
Figure 8.
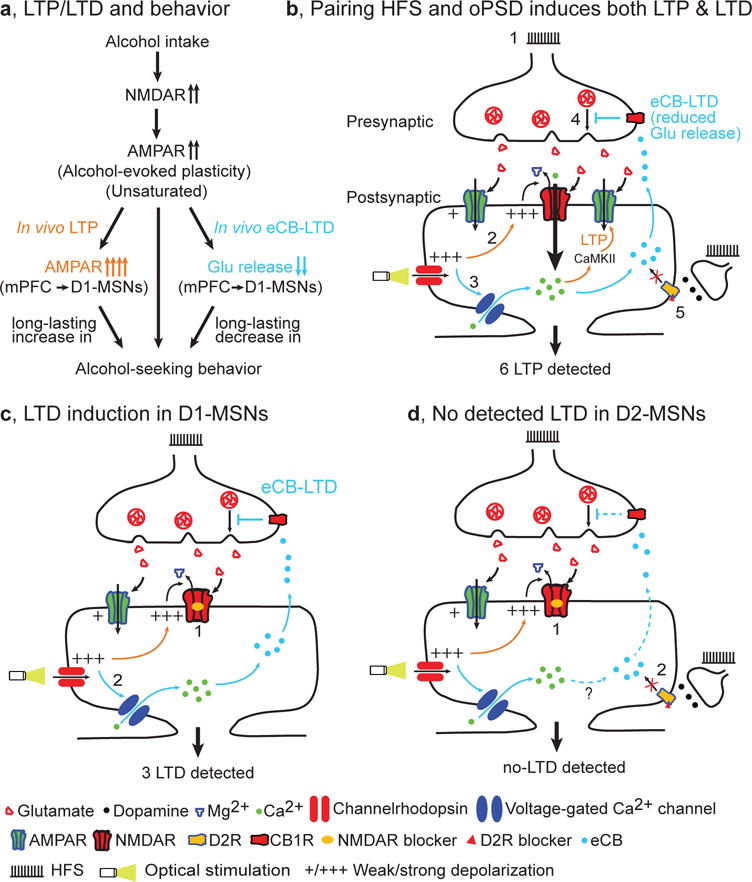
Model of bidirectional and long-lasting control of alcohol-seeking behavior by corticostriatal plasticity. (a) Alcohol intake facilitates NMDAR activity, leading to potentiation of AMPAR activity (unsaturated alcohol-evoked plasticity). This is further potentiated by in vivo LTP induction at mPFC inputs to DMS D1-MSNs, producing a long-lasting enhancement of alcohol-seeking behavior. Conversely, in vivo eCB-LTD induction at the same synapses elicited a long-lasting suppression of this behavior. (b) Paired HFS and oPSD induces both LTP and LTD, but only LTP is detected. 1, HFS causes presynaptic release of glutamate, which activates AMPARs. The resultant weak membrane depolarization is insufficient to remove the Mg2+ blockade of NMDARs and thus fails to induce LTP. 2, Optical stimulation of channelrhodopsin expressed on postsynaptic neurons causes strong membrane depolarization (oPSD). This is sufficient to remove Mg2+ blockade of NMDARs, leading to greater Ca2+ influx, activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling pathways, and consequently AMPAR insertion (LTP induction). 3, oPSD also opens voltage-gated Ca2+ channels, causing Ca2+ influx and production of endocannabinoids; these are retrogradely released into the synaptic cleft, where they activate presynaptic CB1Rs. 4, CB1R activation reduces glutamate release (eCB-LTD induction). 5, D2R antagonists are used to blocking D2R-mediated eCB-LTD, which may occur following eHFS ex vivo or oHFS in vivo. 6, Since the magnitude of LTP (~31%) is greater than that of LTD (~13%), only LTP is detected. (c) LTD induction and detection in D1-MSNs. Since LTP induction is blocked by NMDAR antagonists (1) and eCB-LTD is induced (2), only eCB-LTD is detected (3). (d) No LTD is detected in D2-MSNs because LTP is blocked by NMDAR antagonists (1) and eCB-LTD is blocked by D2R antagonists (2).