

Abstract
Motivation
Drug combination therapy for treatment of cancers and other multifactorial diseases has the potential of increasing the therapeutic effect, while reducing the likelihood of drug resistance. In order to reduce time and cost spent in comprehensive screens, methods are needed which can model additive effects of possible drug combinations.
Results
We here show that the transcriptional response to combinatorial drug treatment at promoters, as measured by single molecule CAGE technology, is accurately described by a linear combination of the responses of the individual drugs at a genome wide scale. We also find that the same linear relationship holds for transcription at enhancer elements. We conclude that the described approach is promising for eliciting the transcriptional response to multidrug treatment at promoters and enhancers in an unbiased genome wide way, which may minimize the need for exhaustive combinatorial screens.
Availability and implementation
The CAGE sequence data used in this study is available in the DDBJ Sequence Read Archive (http://trace.ddbj.nig.ac.jp/index_e.html), accession number DRP001113.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Unlike single-gene disorders, which are regulated by a particular gene, complex diseases like cancer are dependent on multiple cellular functions and processes. Single drug cancer therapies fail to target more than one signalling pathway and increase the chance of developing drug resistance (Hopkins, 2008). Drug combinations are considered a promising strategy to overcome these limitations (Yang et al., 2008). Combinatorial drug treatment offers significant advantages over high-dosage monotherapy, including amplified therapeutic efficacy and reduced risk of drug resistance (Zimmermann et al., 2007).
Many experimental and computational methods have been proposed to identify effective drug combinations. High-throughput (Mathews Griner et al., 2014) and RNA interference (Prahallad et al., 2012) screenings are unbiased experimental strategies that can indicate favorable combinations of FDA approved compounds. On account of the high cost and large combinatorial space, numerous computational approaches have been developed to reduce the need for exhaustive screens (Bansal et al., 2014; Sun et al., 2015). Despite recent advances on prioritizing multidrug therapies for further experimental validation, no existing methodology has been demonstrated to quantify and model the combinatorial drug effects at the transcriptional level.
Most earlier studies looking at the transcriptional response to multidrug therapy use microarrays (Jin et al., 2011; Lee et al., 2012). We recently employed CAGE, a sequencing-based unbiased approach for quantifying promoter expression, after single drug treatment (Kajiyama et al., 2013), and were able to capture moderate cellular responses in the transcriptome even at submaximal drug dosages, which is very difficult to attain with microarray approaches (Kajiyama et al., 2013). Apart from giving promoter based resolution, CAGE has the additional advantage of detecting enhancer expression in the same experiment (Andersson et al., 2014). Transcribed enhancers as detected by CAGE are more likely to be functional than enhancer predictions based on chromatin status (Andersson et al., 2014). With CAGE, it is possible to not only examine whether drug treatment alters enhancer RNA transcription, but also investigate if enhancer expression levels in drug combinations can be explained using individual treatment profiles.
It has been recently shown that protein dynamics after combinatorial treatment of drugs can be accurately described as a linear combination of individual responses (Geva-Zatorsky et al., 2010). Moreover, Pritchard et al. reported that RNAi signatures of drug combinations are mainly a weighted composite of single drug effects (Pritchard et al., 2013). These findings suggest that the same relationship may be true at the transcriptional level. This paper aims to test this hypothesis. To this end, we performed multiple linear regression analyses on promoter and enhancer transcriptional activities after single and combinatorial drug treatment. Without using any prior information about known drug targets and affected pathways, we show that it is possible to describe the transcriptome response at promoters and enhancers with high accuracy in an unbiased global way.
2 Materials and methods
2.1 RNA sample preparation and CAGE data generation
Sample preparation, CAGE data production and basic processing are described in detail in Supplementary Notes. Briefly, human MCF-7 breast cancer cells were treated with drugs individually and in pair wise combinations, and triplicate CAGE libraries were prepared for each drug treatment as described previously (Kanamori-Katayama et al., 2011) and sequenced on HeliScope. CAGE tags were processed and mapped to genomic positions as described previously (Kajiyama et al., 2013) and projected on to FANTOM 5 defined promoter and enhancer regions.
