


Abstract
Etoposide and other topoisomerase II-targeted drugs are important anticancer therapeutics. Unfortunately, the safe usage of these agents is limited by their indiscriminate induction of topoisomerase II-mediated DNA cleavage throughout the genome and by a lack of specificity toward cancer cells. Therefore, as a first step toward constraining the distribution of etoposide-induced DNA cleavage sites and developing sequence-specific topoisomerase II-targeted anticancer agents, we covalently coupled the core of etoposide to oligonucleotides centered on a topoisomerase II cleavage site in the PML gene. The initial sequence used for this ‘oligonucleotide-linked topoisomerase inhibitor’ (OTI) was identified as part of the translocation breakpoint of a patient with acute promyelocytic leukemia (APL). Subsequent OTI sequences were derived from the observed APL breakpoint between PML and RARA. Results indicate that OTIs can be used to direct the sites of etoposide-induced DNA cleavage mediated by topoisomerase IIα and topoisomerase IIβ. OTIs increased levels of enzyme-mediated cleavage by inhibiting DNA ligation, and cleavage complexes induced by OTIs were as stable as those induced by free etoposide. Finally, OTIs directed against the PML-RARA breakpoint displayed cleavage specificity for oligonucleotides with the translocation sequence over those with sequences matching either parental gene. These studies demonstrate the feasibility of using oligonucleotides to direct topoisomerase II-mediated DNA cleavage to specific sites in the genome.
INTRODUCTION
Type II topoisomerases are ubiquitous enzymes that regulate DNA topology by creating transient double-stranded breaks in the DNA (1–6). As a result of their activity, they can alleviate torsional stress generated by the movement of DNA replication forks and transcription complexes, and can resolve DNA knots and tangles generated during recombination and replication (1–6). When type II topoisomerases cleave the DNA, they create a four-base stagger with a 5′ overhang. In order to maintain genomic integrity during this process, active site tyrosine residues covalently attach to the newly generated 5′-termini; these covalent enzyme-cleaved DNA complexes are referred to as cleavage complexes (1–6). It is notable that the two cleavage steps do not occur simultaneously (7–9), and that the anticancer drug etoposide is capable of stabilizing both single and double-stranded cleavage complexes (Figure 1) (8).
Figure 1.

Structure-guided design of an OTI. (A) Schematic illustrating domains of type II topoisomerases used to determine the crystal structure of human topoisomerase IIβ covalently attached to DNA (green) in the presence of etoposide (orange). Domains pictured are TOPRIM (Top), winged helix domain (WHD), tower domain (Tow), and exit gate domain (Ex). (B) Schematic of topoisomerase II function. Protein protomer subunits are shown in blue and gray. T DNA, transport double helix (black); G DNA, gate double helix (green). (C, D) Detail from the crystal structure of a topoisomerase IIβ cleavage complex with two bound etoposide molecules (orange) stabilizing a double-stranded DNA (green) break; PDB code 3QX3. For clarity, in panel C, only the Cα trace of the protein subunits (blue and black lines) and catalytic tyrosines (blue and gray sticks) are shown. In panel D, only the catalytic tyrosine residues that cleave the DNA are shown. The conventional numbering scheme used for DNA cleavage complexes formed by type II topoisomerases is shown. The enzyme cleaves between the -1 and the +1 on each strand. The numbering on the two strands in the double helix is differentiated by the presence or absence of asterisks. The catalytic tyrosine residues are covalently attached to the DNA at the +1 positions. (E, F) Model of a cleavage complex with one bound etoposide molecule stabilizing a single-stranded DNA break. The cleaved DNA strand is indicated by asterisks. The protein subunits shown are the same as those in C and D. (G) Chemical (left) and modeled (right) structure of the etoposide core (DEPT) linked to the pyrimidine base. (H) OTI28 (orange strand) modeled with the modified cytosine base in the +5* position, stabilizing DNA scission at the 23–24 site (–1 to +1) on the cleaved target strand (green). Structural figures were drawn with Pymol (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Human cells express two closely related type II enzymes, topoisomerase IIα and IIβ. Topoisomerase IIα is upregulated in proliferating cells and plays essential roles in DNA replication and chromosome segregation (2,3,6). A recent study also presents strong evidence that this isoform plays important roles in transcription (10). Topoisomerase IIβ concentration is high in all cells irrespective of proliferative status, and the enzyme has demonstrated roles in transcription (11,12). Although it is not essential at the cellular level, topoisomerase IIβ is critical for the proper development of the nervous system (13,14).
