

Key Points
Under hypoxia, KDM3A, but not IRF4, leads myeloma cells to acquire an antiapoptotic phenotype.
KDM3A regulates a long noncoding RNA, MALAT1, leading to upregulation of glycolytic genes under hypoxia.
Abstract
In multiple myeloma (MM), the bone marrow (BM) microenvironment may contain a myeloma cell fraction that has acquired treatment resistance by undergoing an epigenetic gene expression change. Hypoxic stress is an important factor in the BM microenvironment. Recently, we demonstrated that miR-210 was upregulated in hypoxia and downregulated IRF4, which is known as an essential factor in myeloma oncogenesis in normoxia. In the study, we demonstrated that myeloma cells still showed a strong antiapoptotic phenotype despite IRF4 downregulation, suggesting that another antiapoptotic factor might be involved under hypoxic stress. To determine the factor or factors, we conducted gene expression analysis on myeloma cells (primary samples and cell lines) that were exposed to chronic hypoxia and observed upregulation of glycolytic genes and genes encoding H3K9 demethylases in myeloma cells with hypoxia. Among these, KDM3A was most significantly upregulated in all examined cells, and its knockdown induced apoptosis of myeloma cells in chronic hypoxia. Expression of KDM3A was dependent on HIF-1α, which is a transcription factor specifically upregulated in hypoxia. We further demonstrated that an essential target of KDM3A was a noncoding gene, MALAT1, whose upregulation contributed to acquisition of an antiapoptotic phenotype by accumulation of HIF-1α, leading to upregulation of glycolytic genes under hypoxia. This process was independent from IRF4. These results led us to conclude that the hypoxia-inducible HIF-1α-KDM3A-MALAT1 axis also contributes to acquisition of the antiapoptotic phenotype via upregulation of glycolysis-promoting genes. Thus, this axis is a promising therapeutic target against myeloma cells in the BM microenvironment.
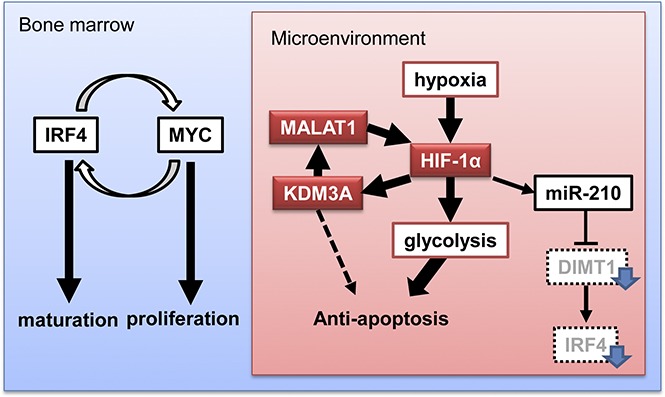
Visual Abstract
Introduction
Multiple myeloma (MM) is a hematological malignancy for which novel drugs have been developed and applied, leading to a dramatic improvement in the overall survival rate.1 However, the tumor entity remains an incurable disease, and therefore, molecular-based studies aimed at establishing a radical new treatment strategy for MM are required. Recently, many studies have demonstrated that heterogeneity of MM can result in treatment resistance in different circumstances. In particular, epigenetic alterations of tumor cells in the microenvironment are known to be strongly associated with treatment resistance.2
Myeloma cells showing treatment resistance can exist in the bone marrow (BM) microenvironment. BM niches are composed of cellular components (blood cells, vascular endothelial cells, osteoblasts, osteoclasts, and stromal cells), soluble factors (cytokines and chemokines), and noncellular factors such as hypoxia.3 A number of studies have investigated myeloma oncogenesis, with a focus on the cellular compartments. For example, Kikuchi et al.4 demonstrated that myeloma-stromal cells play a crucial role in the maintenance of myeloma progenitor cells. Umezu et al.5 reported that stromal cells that are activated by myeloma-producing exosomes promote angiogenesis, leading to myeloma development via a positive feedback mechanism. Thus, these extracellular environments contribute to myeloma progenitor cell survival in BM niches.
Several groups, including our group, have studied the noncellular components of myeloma oncogenesis, focusing particularly on hypoxia.6-9 These reports are based on the following concept: Gene expression in myeloma cells in the BM microenvironment is distinct from that in the other BM regions. The oxygen pressure of BM is lower than that in the peripheral blood, but higher than that in the BM microenvironment, which may result in different gene expression profiles between BM and the BM microenvironment. Indeed, hypoxia causes myeloma cells to acquire different phenotypes when compared with normoxia.6-9 Hypoxia can suppress myeloma cell proliferation, inhibit differentiation, and cause resistance to apoptosis. Although a variety of gene expression changes occur in cancer cells during hypoxia, there may be master regulators that are common to all cancer subtypes; namely, hypoxia-inducible factor-1α (HIF-1α) and HIF-2α.10-14 These transcription factors regulate a variety of downstream target genes, including those involved in cell cycle arrest, angiogenesis, glycolysis, and glucose transport induction. In addition, HIFs might play a crucial role in MM pathogenesis.15 It has been shown that through activation of HIF-1α, hypoxia induces activation of the glycolytic pathway and inactivation of the tricarboxylic acid cycle in myeloma cells via regulation of many related genes.8
IRF4 is the most important oncogenic factor involved in the antiapoptotic phenotype in MM, and it plays a crucial role in cell proliferation and differentiation into plasma cells.16 Recently, we reported that IRF4 expression is decreased in hypoxia-subjected myeloma cells.9 In the present study, we demonstrated that hypoxia-inducible miR-210 upregulation suppressed IRF4, resulting in proliferation and maturation inhibition.9 We further demonstrated that, despite IRF4 downregulation in hypoxia, myeloma cells still exhibited an antiapoptotic phenotype under chronic hypoxia. This suggests that the IRF4-independent axis may contribute to MM acquisition of antiapoptotic phenotypes during hypoxia.
