

Abstract
miRNAs are a class of noncoding RNAs important in posttranscriptional repressors and involved in the regulation of almost every biological process by base paring with target genes through sequence in their seed regions. Genetic variations in the seed regions have vital effects on gene expression, phenotypic variation, and disease susceptibility in humans. The distribution pattern of genetic variation in miRNA seed regions might be related to miRNA function and is worth paying more attention to. We here employed computational analyses to explore the clustering pattern and functional effect of SNPs in human miRNA seed regions. A total of 1879 SNPs were mapped to 1226 human miRNA seed regions. We found that miRNAs with SNPs in their seed region are significantly enriched in miRNA clusters. We also found that SNPs in clustered miRNA seed regions have a lower functional effect than have SNPs in nonclustered miRNA seed regions. Additionally, we found that clustered miRNAs with SNPs in seed regions are involved in more pathways. Overall, our results demonstrate that SNPs in clustered miRNA seed regions can take part in more intricate and complex gene-regulating networks with lower functional cost by functional complementarity. Moreover, our results also broaden current knowledge on the genetic variation in human miRNA seed regions.
1. Introduction
miRNAs are small noncoding RNAs of 20–22 nucleotides (nt) in length in their mature form, processed from a longer hairpin structure, that act as posttranscriptional gene regulators by either mRNA degradation or translational repression [1]. It is estimated that miRNAs regulate more than 30~60% of all protein-coding genes, thus building complex regulatory networks that participate in the control of most biological processes and are related to shaping phenotypic variability and disease development [2, 3]. miRNA-mediated gene regulation depends on perfect matching between the seven nucleotides of its seed region (nucleotides 2 through 8 from the 5′ end of the mature miRNAs) and the target sequence usually located at the 3′ untranslated regions (UTRs) of the regulated genes [4, 5]. With the rapid advance of genome sequencing technologies in recent years, many genetic variants have been identified in miRNA seed regions [6–9]. These genetic variants may disrupt the interactions between miRNAs and their targets or create new targets, rewiring the miRNA regulatory networks and causing diseases. Moreover, previous studies have proven that genetic variations in the seed regions have profound and broad effects on gene expression, phenotypic variation, and disease susceptibility in humans [10–13]. However, there is still little information on the distribution patterns and allele-dependent functional effect of the genetic variations in human miRNA seed regions.
Wang et al. [14] reported that the clustered miRNAs tend to be evolutionarily conserved and miRNAs in the same clusters tend to be coexpressed and regulate overlapping sets of target genes. Therefore, we hypothesized that clustered miRNAs tend to be more tolerant of genetic variations in the seed region due to functional complementary than are those nonclustered miRNAs. In order to test this hypothesis, we investigated the clustering patterns and functional effect of SNPs in human miRNA seed regions in this study. Our results demonstrate that SNPs in clustered miRNA seed regions can take part in more intricate gene-regulating networks with lower functional cost by functional complementarity.
2. Materials and Methods
2.1. Genome-Wide Identification of SNPs in Human miRNA Seed Regions
Genomic coordinates of human mature miRNAs were downloaded and extracted from miRBase version 21 (http://www.mirbase.org/). This release of miRBase comprises 1881 precursor sequences and 2813 mature miRNAs (2588 unique mature miRNA sequences). Genomic locations of miRNA seeds were determined from genomic locations of the 2nd and 8th bases of mature miRNAs. The SNP data for human assemblies GRCh38/hg38 were downloaded from NCBI dbSNP database (version 147, https://www.ncbi.nlm.nih.gov/SNP/). For each miRNA, we collected all SNPs in the seed regions from dbSNP using in-house Perl scripts.