Beyond their cellular functions, type II topoisomerases are also the targets for some of the most widely prescribed anticancer drugs in the world (2,15–17). Drugs such as etoposide, doxorubicin, and mitoxantrone are used routinely to treat breast cancer, germline cancers, leukemias, lymphomas, and a variety of solid tumors (17). These drugs stabilize the cleavage complex by intercalating into the cleaved DNA scissile bonds, thereby blocking ligation of the DNA (7,18). When DNA replication forks or other tracking systems encounter these drug-stabilized enzyme–DNA complexes, they convert the transient DNA breaks into chromosomal damage that requires DNA repair pathways to restore the double helix (2,15–17). Due to their mechanism of action, these drugs are referred to as topoisomerase II poisons.
Despite the fact that all human cells express one or both topoisomerase II isoforms, topoisomerase II-targeting drugs are efficacious against cancer cells primarily for three reasons. First, because cancer cells are generally highly proliferative, they express high levels of topoisomerase IIα (3,17,19), leading to the creation of more drug-stabilized cleavage complexes. Second, because of the high metabolic rate of cancer cells, replication forks and transcription complexes constantly move along the DNA, making it more likely that they will convert cleavage complexes to permanent DNA breaks (20). Third, due to impaired cell cycle checkpoints and DNA damage repair pathways in many cancers, malignant cells often are more susceptible to the DNA damage caused by topoisomerase II-targeted drugs (17,21).
Treatment with topoisomerase II poisons, however, is accompanied by a variety of mechanism-induced (i.e., topoisomerase II-generated) toxicities. These arise because of the difficulty of targeting drugs such as etoposide specifically to cancer cells (2,15,17,22) and because all cell types express one or both topoisomerase II isoforms (4,6). Furthermore, approximately 2–3% of patients treated with regimens that include topoisomerase II-targeted drugs, such as etoposide, mitoxantrone, and doxorubicin, go on to develop secondary leukemias. Primarily, these malignancies are acute myelogenous leukemias (AMLs) associated with rearrangements in the mixed lineage leukemia (MLL) gene at chromosomal band 11q23, and acute promyelocytic leukemias (APLs) associated with rearrangements between the promyelocytic leukemia (PML) gene and the retinoic acid receptor α (RARA) gene [t(15;17)(q22;q12)] (17,23,24). Although the specific mechanism by which drug-induced topoisomerase II-mediated DNA cleavage triggers the leukemic translocation is controversial, considerable circumstantial evidence suggests that sites cleaved by the enzyme go on to generate the translocation breakpoint (10,17,23–26).
A number of cancers are associated with the presence of a driver oncogene that is generated by a mutation or translocation in the DNA (27–29). The ability to rapidly sequence patients’ DNA affords a potential opportunity to develop new generations of topoisomerase II poisons that specifically target the driver mutations in cancer cells of affected individuals. This could introduce DNA strand breaks specifically in the oncogene or could generate cleavage complexes that act as roadblocks for transcription. In either case, disruption of the driver oncogene or interference with its transcription could potentially kill malignant cells that are dependent on the resulting oncoprotein. Consequently, this strategy could result in treatments that are cytotoxic toward cancer but display limited action against normal cells.
As a first step toward developing sequence-specific topoisomerase II poisons with potential for use as anticancer agents, we covalently coupled the active core of etoposide to oligonucleotides centered on a topoisomerase II cleavage site in the PML gene. The initial sequence used for this OTI was identified as part of the translocation breakpoint of a patient with APL who had been treated with mitoxantrone for progressive multiple sclerosis (30). Subsequent OTI sequences were derived from the observed APL breakpoint between PML and RARA. Results indicate that OTIs can be used to direct the sites of etoposide-induced DNA cleavage mediated by topoisomerase IIα and topoisomerase IIβ. Furthermore, OTIs directed against the PML-RARA breakpoint displayed cleavage specificity for oligonucleotides with the translocation sequence over those with sequences matching either parental gene. These studies demonstrate the feasibility of using oligonucleotides to direct topoisomerase II-mediated DNA cleavage to specific sites in the genome.