2.2 Multiple linear regression model
We used multiple linear regression to model the relationship between combinatorial drug response and single drug expression profiles by fitting a linear equation to the observed data. Individual expression profiles were considered as explanatory variables and combinatorial drug action as the response variable. Formally, the model for multiple linear regression, given two drugs A and B, is:
where Fdrugi denotes the logarithmic fold change (log2FC) of drug i (in cpm) against the control condition for i = {A, B}, βj are the regression coefficients for j = {0, 1, 2}, ε is the error variable, and A, B ϵ {Gefitinib, U0126, Wortmannin}. We fitted the linear regression models using the least squares approach in R. Quantile regression, regression tree, multivariable linear regression with interaction term and linear regression using one drug treatment were compared as alternatives to the multiple linear regression method described above (Supplementary Notes). To evaluate the robustness and prediction ability of the regression models on new unseen values of the response variable, we performed 10-fold cross validation. We used three different metrics to assess the performance of the regression models on the test set: Mean Absolute Error (MAE), Pearson correlation and Spearman correlation (Supplementary Notes).
3 Results
3.1 Quantifying promoter expression after treatment with three individual drugs and their combinations
Owing to its multidimensional role in the progression of cancer, the EGFR signaling pathway has been the target of many anti-cancer therapies (Seshacharyulu et al., 2012) (Fig. 1A). When multiple drugs target either parallel signaling pathways or the same signaling pathway at various nodes, they may function synergistically for higher therapeutic efficacy and greater target selectivity (Shahbazian et al., 2012). With this in mind, we recorded the effects of Gefitinib (marketed commercially as Iressa), U0126 and Wortmannin, in human breast cancer MCF-7 cells using the CAGE promoter profiling method. Details about the mode of action of the three drugs are given in Supplementary Notes.
Fig. 1.
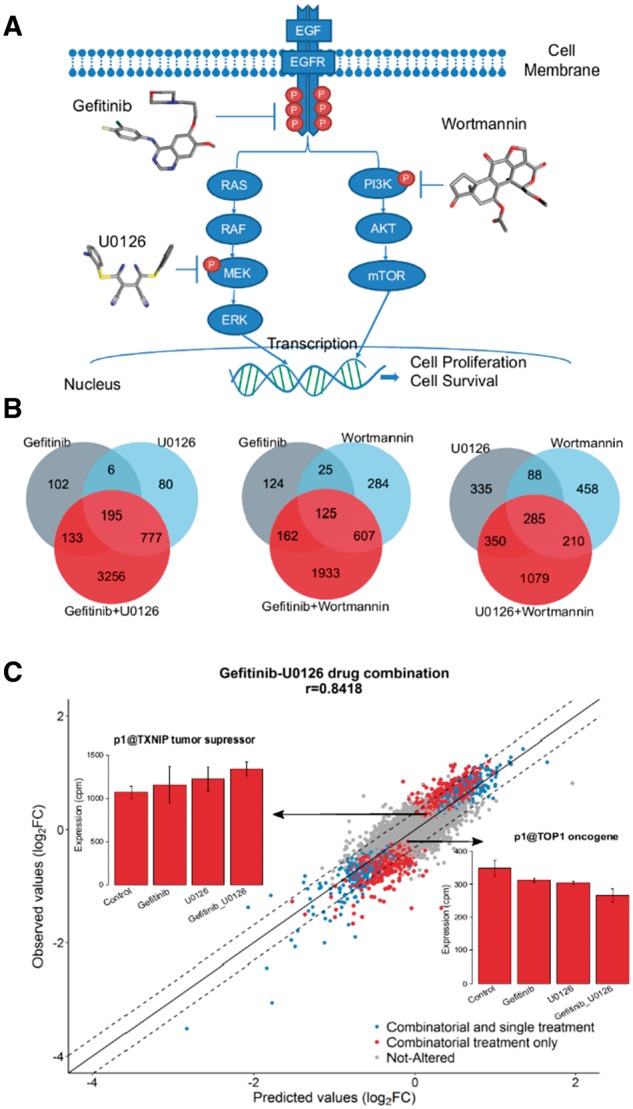
Promoter expression after combinatorial treatment. (A) EGFR pathway: Gefitinib, U0126 and Wortmannin directly inhibit the activity of EGFR, ERK and Akt pathway, respectively. (B) Venn diagrams showing the number of significantly altered promoters after single and combinatorial drug treatments. (C) Scatter plot of observed versus predicted log2FC values for the Gefitinib_U0126 drug combination. Blue dots indicate promoters differentially expressed both in single and combinatorial treatment, red dots denote promoters differentially expressed only in combinatorial treatment and gray dots represent the non-significantly altered promoters. The dashed lines define the bounds for the two standard deviations of the residual error. See also Supplementary Figure S2. Barplots show the expression of tumor suppressor TXNIP and oncogene TOP1 after single and combinatorial treatment of Gefitinib and U0126
We selected moderate concentrations below saturation for each inhibitor and their combinations. CAGE profiles for three replicates were obtained six hours after each drug treatment and assigned to promoter regions. After normalization and filtering of lowly expressed promoters (Supplemental Notes), the resulting dataset consisted of 19 693 promoters, 90% of which (17 768) were located within 500 bp of known RefSeq transcripts. Promoter activities were highly reproducible, achieving 0.9879 mean Pearson correlation coefficient among triplicates (Supplementary Fig. S1).