In the BM microenvironment, various epigenetic alterations that affect treatment resistance might occur in myeloma cells. To achieve full remission of MM, it will be necessary to target specific genes and products that are highly expressed in the BM microenvironment. Thus, in the current study, we sought to determine the most accumulated epigenome factor that contributes to the antiapoptotic phenotype during the hypoxic stress response in myeloma cells.
Materials and methods
Primary MM samples
The study included 15 cases of primary multiple myeloma from Akita University Hospital. Samples were collected under a protocol approved by the Institutional Review Boards of Akita University (no.1313). This study was conducted with written informed consent of the study participants and the approval of these Institutional Review Boards, according to the Declaration of Helsinki, before collection of the specimens. Information about the patient samples is summarized in another published paper.9 Primary MM samples were subjected to hypoxia for 48 hours, and then CD38++ cells were sorted and total RNA was subsequently collected.
Myeloma cell lines
We used 8 well-known MM cell lines with various molecular subtypes: RPMI-8226 (8226), KMS-12-BM (KMS12BM), KMS-11 (KMS11), U266, JJN3, H929, Amo1, and MM.1S. These cell lines were purchased from American Type Culture Collection.
DNA microarray
We analyzed gene expression using an Agilent DNA microarray (Agilent, Santa Clara, CA). The array scanner was an Agilent G2600A SureScan Microarray Scanner System. Experiment protocol was according to Agilent Protocol Ver. 6.7. Data were analyzed by GeneSpring (Agilent), computed normalized signal intensity with 75%-tile normalization. Data were uploaded at GSE80140 (cell lines under normoxia or hypoxia), GSE80545 (patient samples under normoxia or hypoxia), and GSE96858 (normal or scrambled or shKDM3A for U266 under normoxia or hypoxia) in the NCBI database.
Quantitative reverse transcription-polymerase chain reaction analysis
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed by the TaqMan method (Applied Biosystems, Foster City, CA). TaqMan probes of GAPDH (Hs02758991_g1), KDM3A (Hs00218331_m1), IRF4 (Hs02559152_s1), PFKFB3 (Hs00998700_m1), PFKFB4 (Hs00190096_m1), SLC2A1 (Hs00892681_m1), HIF1A (Hs00153153_m1), EPAS1 (Hs01026149_m1), and MALAT1 (Hs00273907_s1) were purchased from Applied Biosystems. Expression levels were separately normalized with GAPDH, and the relative expression level of specific mRNA was presented by 2−ΔΔCt or 2−ΔCt. q-RT-PCR was performed using Light Cycler Nano (Roche, Basel, Switzerland) with the TaqMan method. Total RNA was extracted using TRIzol (Life Technologies, Palo Alto, CA). Reverse transcription was performed using a Transcriptor First Strand cDNA Synthesis Kit (Roche).
Western blot analysis
Western blot analysis was performed according to manufacturer’s protocol. Antibodies of KDM3A (ab107235) and KDM7A (ab36044) were purchased from Abcam (Cambridge, United Kingdom). Antibodies of IRF4 (4964), HIF-1α (3716S), Caspase 3 (9662P), Caspase 9 (9508P), Bim (2819), and PARP (9542P) were purchased from Cell Signaling Technology (Danvers, MA). Tubulin (MS-581-P0) was from NeoMarkers (Fremont, CA).
Transient siRNA transfection
We purchased the following Silencer Select siRNA from Applied Biosystems: siHIF1A (s6539), siEPAS1 (s4698), siKDM3A#1 (s224392), siKDM3A#2 (s224393), and siMALAT1 (n511399). Information of siRNA sequence is listed in supplemental Table 1. Negative control siRNA (scrambled siRNA) was designed and synthesized by Nippon Gene (Toyama, Japan). The transfection of siRNA was used by the Nucleofector II and the Cell Line Nucleofector Kit V (VCA-1003; Amaxa, Koeln, Germany) according to the manufacturer's protocol. Briefly, the cells were resuspended in the nucleofector V solution. One hundred microliters of cell suspension at a density of 1×107/mL mixed with a total of 1.0 μg siRNA were transferred to a cuvette and nucleofected with an Amaxa Nucleofector II apparatus using “S-020” program for MM.1S, “G-015” program for RPMI-8226, “T-001” program for KMS-12-BM, “G-015” program for KMS-11, and “X-005” program for U266.