MATERIALS AND METHODS
Molecular modeling
To guide the placement and length of the chemical linker that joined the active demethylepipodophyllotoxin (DEPT) core of etoposide to the modified base in the oligonucleotides, structures of OTI complexes were modeled using Coot (31), MOE (32), and Maestro (Schrödinger Release February 2017: Maestro, Schrödinger, LLC, New York, NY, 2017). Models were based on the crystal structures of the human topoisomerase IIβ and topoisomerase IIα cleavage complexes with DNA and two etoposide molecules (one at each scissile bond) (PDB code: 3QX3 (18) and PDB code: 5GWK (33), respectively), and the Staphylococcus aureus gyrase–DNA complex with one etoposide molecule (PDB code: 5CDP) (34) (Supplementary Figure S1). The positions of the etoposide molecules in the DNA complexes with human and bacterial enzymes are essentially the same (Supplementary Figure S1).
With two etoposide molecules bound (one at each scissile bond), the DNA gate seems to be wedged open with a relatively small area buried between the two protein subunits at the DNA gate (18,34) (Supplementary Figure S2). However, a much larger area is buried between the two subunits at the DNA gate (Supplementary Figure S2) in the S. aureus DNA gyrase with one etoposide bound, and the common CRsym conformation is observed (34). Given that this conformation has also been observed with eukaryotic type IIA topoisomerases (34,35), the subunits in our modeled topoisomerase IIβ complex with one etoposide were modeled in a CRsym conformation (Figure 1; notice movement of catalytic tyrosines between complex with one and two etoposides relative to movement of the two subunits).
The alkyne moiety of the linker on the C5 position of the modified base projects ∼2.6 Å further than the methyl on the C5 position of a thymine or a methylated cytosine (Supplementary Figure S2A). In modeling studies, the modified base was covalently attached to DEPT with a linker (Figures 1E, F and 2), and was modeled at four positions adjacent to the cleavage site on each strand (Supplementary Figure S3): −2, −1, +1 and +2 on the cleaved strand, and +3*, +4*, +5* and +6* on the non-cleaved (or OTI strand. The rod-like alkyne moiety was observed to clash with protein residues when positioned on the cleaved strand at the −2, −1, +1 or +2 positions. However, modeling the rod-like alkyne moiety on the uncleaved strand at the +3*, +4*, +5* or +6* position did not result in any clashes (Supplementary Figure S3). These models indicate that cleavage is most likely to occur on the target strand rather than the OTI strand itself. Two different lengths of linkers were modeled, but a much longer linker (Figure 1E, F) was needed to enable the linked etoposide core to reach back to the binding site in the DNA from the +3*, +4*, +5* or +6* positions.
Figure 2.

Schematic of the synthesis of OTIs. The synthesis of OTI28 (7) is shown as an example. DEPT (1) was protected with carboxybenzyl (Cbz) at the 4′-OH using benzyl chloroformate and triethylamine in dichloromethane to yield 2. The 4-OH of 2 was reacted with monotosylated diethylene glycol using boron trifluoride etherate in dichloromethane at –20°C to generate 3a–b as a mixture of two epimers. Removal of the Cbz protecting group under hydrogenation reaction conditions with Pd/C in ethanol resulted in 4a–b. The tosyl group was displaced with sodium azide in dimethyl formamide at 60°C to generate 5a–b as a mixture of two epimers. The desired azide coupling partner 5a was purified as a single epimer by chiral chromatography. Copper-catalyzed click chemistry was used to couple 5a to oligonucleotide 6 (which included an alkyne-modified cytosine at position 28) to yield OTI28 (7).
Chemistry
A scheme for the synthesis of OTIs linked to the etoposide core is shown in Figure 2. On the basis of the molecular modeling results, an 8-carbon linker was utilized, which consisted of a terminal alkyne moiety attached to the C5 position of a cytosine or thymine residue to attach an azide-modified 4′-DEPT (1) via copper-catalyzed click chemistry.