Differential expression analysis identified 436, 1058 and 1041 promoters significantly altered by Gefitinib, U0126 and Wortmannin treatment, respectively (FDR 5%, Supplemental Notes). However, after the combinatorial drug treatment the number of significantly affected promoters was notably greater compared to single drug treatment (Fig. 1B), indicating the complicated action of drug combinations in the entire transcriptome.
3.2 Promoter activity under combinatorial drug treatment is efficiently described by a multiple linear regression model
To test the hypothesis that the combinatorial drug response at the transcriptome level could be explained by combining single drug expression profiles, we performed multivariable linear regression analysis, where individual expression profiles were considered as explanatory variables and the combinatorial drug action as the response variable. We applied our analysis to the entire promoter dataset. Table 1 demonstrates the performance of the linear regression model after ten-fold cross validation in all the promoters. The results showed that the linear model could describe the relation between single and combinatorial drug effects at the transcriptome level to a high degree on a genome-wide scale. Importantly, with this approach, it was possible to explain the response of 2963 out of 3256 (91%) promoters significantly differentially expressed by combinatorial treatment only for the Gefitinib-U0126 drug pair (85% and 80% respectively for the Gefitinib-Wortmannin and U0126-Wortmannin combinations). Figure 1C shows the correlation between the regression model‘s estimations and Gefitinib-U0126 observed expression among the different promoter categories (also Supplementary Fig. S2), and highlights two examples of genes differentially expressed only after combinatorial treatment and well described by the linear regression model. Thioredoxin-interacting protein (TXNIP, a tumor suppressor) is an effective target for the treatment of breast cancer (Shen et al., 2015). Its expression was significantly increased in Gefitinib-U0126 combinatorial treatment and it was well captured by the linear model. The same drug pair also downregulated Topoisomerase I (TOP1, an oncogene) (Chen et al., 2015; Zhao et al., 2003) with a combinatorial response very close to the regression estimation. We identified numerous other key genes important for the phenotypic outcome with promoters that were differentially expressed only in combinatorial treatment and efficiently described by the linear regression model (Supplementary Tables S1–S3). Two cases of promoters where single drug treatment influences their expression in opposite direction are illustrated in Supplementary Figure S2B and C. When the two drugs are combined, they cancel out the individual effects in a manner well described by the regression model, emphasizing its capacity to capture any additive effect between single drug treatments whether there is amplification or cancellation of the transcriptome response.
Table 1.
Performance of linear regression analysis, applied in all the promoters
| 19693 promoters | MAE | Pearson correlation | Spearman correlation |
|---|---|---|---|
| Gefitinib_U0126 | 0.1160 | 0.8418 | 0.8284 |
| Gefitinib_Wortmannin | 0.1238 | 0.7453 | 0.7474 |
| U0126_Wortmannin | 0.1152 | 0.7480 | 0.7182 |
Note: The values shown in the table are the mean performance after tenfold cross validation.