Stable shKDM3A constructs and lentivirus infection
We purchased 4 shKDM3A pGFP-C-shLenti Lentiviral plasmids (TL303857) and a control plasmid from OriGene (Rockville, MD). Together with 9.0 µg ViraPower Packaging mix (Invitrogen, Carlsbad, CA), 3.0 µg of vectors was transfected using Lipofectamine 3000 (Invitrogen) into 293FT producer cells. After overnight culture, medium was exchanged to remove transfection reagents. The following day, viral supernatants were harvested 48 to 72 hours after transfection. Then, 1×106 MM cells were prepared with medium changed, and medium-contained virus was added. After 3-day cultures, cells stably expressing siRNAs were sorted for GFP expression using a MoFlo (Agilent). Information of shRNA sequences are listed in supplemental Table 1.
Cell cycle analysis
The cells were suspended in a mixture containing 0.2 mL of 0.9% NaCl and 3.0 mL of 70% EtOH, after which the nuclei were stained with propidium iodide (Sigma-Aldrich Co, St. Louis, MO). The cellular DNA content was then measured using a FACS Canto flow cytometer running the FACS Diva program (BD Biosciences, San Jose, CA).
Statistical analysis
Data were analyzed by either Student t test or paired t test. Bars represent mean ± 95% confidence interval (CI) of 3 independent experiments.
Results
Chronic hypoxia activates genes encoding glycolytic enzymes and H3K9 demethylase in MM
The oxygen pressure in microenvironments (endosteal or vascular niches) is thought to be less than 10 mm Hg.15 We therefore used 1% O2, which equates to 7.6 mm Hg, in our in vitro experiments to mimic the microenvironment. We examined myeloma cell lines under chronic hypoxia (1% O2 for 48 hours) and observed phenotypic changes, such as growth inhibition accompanied by an increase in the proportion of G1-arrested cells, acidification of the medium resulting from accelerated glycolysis, increased drug resistance, and a decrease in CD138 expression.9 To screen for genes whose expression increases with chronic hypoxia in myeloma and to identify IRF4-independent antiapoptosis factors, we conducted a gene expression analysis of cell lines (n = 5) and patient samples (n = 4). Samples were cultured under normoxia and chronic hypoxia (1% O2, 48 hours). A gene set enrichment analysis showed that glycolytic genes and HIF-1α-regulated genes were significantly activated after exposure to hypoxia (supplemental Figure 1A). Glycolysis is thought to play an important role in the survival of myeloma cells.8 To address this in our system, we performed an XTT assay with normoxia and hypoxia in the MM cell line, U266, using the glycolytic inhibitor 3-bromopyruvate.17 We found that inhibition of glycolysis led to an increase in cell death under hypoxic conditions (supplemental Figure 1B-C), suggesting that glycolysis becomes more important during hypoxia in MM cells. We identified genes whose expression increased more than 2-fold in the examined specimens and performed a gene ontology analysis (Figure 1A). The fluctuating gene ontology consisted of many genes related to the glycolytic pathway, dioxygenase activity, and H3K9 demethylases. Both KDM3A and KDM7A were categorized to H3K9 demethylases and dioxygenase activity, and these genes showed more than a 2-fold increase under hypoxic conditions in all cell lines and patient specimens (Figure 1B; supplemental Table 2). We examined the expression of H3K9 methylating enzymes and demethylating enzymes and found that members of the H3K9 methylating enzyme group, including SUV39H1, SUV39H2, and EHMT2 (G9a), showed a decreasing trend during hypoxia (Figure 1C). In particular, SUV39H2 was included in the group of genes whose expression decreased to a fold change <0.5 in all patient samples, together with genes related to the cell cycle (supplemental Figure 2A-B; supplemental Table 3). In contrast, genes in the H3K9 demethylating enzyme group, including KDM3A and KDM7A, showed increased expression (Figure 1C). Although it is known how glycolytic genes function in the hypoxic environment of myeloma, this has yet to be clarified for H3K9 demethylases. Therefore, we sought to determine how these demethylating enzymes are associated with myeloma oncogenesis during hypoxia.
Figure 1.

Glycolysis-associated genes and H3K9 histone demethylases are upregulated under hypoxia in MM. (A) Gene ontology analysis of hypoxia-inducible genes (normoxia vs hypoxia expression, fold change >2.0) of 5 cell lines (RPMI-8226, KMS-11, KMS-12-BM, MM.1S, and U266) and samples from patients MM (n = 4) cultured in normoxia or chronic hypoxia (1% O2) for 48 hours. (B) Commonly upregulated genes (fold change >2.0) in both cell lines and primary samples. (Upper) Diagram showing number of upregulated genes. (Lower) Gene symbols of upregulated genes. Red genes: H3K9 demethylases. (C) Quantitative gene expression change of H3K9 methylases and demethylases under chronic hypoxia. Y-axis: log2-scale FC (fold change). Pt, primary sample.
Upregulation of H3K9 demethylase KDM3A is regulated by HIF-1α during chronic hypoxia
It is known that expression of H3K9 demethylating enzymes KDM3A and KDM7A is induced under stress conditions: hypoxic stress mainly induces KDM3A, and serum starvation mainly induces KDM7A.18-20 We performed a western blot analysis of KDM3A and KDM7A expression in normoxia- and hypoxia-subjected myeloma cell lines and found that expression of KDM3A was more strongly increased than that of KDM7A under hypoxia (Figure 2A-B). The increase in KDM3A expression was time-dependent, but that of KDM7A was not (Figure 2B). These results suggested that KDM3A has a more potent function in chronic hypoxia than KDM7A does. The increase in KDM3A expression during chronic hypoxia was confirmed by qRT-PCR, using all myeloma cell lines (n = 8) and patient specimens (n = 15) (Figure 2C-D).