2.2. Computational Predictions of Allele-Dependent miRNA Target and Percent Overlap
The miRNA sequences were downloaded from the miRBase (version 21). To determine the target gene spectrum for the reference and derived allele of the SNPs in the miRNA seed regions, we predicted targets on the human genome assembly (NCBI36/hg18, March 2006) using the online target prediction program, namely, TargetRank (http://hollywood.mit.edu/targetrank) [15]. The reference targets for a SNP were predicted using TargetRank to analyse the miRNA seed sequence carrying the reference allele of the SNP. On the other hand, the derived targets of a SNP were predicted when the derived allele of the SNP was in the miRNA seed sequence. We defined the common targets (overlapping genes) as predicted targets for both the reference and derived alleles of a SNP. Percent overlap between the reference and derived targets for a SNP in miRNA seeds was determined using cosine similarity [16], which is calculated by the total number of overlapping genes divided by the square root of the product of the number of targets of reference and derived alleles of a SNP. Taking the square root of the number of predicted targets reduces the influence of miRNAs with abnormally large numbers of targets and simultaneously normalizes the result, generating a score between 0 and 1.
2.3. Clustering Analysis of miRNAs with SNPs in Their Seed Region
The clustering information of human miRNAs was obtained from Wang et al. [14] to investigate the clustering patterns of the miRNAs with SNPs in their seed region. Specifically, clustering of miRNA genomic locations is determined if two neighboring miRNAs are located within 10 kb and are in the same strand. Based on this criterion, among all the 1881 precursor sequences and 2813 mature miRNAs annotated in humans, 352 miRNA genes including 634 mature miRNAs were grouped into 99 distinct clusters [14]. The significance of the difference in percentage of clustered miRNAs between miRNAs with or without SNPs in seed regions was calculated using the chi-square test. Additionally, a two-tailed Student's t-test was used for comparisons between the percent overlap of the clustered and nonclustered miRNAs with SNPs in seed regions. P value < 0.05 was considered statistically significantly.
2.4. Function and Pathway Analysis
The function annotation and pathway enrichment of the clustered and nonclustered miRNAs with SNPs in seed regions were performed using miRNA Enrichment Analysis and Annotation (miEAA) tool database (http://www.ccb.uni-saarland.de/mieaa_tool/) [17]. miEAA is a web-based system, which offers miRNA set enrichment analysis similar to gene set enrichment analysis (GSEA).
3. Results and Discussion
3.1. miRNAs with SNPs in Their Seed Region Are Significantly Enriched in Clusters
In total, we identified 1879 SNPs in 1226 (43.6%) human miRNA seed regions after mapping genetic variation onto human miRNA seed regions based on the genomic coordinates of SNPs in dbSNP human Build 147 and miRNAs in miRBase release 21 (Table S1). We found that most of the SNPs (1833 SNPs, 97.5%, Table S1) in miRNA seed regions were rare variants (defined as SNPs with minor allele frequency (MAF) < 5%). Recently, Torruella-Loran et al. [18] studied miRNA genetic variation in human populations and found that the seed regions tend to be depleted of high-frequency variants, which is consistent with our finding. We also found that there are 1587, 749, 340, 102, 31, and 4 miRNAs, which carry zero, one, two, three, four, and five SNPs, respectively, in their seed region (Figure S1, Table S1). This indicates that miRNA seed regions might be not so tolerant of genetic variants since most miRNAs have few or rare SNPs in their seed regions.
Further, we investigated the clustering patterns of the miRNAs with SNPs in their seed regions. An interesting observation is that the miRNAs with SNPs in their seed region are significantly enriched in clusters (Figure 1, Table S2). For the 1226 human miRNAs with SNPs in their seed region, 314 (25.6%) of them are located in miRNA clusters, whereas among the 1587 human miRNAs without SNPs in their seed region, only 320 (20.2%) of them are located in miRNA clusters (P = 6.06 × 10−4, χ 2 test) (Table S2). miRNAs from the same cluster have the tendency to regulate the same sets of target genes and cooperatively repress expression levels of such genes [14]. Therefore, clustered miRNAs tend to be more tolerant of genetic variations in the seed region due to functional complementarity than are nonclustered miRNAs.
Figure 1.