Synthesis of the activated etoposide core, shown in the top portion of Figure 2, started with commercially available DEPT (1) (ABCAM Biochemicals). Compound 1 subsequently was protected with carboxybenzyl (Cbz) at the 4′-OH using benzyl chloroformate and triethylamine in dichloromethane (20). The desired product 2 was obtained in 91% isolated yield. The 4-OH of 2 was reacted with monotosylated diethylene glycol (21) using boron trifluoride etherate in dichloromethane at −20°C to generate 3a–b in 72% yield as a mixture of two epimers. Removal of the Cbz protecting group under hydrogenation reaction conditions with Pd/C in ethyl acetate resulted in 4a–b in 94% yield. The tosyl group was displaced with sodium azide in dimethyl formamide at 60°C and generated 5a–b in 67% yield as a mixture of two epimers. The desired azide coupling partner 5a was purified as a single epimer by chiral chromatography.
The synthesis of OTIs, exemplified by 7, is shown in the bottom portion of Figure 2. Oligonucleotide 6, which included an alkyne-modified cytosine (purchased from Jena Biosciences) at position 28, was synthesized by well-established solid phase methods. Copper-catalyzed click chemistry was employed to synthesize OTI28 (7) using a preformed complex of copper bromide and Tris(benzyltriazolylmethyl)amine (TBTA) in a mixture of dimethyl sulfoxide (DMSO), tert-butanol and water. A final isolated yield of 46% was obtained after HPLC purification. Subsequent OTIs were synthesized in a similar manner with an alkyne-modified thymine (Integrated DNA Technologies) at positions 23, 29 or 33 as the point of attachment for the linked DEPT, yielding OTI23, OTI29, and OTI33, respectively. Three additional OTIs of different lengths, a 50-mer, 30-mer and 20-mer, were directed against a patient-derived PML-RARA breakpoint sequence (see below). The linked etoposide core in the 50-mer was positioned at the same site as OTI29.
Data confirming the structure and purity of all OTIs and intermediate small molecules can be found in the Appendix. Purity ranged from 90% to 99.9%.
Enzymes and materials
Recombinant wild-type human topoisomerase IIα and topoisomerase IIβ and wild-type yeast topoisomerase II and the etoposide-resistant H1011Y mutant yeast type II enzyme were expressed in Saccharomyces cerevisiae JEL-1Δtop1 and purified as described previously (36–39). The drug resistance mutation was originally published as H1012Y (38). The numbering of the mutation was changed to H1011Y in 2001 (40) to accommodate a reported error in the original amino acid sequence of S. cerevisiae topoisomerase II (41). Human enzymes were stored at −80°C as 1.5 mg/ml stocks in 50 mM Tris–HCl, pH 7.9, 0.1 mM EDTA, 750 mM KCl, and 40% glycerol, and yeast enzymes were stored as 2 mg/ml stocks in a similar buffer. Analytical grade etoposide was purchased from Sigma-Aldrich and stored at room temperature as a 40 mM stock solution in 100% DMSO.
Oligonucleotides and OTIs
Three DNA sequences were used to create a series of oligonucleotide duplexes. The first sequence was a 50-mer that encompassed bases 1461–1510 of intron 6 of PML, and contained a previously identified, 8-base topoisomerase II cleavage hotspot (bases 1482–1489) associated with the generation of therapy-related APLs (t-APL) (42). The hotspot contains a strong topoisomerase II-mediated cleavage site that corresponds to position 24–25 on the top (or target) strand of the oligonucleotide and to 26–27 on the bottom strand (either an unmodified oligonucleotide or an OTI). Top strand: 5′-CTTTGTTCCTCATTCTGACTGAGCCCTAGCCTTGGTCACACACTGAGCAG-3′. Bottom strand: 5′-CTGCTCAGTGTGTGACCAAGGCTAGGGCTCAGTCAGAATGAGGAACAAAG-3′. OTI28, OTI29, OTI33, and OTI23 had the same sequence as the PML 50-mer bottom strand, except that the active core of etoposide (DEPT) was linked at positions 28, 29, 33 and 23, respectively. A 50-mer bottom strand that contained the linker with no attached drug (LIN28) or a tetrahydrofuran (Eurofins MWG Operon) abasic site analog (AP28) at position 28 also were synthesized.