Figure 2.

KDM3A is upregulated via HIF-1α activation in hypoxia-stressed MM. (A) Western blot analysis of KDM3A and KDM7A for indicated cell lines cultured in normoxia or hypoxia (1% O2) for 48 hours. H, hypoxia; N, normoxia. (B) Western blot analysis of KDM3A and KDM7A in U266 cell line cultured in hypoxia (1% O2) for 0, 3, 6, 12, 24, and 48 hours. (C) qRT-PCR of KDM3A for indicated cell lines cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. ***P < .001. Student t test was used to test for significance. (D) qRT-PCR of KDM3A for primary myeloma samples (n = 15) cultured in normoxia or hypoxia (1% O2) for 48 hours. ***P < .001. Student t test was used to test for significance. (E) qRT-PCR of KDM3A for KMS-11 and U266 cell lines transiently transduced with siHIF1A and/or siHIF2A and control scrambled siRNA (Scr) and cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. **.001 ≤ P < .01. Student t test was used to test for significance. (F) Western blot analysis of KDM3A for KMS-11 and U266 cell lines transiently transduced with siHIF1A and/or siHIF2A and control scrambled siRNA and cultured in normoxia or hypoxia (1% O2) for 48 hours. NS, not significant.
It is known that KDM3A has a hypoxia response element within its promoter, and induction of KDM3A by hypoxia is HIF-dependent.21-24 Therefore, to determine the relationship between HIFs (HIF-1α and HIF-2α) and KDM3A, we knocked down HIF-1α or HIF-2α in U266 and KMS-11 cell lines. We confirmed that HIF-1α and HIF-2α expression was reduced to at least half of their normal levels (supplemental Figure 3). Knockdown of HIF-1α, but not HIF-2α, suppressed the expression of KDM3A (Figure 2E-F) and glycolytic genes, such as SLC2A1 (GLUT1; supplemental Figure 3). These results suggested that the increase in KDM3A expression during hypoxia is caused by activation of HIF-1α, but not HIF-2α. It has been shown that HIF-1α can bind to the KDM3A promoter region in the human breast cancer cell line MCF7.23 In the current study, we also found that HIF-1α binds to KDM3A in KMS-11 cells (supplemental Figure 4A), suggesting that hypoxia-induced HIF-1α activation can induce KDM3A expression in myeloma cells. We also examined this relationship in the clinical specimen datasets (GSE2113 and GSE6477)25,26 and found a significant positive correlation in GSE2113 (P < .001) and a positive correlation trend in GSE6477 (P = .07) for KDM3A and HIF1A (supplemental Figure 4B). Furthermore, we investigated whether any stimulation from the microenvironment other than hypoxia affects the expression of KDM3A. However, serum starvation, IL-6, and coculture with the stromal cell line HS-5 did not notably affect the expression of KDM3A (supplemental Figure 4C-D). Although it has been reported that KDM3A, an H3K9 demethylase containing a jumonji domain, plays an important oncogenic role in cancer,27-29 the exact role of KDM3A in the hypoxic stress response of myeloma has not been reported. We therefore sought to investigate the role of KDM3A in hypoxia.
Knockdown of KDM3A induces apoptosis in myeloma cells under hypoxia
To analyze the function of KDM3A in myeloma cells, we transduced a GFP-scramble control and 4 GFP-shKDM3A into the U266 cell line, using a lentiviral vector. The resulting transfectants were then sorted using GFP markers to obtain stable transfectants expressing each shRNA. Of the 4 stable knockdown transfectants, qRT-PCR revealed that “shKDM3A#C” showed the strongest suppression of KDM3A (Figure 3A). Western blot data for KDM3A protein expression supported the qRT-PCR data (Figure 3B). Changes in expression were also examined under hypoxic conditions to identify a reduction compared with the hypoxia-subjected GFP-scramble cells (Figure 3C-D). In addition, because KDM3A has been reported to positively regulate IRF4,29 we examined IRF4 expression. Although IRF4 expression is reduced by hypoxia, its expression was not affected by shKDM3A in any of the myeloma cell lines (U266, RPMI-8226, and KMS-11; Figure 3D; supplemental Figure 5A). There was no effect of siKDM3A knockdown on IRF4 expression in 5 additional myeloma cell lines that we examined (supplemental Figure 5B). These results suggest that KDM3A does not regulate IRF4 in either normoxia or hypoxia.
Figure 3.