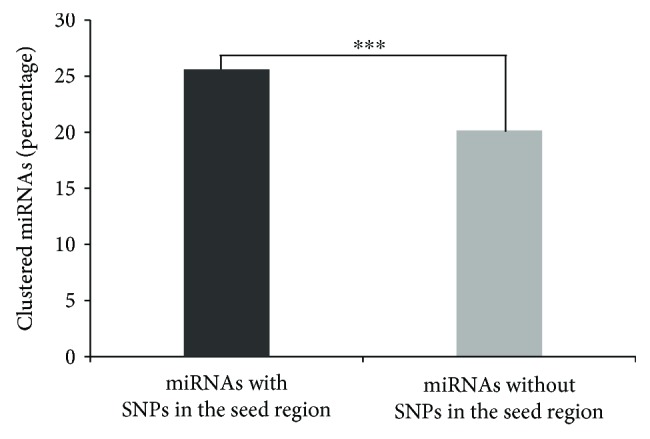
Clustering patterns of the miRNAs with SNPs in their seed region. Percentage of clustered miRNAs in miRNAs with or without variations in their seed region. miRNAs with SNPs in their seed region were significantly enriched in clusters. The y-axis is the percentage of miRNAs that are located in clusters in that group. ∗∗∗ P < 0.001.
3.2. SNPs in Clustered miRNA Seed Regions Have a Lower Functional Effect
We speculated that the functional effect of genetic variation in clustered miRNA seed regions may be relatively low compared with that in nonclustered miRNA seed regions. The functional effect or “cost” of SNPs in miRNA seed regions involves the loss of regulatory control over previously targeted mRNAs and/or the acquisition of novel regulatory control over previously untargeted mRNAs [16].
In order to test this hypothesis, we calculated the percent overlap (cosine similarity) of predicted targets for the reference and derived allele of each SNP by TargetRank. A lower percent overlap indicates large overall differences between the targets of reference and derived allele. The average percent overlap between reference target sets and derived target sets for all the 1879 SNPs is only 15.8% (Table S1). By considering the importance of miRNAs in regulating gene expression, this result suggests that as few as one nucleotide substitution within the seed region of miRNAs will cause a significant functional effect.
Then, we compared the percent overlap of SNPs in clustered miRNA seed regions with that of SNPs in nonclustered miRNA seed regions. We found that the percent overlap of SNPs in clustered miRNA seed regions is much higher than that of SNPs in nonclustered miRNA seed regions (Student's t-test; P < 0.05) (Figure 2). Therefore, it indicates that SNPs in clustered miRNA seed regions will produce a lower functional effect, which may be a result of functional complementarity and relaxed selection or adaptive evolution.
Figure 2.
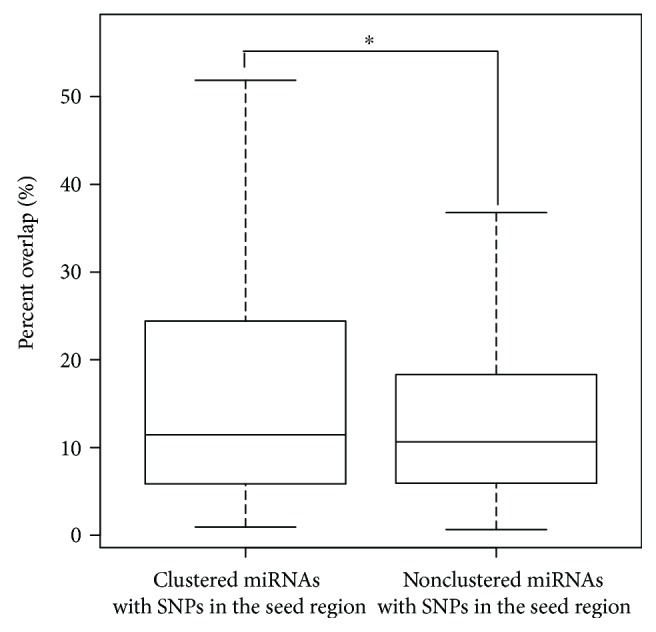
Difference in the functional effect of SNPs in clustered and nonclustered miRNA seed regions. ∗ P < 0.05.