The second sequence was a 50-mer that spanned a previously identified translocation between PML and RARA in a patient with t-APL (30). Top strand: 5′-CTTTGTTCCTCATTCTGACTGAGCCCTA/GTCTGCCATCCTAACCTTCCAT-3′, made up of bases 1461–1488 of intron 6 of PML (before the slash) and bases 12039–12060 of intron 2 of RARA (after the slash) The bottom strand was fully complementary. Bottom strand: 5′-ATGGAAGGTTAGGATGGCAGAC\TAGGGCTCAGTCAGAATGAGGAACAAAG-3′. Sequences from RARA are located to the left of the backslash and sequences from PML are located to the right of the backslash. A 50-mer OTI with the bottom strand sequence was synthesized and contained a linked DEPT at position 29. Two additional OTIs based on the same sequence, a 30-mer (5′-TAAGACTGACTCGGGATCAGACGGTAGGAT-3′) and a 20-mer (5-CTGACTCGGGATCAGACGGT-3′), were synthesized. These sequences contained 10 or 15 fewer bases from each of the 5′- and 3′-termini of the 50-mer, respectively.
The third sequence was a 50-mer that corresponded to the parental RARA sequence that spanned a patient-derived translocation (at 12038–12039, corresponding to 28–29 on the oligonucleotide) and that included bases 12011–12060 of intron 2. Top strand: 5′-CAGAAAGGGGCAACTTCATCAGACACCCGTCTGCCATCCTAACCTTCCAT-3′. All the non-OTI oligonucleotides were synthesized by Sigma-Aldrich unless specified.
DNA cleavage
DNA cleavage reactions were carried out by a modification of the procedure of Deweese et al. (43). The top (target) strand of each double-stranded oligonucleotide was labeled on its 5′-terminus in 30 μl reactions containing 200 pmol of oligonucleotide, 1 μl of T4 polynucleotide kinase (PNK) (New England BioLabs), 3 μl of T4 PNK buffer as supplied by the manufacturer, and 2 μl of [γ-32P]ATP (∼5000 Ci/mmol, Perkin Elmer). Reactions were incubated at 37°C for 60 min, with an additional 1 μl of T4 PNK added after the first 30 min. Radiolabeled oligonucleotides were purified with Qiagen Mini Quick Spin columns according to the manufacturer's instructions. Top/target and bottom/OTI oligonucleotides were annealed by incubating them at a 1:1 ratio (0.5 pmol/μl) at 70°C for 10 min, followed by a gradual cool down to room temperature.
DNA cleavage reaction mixtures contained 2 pmol of double-stranded oligonucleotides and 440 nM human topoisomerase IIα, 415 nM human topoisomerase IIβ, 600 nM wild-type yeast topoisomerase II, or 1340 nM H1011Y mutant yeast topoisomerase II in a final volume of 20 μl of human [10 mM Tris–HCl pH 7.9, 5 mM MgCl2, 100 mM KCl, 0.1 mM EDTA and 2.5% (v/v) glycerol] or yeast [50 mM Tris–HCl pH 7.9, 25 mM MgCl2, 500 mM NaCl, 0.5 mM NaEDTA, pH 8.0, 12.5% (v/v) glycerol] cleavage buffer. (Levels of yeast enzymes in DNA cleavage assays were adjusted to yield similar levels of background DNA cleavage.) Reactions were initiated by the addition of enzyme. Samples were incubated at 37°C for 10 min (topoisomerase IIα or yeast topoisomerase II) or 1 min (topoisomerase IIβ), and reactions were stopped by the addition of 2 μl of 10% sodium dodecyl sulfate (SDS) and 2 μl of 250 mM disodium ethylenediaminetetraacetic acid (Na2EDTA). Samples were incubated with proteinase K (2 μl of 0.8 mg/ml) for 30 min at 37°C to digest the type II enzyme. DNA cleavage products were precipitated with ethanol, mixed with 5 μl of 80% (v/v) formamide, 10% TBE (100 mM Tris–borate and 2 mM EDTA), and 10% agarose loading dye [60% sucrose (w/v), 10 mM Tris–HCl, 0.5% bromophenol blue, 0.5% xylene cyanol] and heated to 75°C for 2 min. DNA samples were resolved on 14% denaturing polyacrylamide gels and were visualized and quantified using a Bio-Rad Molecular Imager.
