrane structure as for the truncated DHODH, using the PyMol software (PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). The equilibration and production MD of the TM-helix was performed according to the methodology used for the other systems. Subsequently, the full-length DHODH structure was constructed by attaching the equilibrated membrane-bound TM-helix to the structure of the truncated protein taken after its helices had been aligned with the membrane. MODLOOP (Fiser and Sali, 2003, Fiser et al., 2000) was used to position the bridging residues (31-35). Following steepest-descent energy minimization, the system was equilibrated in the NVE ensemble with position restraints, then the protein was pulled towards the membrane system using the same method as for the truncated protein. Afterwards, the system was equilibrated in the NPT ensemble and ran for 200 ns of production MD. All steps were made in triplicate (n=3) and the results for each run shown in Figure S4. Images were prepared using UCSF Chimera v1.11.02 (Pettersen et al., 2004).
Quantification and Statistical Analysis
Enzymatic activity assays (Figure S2A) were performed three times (n=3) with four technical replicates each time. The Vmax values were averaged for each set of technical replicates. The average and standard deviation for the three separate measurements were then calculated and plotted using Microsoft Excel. No additional statistical tests were performed.
Acknowledgments
The authors would like to thank Timothy M. Allison and Carol V. Robinson, University of Oxford, and David Drew, Stockholm University, for their valuable comments on the manuscript. The Robinson lab is gratefully acknowledged for use of the DT-IMS instrument. Thanks to Stephen Ambrose, University of Oxford, for rendering of the nESI-capillary in Figure 1C. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project SNIC 2017/7–15 and SNIC 2017/1–188. ML is supported by an Ingvar Carlsson Award from the Swedish Foundation for Strategic Research and a KI faculty-funded Career Position. J.G. is a Junior Research Fellow of The Queen's College, University of Oxford. E.G.M. holds a Marie Skłodowska Curie International Career Grant from the European Commission and the Swedish Research Council (2015-00559). S.L. and D.P.L. acknowledge support from the Swedish Research Council (VR), Cancerfonden, Barncancerfonden, and Karolinska Institutet. This work is supported by an EPSRC Institutional Sponsorship 2016 award (EP/P511377/1).
Author Contributions
E.G.M. and M.L. designed the study with support from S.L. and D.P.L. J.C.P. and E.G.M. planned and performed all computational analyses. J.G. optimized the Orbitrap Fusion platform for native mass spectrometry and performed IMMS measurements. M.L., J.G., and M.J.G.W.L. performed all other MS experiments. A.O. purified and G.P. and I.M.M.L. characterized protein. M.S. performed CD measurements. E.G.M., M.L., D.P.L., and S.L. analyzed the data, and E.G.M. and M.L. wrote the manuscript with input from all authors.
Declaration of Interests
The authors declare no competing interests.
Published: January 18, 2018
Footnotes
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.chembiol.2017.12.012.
Contributor Information
David P. Lane, Email: david.lane@ki.se.
Sonia Laín, Email: sonia.lain@ki.se.
Erik G. Marklund, Email: erik.marklund@kemi.uu.se.
Michael Landreh, Email: michael.landreh@ki.se.
Supplemental Information
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- Allison T.M., Reading E., Liko I., Baldwin A.J., Laganowsky A., Robinson C.V. Quantifying the stabilizing effects of protein–ligand interactions in the gas phase. Nat. Commun. 2015;6:8551. doi: 10.1038/ncomms9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H., Nasiri H.R., Niedergesäß V., Kerscher S., Schwalbe H., Brandt U. Tracing the tail of ubiquinone in mitochondrial complex I. Biochim. Biophys. Acta. 2012;1817:1776–1784. doi: 10.1016/j.bbabio.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera N.P., Di Bartolo N., Booth P.J., Robinson C.V. Micelles protect membrane complexes from solution to vacuum. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- Basso L.G.M., Santos Mendes L.F., Costa-Filho A.J. The two sides of a lipid-protein story. Biophys. Rev. 2016;8:179–191. doi: 10.1007/s12551-016-0199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara C., Nöll A., Morgner N., Degiacomi M.T., Tampé R., Robinson C.V. A subset of annular lipids is linked to the flippase activity of an ABC transporter. Nat. Chem. 2015;7:255–262. doi: 10.1038/nchem.2172. [DOI] [PubMed] [Google Scholar]