3.3. Clustered miRNAs with SNPs in Seed Regions Are Involved in More Pathways
Additionally, we applied a gene set enrichment analysis to understand on which pathways the clustered and nonclustered miRNAs with SNPs in seed regions participate using miEAA [17]. Table 1 gives most enriched pathway information with P value < 0.01. Clustered miRNAs with SNPs in seed regions are involved in more pathways, such as Alzheimer disease amyloid secretase pathway, and TCR and IL signaling pathways, whereas nonclustered miRNAs with SNPs in seed regions are only enriched in TCR and Id signaling pathways (P < 0.01). From the point of evolution, a gene can allow more variations and obtain new function during evolution through gene duplication [19]. We guess that compared with nonclustered miRNAs, clustered miRNAs with SNPs in seed regions can take part in more intricate and complex regulating networks by obtaining SNPs in seed regions and functional complementarity of the members in the same cluster.
Table 1.
Pathway enrichment analysis of clustered or nonclustered miRNAs with SNPs in seed regions by using miEAA.
| Term | P value |
|---|---|
| Clustered miRNAs with SNPs in seed regions | |
| Alzheimer disease amyloid secretase pathway (P00003) | 0.0010 |
| Selenium (WP15) | 0.0020 |
| TCR signaling pathway (WP69) | 0.0028 |
| IL-3 signaling pathway (WP286) | 0.0031 |
| IL-1 signaling pathway (WP195) | 0.0036 |
| Endochondral ossification (WP474) | 0.0047 |
| IL-5 signaling pathway (WP127) | 0.0056 |
| Natural killer cell-mediated cytotoxicity (hsa04650) | 0.0062 |
| G protein signaling pathways (WP35) | 0.0066 |
| IL-6 signaling pathway (WP364) | 0.0075 |
| IL-2 signaling pathway (WP49) | 0.0078 |
| EPO receptor signaling (WP581) | 0.0081 |
| Ubiquitin proteasome pathway (P00060) | 0.0098 |
| Nonclustered miRNAs with SNPs in seed regions | |
| TCR signaling pathway (WP69) | 0.0074 |
| Id signaling pathway (WP53) | 0.0097 |
4. Conclusions
In this study, we took advantage of microRNA gene location and genetic variability obtained from miRBase 21 and dbSNP database to systematically identify all substitutions located in human miRNA seed regions and explore the clustering pattern and functional effect of these SNPs. In total, we have identified 1879 SNPs in 1226 human miRNA seed regions. We found that miRNAs with SNPs in their seed region are significantly enriched in clusters. We also found that functional cost of genetic variations in clustered miRNA seed regions was relatively low due to functional complementarity compared with that of variations in nonclustered miRNA seed regions. Additionally, we found that clustered miRNAs with SNPs in seed regions are involved in more pathways. Taken together, our results broaden current knowledge on the genetic variation in human miRNA seed regions and demonstrate that SNPs in clustered miRNA seed regions can take part in more intricate and complex networks with lower functional cost by functional complementarity.
Acknowledgments
Funding for this study was provided by the National Science Foundation of China (81601175 and 31601032), the Natural Science Foundation of Guangdong Province (2016A030310401), the Guangdong Medical Research Foundation (A2016539), and the Scientific Research Staring Foundation of Southern Medical University (PY2015N002 and PY2015N006).
Disclosure
The funders had no further role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Conflicts of Interest
All authors have reported no financial interests or potential conflicts of interest regarding the publication of this article.
Supplementary Materials
Figure S1: SNPs in human miRNA seed regions. Number of miRNAs that carry zero, one, two, three, four, and five SNPs, respectively, in their seed region.
{kind=link}
Table S1: genome-wide identification of SNPs in human miRNA seed regions.