Permutation analysis (Supplementary Notes) confirmed the statistical significance of the results (P-value < 2.2e-16 and Supplementary Fig. S3). The distributions of the permutation tests revealed the dominance of one drug in the combination, which is additionally supported by the coefficient of the regression models in the linear predictor function (Supplementary Table S4). When we further tried to fit the linear regression model using only the dominant drug profile, the performance was inferior to using both explanatory variables (P-value < 3e-4 for all combinations). The inclusion of an interaction term between the single drugs did not increase the adjusted r-squared statistic for the three models and did not result in statistically significant improvement. Furthermore, alternative regression models, namely quantile regression and regression tree (Supplementary Notes), did not demonstrate notably higher performance than multivariable linear regression (Supplementary Tables S5–S7).
3.3 Enhancer activity under combinatorial drug treatment is efficiently described by a multiple linear regression model
CAGE identifies transcribed enhancers at high nucleotide resolution by detecting bidirectionally transcribed enhancer RNAs (eRNAs) (Andersson et al., 2014), having an almost balanced transcription on both strands and being consistent with nucleosome borders. The enhancer set analyzed here consisted of 1028 enhancers after normalization and filtering, with mean Pearson correlation among replicates 0.74 (Supplementary Fig. S4). Principal component analysis showed (Supplementary Fig. S5) that the different drug conditions and samples after filtering can be separated in the first two principal components, suggesting not only that the drug treatment modifies enhancer expression but also that different drug agents have a distinct impact.
Having observed that the treatment alters enhancer expression, we further explored whether enhancer expression in drug combinations can be modeled using individual profiles. Thus, we performed the multivariable linear regression analysis also in the enhancer dataset. Table 2 demonstrates the performance of the linear regression model after ten-fold cross validation in all the three drug combinations. Notably, there is clear evidence that single drug therapy can be used to explain the combinatorial treatment in enhancers.
Table 2.
Performance of linear regression analysis, applied in all the enhancers
| 1028 enhancers | MAE | Pearson correlation | Spearman correlation |
|---|---|---|---|
| Gefitinib_U0126 | 0.3168 | 0.6900 | 0.6933 |
| Gefitinib_Wortmannin | 0.3150 | 0.6045 | 0.5722 |
| U0126_Wortmannin | 0.2920 | 0.6244 | 0.6119 |
Note: The values shown in the table are the mean performance after ten-fold cross validation.
Permutation tests validated the statistical significance of the results and verified the contribution of both single-drug profiles in the regression model (P-value< 2.2e-16, Supplementary Fig. S6) for all drug combinations. The distributions of the permutations, as well as the coefficient of the regression function, confirmed the dominance of one drug in the description of combinatorial expression again (Supplementary Table S8). The dominant drug was the same in promoters and enhancers in all combinations.
The samples of our study are too few to infer reliable functional promoter–enhancer pairs based on these samples alone. However, we identified several candidate functional pairs that were highly correlated across the samples of our study and also correlated across all the FANTOM5 phase 1 samples (Supplementary Table S9).
4 Discussion
Although prioritizing effective drug combinations has been the subject of extensive studies, the effects of drug combinations on the transcriptional activity of enhancers and promoters have not yet been investigated. Here we analyzed the pair-wise combined impact of Gefitinib, U0126 and Wortmannin on human breast cancer MCF-7 cells using single-molecule CAGE technology, and showed that promoter and enhancer expression under combinatorial treatment can be efficiently explained by a linear regression model using as input single drug profiles. Our discovery facilitates the approximate control of the transcriptional response to multidrug therapies and minimizes the need for exhaustive screens.
Current approaches in combinatorial drug prediction have two main limitations: (i) they utilize microarray expression profiling and therefore focus only on gene expression, and have a lower dynamic range in comparison to using a sequencing based approach; (ii) they consider only a small set of either the significantly altered genes by the individual inhibitors or the genes located downstream of the target pathways. Thus such methods fail to capture the complicated action of drug combinations in the entire transcriptome. In contrast, (i) our analysis is the first to study the effect of drug combinations on promoter as well as enhancer expression by taking advantage of CAGE profiling; (ii) We model the transcriptional relationship for drug combinations on a genome-wide scale. Consequently, we manage to describe with high accuracy the transcriptome response at promoters and enhancers, without prior knowledge of drug targets and pathways. CAGE also has the additional advantage that it can detect distinct effects of single or combination treatment on different promoters belonging to the same gene (an example is given in Supplementary Fig. S7).