- Benesch J.L., Ruotolo B.T. Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Beveridge R., Migas L.G., Payne K.A.P., Scrutton N.S., Leys D., Barran P.E. Mass spectrometry locates local and allosteric conformational changes that occur on cofactor binding. Nat. Commun. 2016;7:12163. doi: 10.1038/ncomms12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M.F., Hall Z., Giles K., Hoyes J., Robinson C.V., Ruotolo B.T. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal. Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Cubrilovic D., Zenobi R. Influence of dimethylsulfoxide on protein-ligand binding affinities. Anal. Chem. 2013;85:2724–2730. doi: 10.1021/ac303197p. [DOI] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- Fang J., Uchiumi T., Yagi M., Matsumoto S., Amamoto R., Saito T., Takazaki S., Kanki T., Yamaza H., Nonaka K. Protein instability and functional defects caused by mutations of dihydro-orotate dehydrogenase in Miller syndrome patients. Biosci. Rep. 2012;32:631–639. doi: 10.1042/BSR20120046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J., Uchiumi T., Yagi M., Matsumoto S., Amamoto R., Takazaki S., Yamaza H., Nonaka K., Kang D. Dihydroorotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013;33:e00021. doi: 10.1042/BSR20120097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A., Sali A. ModLoop: automated modelling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- Fiser A., Do R.K., Sali A. Modelling loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J., Donlan J.A.C., Liko I., Hopper J.T.S., Gupta K., Housden N.G., Struwe W.B., Marty M.T., Mize T., Bechara C. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat. Methods. 2016;13:333–336. doi: 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K., Donlan J.A., Hopper J.T., Uzdavinys P., Landreh M., Struwe W.B., Drew D., Baldwin A.J., Stansfeld P.J., Robinson C.V. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature. 2017;541:421–424. doi: 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z., Robinson C.V. Do charge state signatures guarantee protein conformations? J. Am. Soc. Mass Spectrom. 2012;23:1161–1168. doi: 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- Hanson C.L., Ilag L.L., Malo J., Hatters D.M., Howlett G.J., Robinson C.V. Phospholipid complexation and association with apolipoprotein C-II: insights from mass spectrometry. Biophys. J. 2003;85:3802–3812. doi: 10.1016/S0006-3495(03)74795-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- Hockney R.W., Goel S.P., Eastwood J.W. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974;14:148–158. [Google Scholar]
- Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Hurley J.H. Membrane binding domains. Biochim. Biophys. Acta. 2006;1761:805–811. doi: 10.1016/j.bbalip.2006.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt D.E., Sutton A.E., Clardy J. Brequinar derivatives and species-specific drug design for dihydroorotate dehydrogenase. Bioorg. Med. Chem. Lett. 2006;16:1610–1615. doi: 10.1016/j.bmcl.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Ilag L.L., Ubarretxena-Belandia I., Tate C.G., Robinson C.V. Drug binding revealed by tandem mass spectrometry of a protein-micelle complex. J. Am. Chem. Soc. 2004;126:14362–14363. doi: 10.1021/ja0450307. [DOI] [PubMed] [Google Scholar]
- Jämbeck J.P.M., Lyubartsev A.P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B. 2012;116:3164–3179. doi: 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J.P.M., Lyubartsev A.P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 2012;8:2938–2948. doi: 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- Kaltashov I.A., Mohimen A. Estimates of protein surface areas in solution by electrospray ionization mass spectrometry. Anal. Chem. 2005;77:5370–5379. doi: 10.1021/ac050511+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J.B., Venable R.M., Freites J.A., O’Connor J.W., Tobias D.J., Mondragon-Ramirez C., Vorobyov I., MacKerell A.D., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight C.J., Hub J.S. MemGen: a general web server for the setup of lipid membrane simulation systems. Bioinformatics. 2015;31:2897–2899. doi: 10.1093/bioinformatics/btv292. [DOI] [PubMed] [Google Scholar]
- Konijnenberg A., Yilmaz D., Ingólfsson H.I., Dimitrova A., Marrink S.J., Li Z., Vénien-Bryan C., Sobott F., Koçer A. Global structural changes of an ion channel during its gating are followed by ion mobility mass spectrometry. Proc. Natl. Acad. Sci. USA. 2014;111:17170–17175. doi: 10.1073/pnas.1413118111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konijnenberg A., van Dyck J.F., Kailing L.L., Sobott F. Extending native mass spectrometry approaches to integral membrane proteins. Biol. Chem. 2015;396:991–1002. doi: 10.1515/hsz-2015-0136. [DOI] [PubMed] [Google Scholar]
- Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Laganowsky A., Reading E., Allison T.M., Ulmschneider M.B., Degiacomi M.T., Baldwin A.J., Robinson C.V. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M., Robinson C.V. A new window into the molecular physiology of membrane proteins. J. Physiol. 2015;593:355–362. doi: 10.1113/jphysiol.2014.283150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M., Costeira-Paulo J., Gault J., Marklund E.G., Robinson C.V. The effects of detergent micelles on lipid binding to proteins in electrospray ionization mass spectrometry. Anal. Chem. 2017;89:7425–7430. doi: 10.1021/acs.analchem.7b00922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.L., Buck M. Computational modeling reveals that signaling lipids modulate the orientation of K-Ras4A at the membrane reflecting protein topology. Structure. 2017;25:679–689. doi: 10.1016/j.str.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Neidhardt E.A., Grossman T.H., Ocain T., Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- Lössl P., van de Waterbeemd M., Heck A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016;35:2634–2657. doi: 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M.W., Jorgensen W.L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000;112:8910. [Google Scholar]