Knockdown of KDM3A leads myeloma cells to induce apoptosis under chronic hypoxia in MM. (A) qRT-PCR of KDM3A for U266 cell line stably transduced with shKDM3A#A, #B, #C, #D, and control scrambled shRNA (Scr). Bars represent mean ± 95% CI of 3 independent experiments. ***P < .001. Student t test was used to test for significance. (B) Western blot analysis of KDM3A for U266 cell line stably transduced with shKDM3A#A, #B, #C, #D, and control scrambled shRNA (Scr). (C) qRT-PCR of KDM3A for U266 cell line stably transduced with shKDM3A#C, #D, and control Scr and cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. ***P < .001. Student t test was used to test for significance. (D) Western blot analysis of KDM3A and IRF4 for U266 cell line stably transduced with shKDM3A#C, #D, and control Scr and cultured in normoxia or hypoxia (1% O2) for 48 hours. H, hypoxia; N, normoxia. (E) Cell count of U266 cells stably transduced with shKDM3A#C or control Scr and cultured in normoxia or hypoxia (1% O2; 24, 48, 72, and 96 hours). Cells (0.5 × 106) were cultured at 0 hours. Bars represent mean ± 95% CI of 3 independent experiments. **.001 ≤ P < .01. Student t test was used to test for significance. (F) Cell cycle analysis of U266 cells stably transduced with shKDM3A#C or control Scr and cultured in normoxia or hypoxia (1% O2; 48, 72, and 96 hours). X-axis: intensity of propidium iodide (PI). Y-axis, cell count. (G) SubG1 cells (%) of U266 cell line stably transduced with shKDM3A#C or control Scr and cultured in normoxia or hypoxia (1% O2; 48, 72, and 96 hours). Bars represent mean ± 95% CI of 3 independent experiments. **.001 ≤ P < .01; ***P < .001. Student t test was used to test for significance. (H) western blot analysis of PARP, cleaved PARP, Caspase 3, cleaved Caspase 3, Caspase 9, cleaved Caspase 9, and Bim for U266 cells stably transduced with shKDM3A#C or control scrambled shRNA (Scr) and cultured in hypoxia (1% O2; 0, 48, 72, 96 hours).
Next, we used the stable transfectant shKDM3A#C cells to investigate whether shKDM3A causes different hypoxic stress susceptibility in vitro. We found that although there was no significant difference in the proliferation of GFP-scramble control-transduced and shKDM3A-transduced cells in normoxia, proliferation was significantly inhibited in shKDM3A-transduced U266 cells in hypoxia (Figure 3E). To determine whether this proliferation inhibition was caused by cell death, cell cycle arrest, or both, we conducted a cell cycle assay. SubG1 was significantly increased after shKDM3A transduction compared with that of the GFP-scramble control in hypoxia, although there was no significant difference in cell cycle phases (G1, S, G2/M) (Figure 3F-G). Thus, it seems likely that the inhibition of cell proliferation was not a result of cell cycle arrest, but possibly a result of the induction of apoptosis. As expected, shKDM3A-transduced cells showed an increase in the expression of cleaved Caspase 3, cleaved Caspase 9, cleaved PARP, and Bim after hypoxic exposure compared with that in GFP-control-transduced cells (Figure 3H). Moreover, we performed an apoptosis assay on KDM3A knockdown U266 cells (shKDM3A#C and #D) and found that hypoxia increased the number of Annexin-V-positive cells in the KDM3A knockdown U266 cells (supplemental Figure 6A). Apoptosis during hypoxia was partially inhibited by the addition of the caspase inhibitor Z-VAD (supplemental Figure 6B). Furthermore, we showed that shKDM3A#C-transduced RPMI-8226 cells also showed an increase in apoptosis during hypoxia (supplemental Figure 6C). These results indicated that apoptotic cell death is induced in KDM3A knockdown myeloma cells during hypoxia. However, no difference was observed between the behavior of control U266 and KDM3A knockdown U266 (shKDM3A#C) cells in a xenograft mouse model using immunodeficient mice (supplemental Figure 6D). Together, these results suggested that upregulation of KDM3A contributes to myeloma cell survival in hypoxic circumstances, such as in BM niches.
A KDM3A-MALAT1 positive feedback loop contributes to a continuous HIF-1α-glycolysis axis
IRF4 has been identified as a KDM3A target gene in myeloma cells.29 However, our data suggested that IRF4 is not an essential target of KDM3A, at least in hypoxia, for the following reasons. First, although KDM3A expression was very weak in normoxia, but markedly increased in hypoxia, IRF4 expression was suppressed by hypoxia. Second, shKDM3A and siKDM3A did not affect IRF4 expression during either normoxia or hypoxia (Figure 3D; supplemental Figure 5). This led us to investigate the expression of other targets of KDM3A during hypoxia. We performed a comprehensive gene expression analysis on the shKDM3A-transduced U266 cell line and identified 290 genes that showed increased expression in hypoxia and were downregulated by shKDM3A in hypoxia (Figure 4A). These genes included those already known to act as oncogenes in myeloma, such as IGF1 and MAF (supplemental Table 5). Of the remaining candidate genes, the long noncoding RNA MALAT1 was of particular interest because it has been reported to be a poor prognostic factor in myeloma30,31 and is involved in various antiapoptotic pathways in myeloma oncogenesis.32 As expected, MALAT1 was upregulated during hypoxia in all 8 myeloma cell lines (Figure 4B). In the patient specimens, 11 of 15 showed an increase in MALAT1 expression during hypoxia (supplemental Figure 7A). The transient transduction of siKDM3A in hypoxia caused a more significant reduction in MALAT1 expression than in normoxia (Figure 4C).
Figure 4.