Table S2: chi-square test of clustering patterns of miRNAs with SNPs in their seed region.
References
- 1.Krol J., Loedige I., Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature Reviews Genetics. 2010;11(9):597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 2.Siomi H., Siomi M. C. Posttranscriptional regulation of microRNA biogenesis in animals. Molecular Cell. 2010;38(3):323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Filipowicz W., Bhattacharyya S. N., Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Reviews Genetics. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 4.Lewis B. P., Burge C. B., Bartel D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 5.Bartel D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya A., Ziebarth J. D., Cui Y. PolymiRTS database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Research. 2013;42(Database issue):D86–D91. doi: 10.1093/nar/gkt1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong J., Liu C., Liu W., et al. An update of miRNASNP database for better SNP selection by GWAS data, miRNA expression and online tools. Database. 2015;2015, article bav029 doi: 10.1093/database/bav029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zorc M., Obsteter J., Dovc P., Kunej T. Genetic variability of microRNA genes in 15 animal species. Journal of Genomics. 2015;3:51–56. doi: 10.7150/jgen.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhartiya D., Laddha S. V., Mukhopadhyay A., Scaria V. miRvar: a comprehensive database for genomic variations in microRNAs. Human Mutation. 2011;32(6):E2226–E2245. doi: 10.1002/humu.21482. [DOI] [PubMed] [Google Scholar]
- 10.Borel C., Antonarakis S. E. Functional genetic variation of human miRNAs and phenotypic consequences. Mammalian Genome. 2008;19(7-8):503–509. doi: 10.1007/s00335-008-9137-6. [DOI] [PubMed] [Google Scholar]
- 11.Iliff B. W., Riazuddin S. A., Gottsch J. D. A single-base substitution in the seed region of miR-184 causes EDICT syndrome. Investigative Ophthalmology & Visual Science. 2012;53(1):348–353. doi: 10.1167/iovs.11-8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mencía Á., Modamio-Høybjør S., Redshaw N., et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nature Genetics. 2009;41(5):609–613. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharya A., Cui Y. SomamiR 2.0: a database of cancer somatic mutations altering microRNA-ceRNA interactions. Nucleic Acids Research. 2016;44(D1):D1005–D1010. doi: 10.1093/nar/gkv1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y., Luo J., Zhang H., Lu J. microRNAs in the same clusters evolve to coordinately regulate functionally related genes. Molecular Biology and Evolution. 2016;33(9):2232–2247. doi: 10.1093/molbev/msw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen C. B., Shomron N., Sandberg R., Hornstein E., Kitzman J., Burge C. B. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007;13(11):1894–1910. doi: 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill C. G., Jabbari N., Matyunina L. V., McDonald J. F. Functional and evolutionary significance of human MicroRNA seed region mutations. PLoS One. 2014;9(12, article e115241) doi: 10.1371/journal.pone.0115241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backes C., Khaleeq Q. T., Meese E., Keller A. miEAA: microRNA enrichment analysis and annotation. Nucleic Acids Research. 2016;44(W1):W110–W116. doi: 10.1093/nar/gkw345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torruella-Loran I., Laayouni H., Dobon B., et al. MicroRNA genetic variation: from population analysis to functional implications of three allele variants associated with cancer. Human Mutation. 2016;37(10):1060–1073. doi: 10.1002/humu.23045. [DOI] [PubMed] [Google Scholar]
- 19.Pegueroles C., Laurie S., Mar Albà M. Accelerated evolution after gene duplication: a time-dependent process affecting just one copy. Molecular Biology and Evolution. 2013;30(8):1830–1842. doi: 10.1093/molbev/mst083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: SNPs in human miRNA seed regions. Number of miRNAs that carry zero, one, two, three, four, and five SNPs, respectively, in their seed region.
Table S1: genome-wide identification of SNPs in human miRNA seed regions.
Table S2: chi-square test of clustering patterns of miRNAs with SNPs in their seed region.