- Malmquist N.A., Baldwin J., Phillips M.A. Detergent-dependent kinetics of truncated Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2007;282:12678–12686. doi: 10.1074/jbc.M609893200. [DOI] [PubMed] [Google Scholar]
- Marcoux J., Wang S.C., Politis A., Reading E., Ma J., Biggin P.C., Zhou M., Tao H., Zhang Q., Chang G. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund E.G., Degiacomi M.T., Robinson C.V., Baldwin A.J., Benesch J.L. Collision cross sections for structural proteomics. Structure. 2015;23:791–799. doi: 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Marty M.T., Baldwin A.J., Marklund E.G., Hochberg G.K., Benesch J.L., Robinson C.V. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 2015;87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier-Lehmann H., Vidalain P.O., Tangy F., Janin Y.L. On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 2013;56:3148–3167. doi: 10.1021/jm301848w. [DOI] [PubMed] [Google Scholar]
- Nørager S., Jensen K.F., Björnberg O., Larsen S. E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure. 2002;10:1211–1223. doi: 10.1016/s0969-2126(02)00831-6. [DOI] [PubMed] [Google Scholar]
- Nosé S., Klein M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983;50:1055–1076. [Google Scholar]
- Parrinello M. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182. [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Politis A., Schmidt C. Structural characterisation of medically relevant protein assemblies by integrating mass spectrometry with computational modelling. J. Proteomics. 2017 doi: 10.1016/j.jprot.2017.04.019. [DOI] [PubMed] [Google Scholar]
- Rawls J., Knecht W., Diekert K., Lill R., Löffler M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur. J. Biochem. 2000;267:2079–2087. doi: 10.1046/j.1432-1327.2000.01213.x. [DOI] [PubMed] [Google Scholar]
- Savitsky P., Bray J., Cooper C.D., Marsden B.D., Mahajan P., Burgess-Brown N.A., Gileadi O. High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C., Sühnel J. A molecular dynamics simulation of the flavin mononucleotide-RNA aptamer complex. Biopolymers. 1999;50:287–302. doi: 10.1002/(SICI)1097-0282(199909)50:3<287::AID-BIP5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Sterling H.J., Prell J.S., Cassou C.A., Williams E.R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011;22:1178–1186. doi: 10.1007/s13361-011-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohalm M., Kavan D., Novak P., Volny M., Havlicek V. mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 2010;82:4648–4651. doi: 10.1021/ac100818g. [DOI] [PubMed] [Google Scholar]
- Sykes D.B., Kfoury Y.S., Mercier F.E., Wawer M.J., Law J.M., Haynes M.K., Lewis T.A., Schajnovitz A., Jain E., Lee D. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186. doi: 10.1016/j.cell.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A., Knecht W., Fries M., Löffler M. Recombinant expression of N-terminal truncated mutants of the membrane bound mouse, rat and human flavoenzyme dihydroorotate dehydrogenase. Eur. J. Biochem. 2001;268:1861–1868. [PubMed] [Google Scholar]
- Vadas O., Jenkins M.L., Dornan G.L., Burke J.E. Using hydrogen-deuterium exchange mass spectrometry to examine protein-membrane interactions. Methods Enzymol. 2017;583:143–172. doi: 10.1016/bs.mie.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Walse B., Dufe V.T., Svensson B., Fritzson I., Dahlberg L., Khairoullina A., Wellmar U., Al-Karadaghi S. The structures of human dihydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites. Biochemistry. 2008;47:8929–8936. doi: 10.1021/bi8003318. [DOI] [PubMed] [Google Scholar]
- Wang S.C., Politis A., Di Bartolo N., Bavro V.N., Tucker S.J., Booth P.J., Barrera N.P., Robinson C.V. Ion mobility mass spectrometry of two tetrameric membrane protein complexes reveals compact structures and differences in stability and packing. J. Am. Chem. Soc. 2010;132:15468–15470. doi: 10.1021/ja104312e. [DOI] [PubMed] [Google Scholar]
- Whited A.M., Johs A. The interactions of peripheral membrane proteins with biological membranes. Chem. Phys. Lipids. 2015;192:51–59. doi: 10.1016/j.chemphyslip.2015.07.015. [DOI] [PubMed] [Google Scholar]
- Zhou M., Morgner N., Barrera N.P., Politis A., Isaacson S.C., Matak-Vinkovic D., Murata T., Bernal R.A., Stock D., Robinson C.V. Mass spectrometry of intact V-Type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Politis A., Davies R.B., Liko I., Wu K.-J., Stewart A.G., Stock D., Robinson C.V. Ion mobility–mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat. Chem. 2014;6:208–215. doi: 10.1038/nchem.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Vinothkumar K.R., Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536:354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.