KDM3A upregulates MALAT1, which contributes to glycolytic activation via HIF-1α stabilization under hypoxia in myeloma cells. (A) Number of probes under indicated conditions: blue area shows region of genes upregulated under hypoxia and regulated by KDM3A only under hypoxia (cell line: U266, data uploaded at GSE96858). (B) qRT-PCR of MALAT1 for indicated cell lines cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. **.001 ≤ P < .01; ***P < .001. Student t test was used to test for significance. (C) qRT-PCR of MALAT1 for indicated cell lines transiently transduced with siKDM3A#1, #2, and control scrambled siRNA (Scr) and cultured in normoxia or hypoxia (1% O2) for 72 hours. Bars represent mean ± 95% CI of 3 independent experiments. *.01 ≤ P < .05; **.001 ≤ P < .01; ***P < .001. Student t test was used to test for significance. (D) qRT-PCR of MALAT1 for KMS-11 cell line transiently transduced with siMALAT1 or control Scr and cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. ***P < .001. Student t test was used to test for significance. (E) Western blot analysis of HIF-1α for KMS-11 cell line transiently transduced with siMALAT1 or control Scr and cultured in normoxia or hypoxia (1% O2) for 48 hours. (F) qRT-PCR of PFKFB3, PFKFB4, and SLC2A1 for KMS-11 cell line transiently transduced with siMALAT1 or control Scr and cultured in normoxia or hypoxia (1% O2) for 48 hours. Bars represent mean ± 95% CI of 3 independent experiments. **.001 ≤ P < .01; ***P < .001. Student t test was used to test for significance.
Next, we sought to determine whether KDM3A can bind to the promoter region of MALAT1 in the myeloma cell line KMS-11, using ChIP-PCR (supplemental Figure 7B). We found that siKDM3A reduced H3K9me2 levels of MALAT1, as previously reported (supplemental Figure 7C).33 These results suggested that, in myeloma cells, KDM3A may promote MALAT1 transcription via histone demethylation at the MALAT1 promoter. The expression of MALAT1 was not decreased by either siHIF1A or siHIF2A, but rather was increased by siHIF1A under hypoxia (supplemental Figure 7D). This result suggested that KDM3A, but not HIF-1α, directly controls MALAT1 in myeloma cells under hypoxic conditions.
Finally, we investigated the targets of MALAT1 in hypoxia. Because hypoxia-exposed myeloma cell lines exhibit an elevation of glycolytic enzymes (Figure 1A; supplemental Figure 1A), we investigated changes in the expression of glycolytic enzymes after knockdown of MALAT1. siMALAT1 transduction decreased the expression of MALAT1, even during hypoxia (Figure 4D). Moreover, we found that under hypoxic conditions, siMALAT1 transduction inhibited the accumulation of HIF-1α and significantly inhibited the glycolytic genes PFKFB3, PFKFB4, and SLC2A1 (GLUT1), which are thought to be regulated by HIF-1α in myeloma cells (Figure 4E-F).8 Suppressed proliferation and increased apoptosis was observed in KMS-11 cells after siMALAT1 transduction under hypoxic conditions (supplemental Figure 7E-F). These results strongly suggested that hypoxia-inducible KDM3A enhances the expression of glycolytic genes through the control of MALAT1 in MM, and that the HIF-1α-KDM3A-MALAT1 axis is essential for the antiapoptotic properties of MM in the BM microenvironment (Figure 5).
Figure 5.

Schematic diagram: the HIF-1α/KDM3A/MALAT1 positive feedback loop has an oncogenic role under hypoxia in MM. HIF-1α upregulates KDM3A as well as glycolytic genes under hypoxia. This histone modulator maintenance of the expression of MALAT1 long noncoding RNA leads to accumulation of HIF-1α. This HIF-1α-KDM3A-MALAT1 positive feedback loop contributes to survival of MM cells under hypoxia via enhancing genes controlling the glycolysis pathway.
Discussion
There have been a number of reports on the role of KDM3A in oncogenesis. For example, Osawa et al.19 reported that KDM3A is induced by hypoxia or by starvation, leading to the production of angiogenic factors by tumor cells and promotion of macrophage infiltration into tumors. It has also been reported that in solid tumors (such as breast cancer and rectal cancer), high KDM3A expression is associated with a poor prognosis.27,28 As described earlier, KDM3A serves as an oncogenic factor in hypoxia-subjected tumors. Although KDM3A is known to be induced by hypoxia in a variety of cancers, no association has been reported between hypoxia and KDM3A in MM.