The results show that promoter and enhancer transcription levels follow a linear relationship after combinatorial drug treatment. This finding is in agreement with previous studies about protein dynamics (Geva-Zatorsky et al., 2010) and RNAi screenings(Pritchard et al., 2013). In enhancers, even though the correlations are lower than in promoters, we also see strong evidence that drug combinations can be modeled as a linear relationship of individual responses. The lower performance of the linear regression in enhancers compared to promoters can be attributed to the appreciably lower expression and higher noise level of eRNAs, as shown by randomly sampling promoters with similar transcriptional intensity as the enhancers and re-applying linear regression for the three drug pairs (Supplementary Fig. S8 and Supplementary Notes).
Although our findings suggest that a linear model is sufficient for describing the transcriptional effects of combinatorial drug treatment with high accuracy at the genome wide level, not all promoters were well described by a linear combination of the individual drugs involved (Supplementary Notes, Supplementary Fig. S9). Interestingly, additional analysis of promoters not following the linear pattern showed that these promoters are likely to belong to transcription factors (Supplementary Notes, Supplementary Figs S10–S11 and Supplementary Tables S10–S17) and that their genomic regions are enriched for binding sites recognized by specific transcription factors (Supplementary Notes and Supplementary Tables S18–S21). These findings may indicate that the promoters of transcription factors, as well as the targets of a subset of transcription factors, are less likely to behave in a linear, additive way than the promoter set as a whole.
To generalize the approach described here, future studies may explore additional drug combinations, concentrations, treatment durations and cell types. Future work may also examine whether the linear relationship carries over combinations with three or four different drug agents. The integration of this information will lead to a more extensive understanding of the joint action of drugs and will enable the construction of more reliable and robust models for the quantitative analysis of combinatorial drug treatment.
Supplementary Material
Acknowledgements
We thank Ms Noriko Yumoto for cell culture and sample preparation of MCF-7 cells.
Funding
The research reported in this work was supported by RIKEN CLST Center Director‘s Strategic Program MNC. Additional funding was provided by King Abdullah University of Science and Technology (KAUST), JSPS KAKENHI Grant No.15KT0084 and RIKEN Epigenome and Single Cell Project Grants to MO-H.
Conflict of Interest: none declared.
References
- Andersson R. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M. et al. (2014) A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol., 32, 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. et al. (2015) Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene, 34, 4019–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N. et al. (2010) Protein dynamics in drug combinations: a linear superposition of individual-drug responses. Cell, 140, 643–651. [DOI] [PubMed] [Google Scholar]
- Hopkins A.L. (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol., 4, 682–690. [DOI] [PubMed] [Google Scholar]
- Jin G. et al. (2011) An enhanced Petri-Net model to predict synergistic effects of pairwise drug combinations from gene microarray data. Bioinformatics, 27, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiyama K. et al. (2013) Capturing drug responses by quantitative promoter activity profiling. CPT Pharmacometrics Syst. Pharmacol., 2, e77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori-Katayama M. et al. (2011) Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res., 21, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H. et al. (2012) CDA: combinatorial drug discovery using transcriptional response modules. PLoS One, 7, e42573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews Griner L.A. et al. (2014) High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA, 111, 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A. et al. (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature, 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Pritchard J.R. et al. (2013) Defining principles of combination drug mechanisms of action. Proc. Natl. Acad. Sci. USA, 110, E170–E179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshacharyulu P. et al. (2012) Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets, 16, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian D. et al. (2012) Vertical pathway targeting in cancer therapy. Adv. Pharmacol., 65, 1–26. [DOI] [PubMed] [Google Scholar]
- Shen L. et al. (2015) Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA, 112, 5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. et al. (2015) Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat. Commun., 6, 8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. et al. (2008) Finding multiple target optimal intervention in disease-related molecular network. Mol. Syst. Biol., 4, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C. et al. (2003) Elevated expression levels ofNCOA3, TOP1, andTFAP2C in breast tumors as predictors of poor prognosis. Cancer, 98, 18–23. [DOI] [PubMed] [Google Scholar]
- Zimmermann G.R. et al. (2007) Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov. Today, 12, 34–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.