KDM3A is an H3K9 demethylating enzyme containing a jumonji domain. Demethylated H3K9 is known to increase transcriptional activity,34 and several transcriptional targets of KDM3A in cancer have been identified.24,29,35 However, most of the “hypoxia-specific” transcriptional targets of KDM3A are unknown in myeloma oncogenesis. Our data suggested that MALAT1 could be a target of KDM3A in myeloma oncogenesis under hypoxia. MALAT1 (long noncoding RNA) is a noncoding gene that plays an important oncogenic role in various cancers.36,37 It is a poor prognostic factor in myeloma oncogenesis, and its expression increases with disease progression.30-32 It is known that KDM3A promotes MALAT1 transcription by demethylating histones at its promoter region in neuroblastoma, and its upregulation could contribute to metastasis.33 Furthermore, it has been shown that KDM3A-MALAT1-MAPK signaling contributes to cell proliferation in gastric cancer.38 Recently, Luo et al.39 reported that MALAT1 induces glycolytic enzymes by inhibiting degradation of HIF-1α. Glycolytic regulatory genes have also been identified as promising therapeutic targets for myeloma.8 These reports support our idea that MALAT1 could maintain and promote its own expression through KDM3A-mediated positive feedback, leading to the promotion of glycolytic gene expression (Figure 5). KDM3A-MALAT1-targeting therapy can target myeloma cells, which are present in the BM niches. However, our experiments showed that the knockdown efficiency of MALAT1 by siRNA was worse than that of HIF1A and KDM3A. One possible reason for this is that mRNA localized in the nucleus, such as that of MALAT1, may not be susceptible to RNA interference by siRNA.40 Further functional analysis of MALAT1 in MM will be the subject of future studies.
Our findings suggested that KDM3A behaves in an oncogene-like manner in hypoxic niches and that it protects itself from extracellular stimulation from the BM microenvironment. It has been reported that myeloma cells change their surface antigens and metabolic pathways in response to hypoxic stress, and they also acquire drug resistance.7,8 In addition, we recently reported that miR-210, whose expression is markedly elevated in a hypoxic environment, downregulates IRF4 through the direct regulation of an 18S rRNA base methyltransferase, DIMT1.9 These findings led us to conclude that miR-210 suppresses IRF4 expression under chronic hypoxia and prevents cell proliferation and cell maturation. Expression of IRF4 is suppressed in hypoxic environments,7,9 although IRF4 is a key factor in promoting myeloma oncogenesis.16 Therefore, it is likely that during hypoxia, factors that contribute to myeloma cell survival may act independent of IRF4.
Oxygen pressure is different between the hypoxia niche and other regions, including in the bone marrow.15 Here, we propose that a myeloma cell exists in the hypoxia niche as a “hypoxia niche myeloma cell” (HNMC), and in other bone marrow regions as a “bone marrow myeloma cell” (BMMC; supplemental Figure 8). Different oxygen pressure against HNMCs and BMMCs may expose myeloma cells to different kinds of apoptosis-inducible stress. BMMCs might be exposed to endoplasmic reticulum stress via excessive synthesis of proteins, such as immunoglobulin. In such cases, proteasome inhibitors such as bortezomib and carfilzomib might be effective. However, proteasome inhibitors may not be effective for myeloma cells subjected to lower endoplasmic reticulum stress,41 such as HNMCs. Moreover, IRF4 is highly expressed in BMMCs,9 leading to an antiapoptotic phenotype in myeloma cells. Therefore, immunomodulatory drugs, such as lenalidomide and pomalidomide, which target the Ikaros-IRF4 pathway, would be effective for these cells,42-45 but not for myeloma cells that express lower levels of IRF4, such as HNMCs. Accordingly, to achieve full remission, treatments that target different molecules mainly present in BMMCs and HNMCs will be necessary. Our findings provide reasons to use a combination of proteasome inhibitors and/or immunomodulatory drugs in addition to hypoxia-targeting drugs.46
In conclusion, we have shown that KDM3A strongly correlates with HIF-1α dependence in myeloma cells after hypoxic exposure, contributing to the survival of hypoxia-subjected myeloma cells. KDM3A contributes to the maintenance of MALAT1 expression, which may lead to continuous HIF-1α expression during hypoxia. These results identified KDM3A as a potential therapeutic target for myeloma cells adapted to the BM microenvironment. Further studies aimed at identifying the precise functional mechanism of KDM3A in myeloma oncogenesis will be required, and the development of a KDM3A inhibitor would also be desirable.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank H. Kataho, Y. Chiba, and Y. Abe for their outstanding technical assistance. The authors also thank Y. Michishita and Y. Hatano (Noshiro Kousei Medical Center, Noshiro, Japan), H. Saitoh (Akita Red Cross Hospital, Akita, Japan), K. Teshima, H. Ohyagi, M. Kume (Hiraka General Hospital, Yokote, Japan), Y. Kawabata, and A. Kitabayashi (Akita Kousei Medical Center, Akita, Japan) for collection of primary MM samples. The authors thank S. Yamamoto and Y. Fujita (Daiichi-Sankyo, Tokyo, Japan) for constructive discussion.
This study was supported by a Take a New Challenge for Drug Discovery program (Daiichi-Sankyo).
Authorship
Contribution: S.I., A.K., F.A., and N.T. performed experiments and analyzed data; and S.I. and H.T. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: H.T. and S.I. received research support from Daiichi-Sankyo. The remaining authors declare no competing financial interests.
Correspondence: Hiroyuki Tagawa, Department of Hematology, Nephrology, and Rheumatology, Akita University Graduate School of Medicine, 1-1-1 Hondo, Akita 0108543, Japan; e-mail: htagawa0279jp@yahoo.co.jp.
References
- 1.Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208. [DOI] [PubMed] [Google Scholar]
- 2.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54(5):716-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawano Y, Moschetta M, Manier S, et al. . Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160-172. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi J, Koyama D, Wada T, et al. . Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J Clin Invest. 2015;125(12):4375-4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azab AK, Hu J, Quang P, et al. . Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782-5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawano Y, Kikukawa Y, Fujiwara S, et al. . Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int J Oncol. 2013;43(6):1809-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maiso P, Huynh D, Moschetta M, et al. . Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015;75(10):2071-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda S, Kitadate A, Abe F, et al. . Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4 oncogenic axis in multiple myeloma. Cancer Sci. 2017;108(4):641-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmeliet P, Dor Y, Herbert JM, et al. . Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485-490. [DOI] [PubMed] [Google Scholar]
- 11.Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001;276(11):7919-7926. [DOI] [PubMed] [Google Scholar]
- 12.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23(1):359-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med. 2004;199(1):113-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Storti P, Bolzoni M, Donofrio G, et al. . Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia. 2013;27(8):1697-1706. [DOI] [PubMed] [Google Scholar]
- 15.Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino AC. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia. 2011;25(10):1533-1542. [DOI] [PubMed] [Google Scholar]
- 16.Shaffer AL, Emre NC, Lamy L, et al. . IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakano A, Miki H, Nakamura S, et al. . Up-regulation of hexokinaseII in myeloma cells: targeting myeloma cells with 3-bromopyruvate. J Bioenerg Biomembr. 2012;44(1):31-38. [DOI] [PubMed] [Google Scholar]
- 18.Osawa T, Muramatsu M, Wang F, et al. . Increased expression of histone demethylase JHDM1D under nutrient starvation suppresses tumor growth via down-regulating angiogenesis. Proc Natl Acad Sci USA. 2011;108(51):20725-20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osawa T, Tsuchida R, Muramatsu M, et al. . Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 2013;73(10):3019-3028. [DOI] [PubMed] [Google Scholar]
- 20.Osawa T, Shibuya M. Targeting cancer cells resistant to hypoxia and nutrient starvation to improve anti-angiogeneic therapy. Cell Cycle. 2013;12(16):2519-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellmann S, Bettkober M, Zelmer A, et al. . Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun. 2008;372(4):892-897. [DOI] [PubMed] [Google Scholar]
- 22.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem. 2008;283(52):36542-36552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollard PJ, Loenarz C, Mole DR, et al. . Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J. 2008;416(3):387-394. [DOI] [PubMed] [Google Scholar]
- 24.Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol. 2010;30(1):344-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattioli M, Agnelli L, Fabris S, et al. . Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene. 2005;24(15):2461-2473. [DOI] [PubMed] [Google Scholar]
- 26.Chng WJ, Kumar S, Vanwier S, et al. . Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67(7):2982-2989. [DOI] [PubMed] [Google Scholar]
- 27.Uemura M, Yamamoto H, Takemasa I, et al. . Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res. 2010;16(18):4636-4646. [DOI] [PubMed] [Google Scholar]
- 28.Wade MA, Jones D, Wilson L, et al. . The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 2015;43(1):196-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohguchi H, Hideshima T, Bhasin MK, et al. . The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat Commun. 2016;7:10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho SF, Chang YC, Chang CS, et al. . MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer. 2014;14(1):809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handa H, Kuroda Y, Kimura K, et al. . Long non-coding RNA MALAT1 is an inducible stress response gene associated with extramedullary spread and poor prognosis of multiple myeloma. Br J Haematol. 2017;179(3):449-460. [DOI] [PubMed] [Google Scholar]
- 32.Ronchetti D, Agnelli L, Taiana E, et al. . Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget. 2016;7(12):14814-14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tee AE, Ling D, Nelson C, et al. . The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. 2014;5(7):1793-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barski A, Cuddapah S, Cui K, et al. . High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823-837. [DOI] [PubMed] [Google Scholar]
- 35.Cho HS, Toyokawa G, Daigo Y, et al. . The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int J Cancer. 2012;131(3):E179-E189. [DOI] [PubMed] [Google Scholar]
- 36.Ji P, Diederichs S, Wang W, et al. . MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031-8041. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimoto R, Mayeda A, Yoshida M, Nakagawa S. MALAT1 long non-coding RNA in cancer. Biochim Biophys Acta. 2016;1859(1):192-199. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Liu Z, Yuan C, et al. . Elevated JMJD1A is a novel predictor for prognosis and a potential therapeutic target for gastric cancer. Int J Clin Exp Pathol. 2015;8(9):11092-11099. [PMC free article] [PubMed] [Google Scholar]
- 39.Luo F, Liu X, Ling M, et al. . The lncRNA MALAT1, acting through HIF-1α stabilization, enhances arsenite-induced glycolysis in human hepatic L-02 cells. Biochim Biophys Acta. 2016;1862(9):1685-1695. [DOI] [PubMed] [Google Scholar]
- 40.Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002;8(7):855-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leung-Hagesteijn C, Erdmann N, Cheung G, et al. . Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu G, Middleton RE, Sun H, et al. . The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krönke J, Udeshi ND, Narla A, et al. . Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gandhi AK, Kang J, Havens CG, et al. . Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol. 2014;164(6):811-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjorklund CC, Lu L, Kang J, et al. . Rate of CRL4(CRBN) substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015;5(10):e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J, Van Valckenborgh E, Xu D, et al. . Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol Cancer Ther. 2013;12(9):1763-1773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.