All double-stranded oligonucleotides contained unmodified (non-OTI) top/target strands that were radiolabeled at the 5′ end. Bottom strands were either unmodified (non-OTI) or contained linked etoposide core (OTI), linker alone, or a tetrahydrofuran (abasic site analog). In some cases, unmodified double-stranded oligonucleotides were treated with 0–500 μM etoposide.
DNA ligation
DNA ligation assays were carried out with topoisomerase IIα by a modification of the procedure of Byl et al. (44). DNA cleavage/ligation equilibria were established as in the previous section. Reactions contained either an unmodified double-stranded oligonucleotide in the presence of 500 μM etoposide or an unmodified top (i.e. target) strand hybridized to an OTI28 bottom strand. Ligation was initiated by placing the reaction mixtures on ice. The temperature shift allows ligation but prevents the formation of new cleavage complexes (45). Ligation reactions were stopped by the addition of 2 μl of 10% SDS followed by 2 μl of 250 mM Na2EDTA. Samples were processed and analyzed as described above. The percent DNA cleavage at time zero was set to 100%, and the rate of ligation was determined by quantifying the loss of cleaved DNA over time.
Persistence of cleavage complexes
DNA cleavage/ligation equilibria were established using topoisomerase IIα as described above. Persistence reactions were carried out by a modification of the procedure of Bandele and Osheroff (46). Reactions contained either an unmodified double-stranded oligonucleotide in the presence of 500 μM etoposide or an unmodified top strand hybridized to an OTI28 bottom strand. Assay mixtures were diluted 20-fold with cleavage buffer and incubated at 37°C for up to 120 min. Reactions were stopped as above, and samples were processed and analyzed as described above. The percent DNA cleavage at time zero was set to 100%, and the stability of the cleavage complexes was determined by quantifying the loss of cleaved DNA over time.
RESULTS
Structure-based design of OTIs that contain the core of etoposide
When cells are treated with etoposide, the drug induces topoisomerase II-mediated DNA cleavage at millions of sites throughout the human genome (10). Therefore, in an effort to design an etoposide-based compound with specificity for cancer-associated DNA sequences, we covalently attached the active core of etoposide (DEPT) to a series of oligonucleotides (Figures 1 and 2). DEPT lacks the C4 sugar moiety of etoposide but retains a C4 hydroxyl group that can be activated for coupling reactions. As determined by saturation transfer difference nuclear magnetic resonance spectroscopy, the sugar moiety does not interact with topoisomerase II (47,48). Furthermore, its removal has a negligible effect on the activity of the drug toward the type II enzyme (47,48). For the purposes of the paper, the linked DEPT will be referred to as the etoposide core.
OTIs were designed to generate single-stranded, topoisomerase II-mediated cleavage on the strand opposite the etoposide core attachment site (Figure 1) (8). To this end, crystal structures of human topoisomerase IIβ (18) and IIα (33) with two etoposide molecules and the bacterial Staphylococcus aureus DNA gyrase with one or two etoposide molecules (34) served as the basis for modeling studies (Supplementary Figure S1). As a result of these studies, a linker consisting of a terminal alkyne moiety was attached to the C5 position of a cytosine or thymine residue and was coupled to a 4-azide-modified DEPT via copper-catalyzed click chemistry (Figure 2). The modeling studies indicated that the linked DEPT was capable of intercalating into a cleaved site on the opposite strand (shown for OTI28 in Figures 1 and 4 and Supplementary Figures S3 and S4).
Figure 4.

Molecular models of DNA cleavage complexes formed with OTI28. Models were based on the crystal structure of topoisomerase IIβ (34). (A) Cleavage between bases 24–25 is depicted on the target (top) strand (green). (B) Cleavage between bases 23–24 is depicted on the target strand (green). The bottom (OTI) strand is shown in orange. The tethered etoposide core is shown in yellow (carbons, yellow; nitrogen, blue; oxygen, red). A Cα trace is shown for the two topoisomerase II subunits (blue and black lines) in the top panels. The bottom panels include a semi-transparent molecular surface, illustrating that the linker does not clash with the protein. The sequence diagram (middle) shows the position of the tethered etoposide core on OTI28 (yellow box). Black arrows indicate the cleavage sites.
An OTI directed against the PML gene enhances DNA cleavage mediated by human type II topoisomerases
As a proof of concept that a covalently attached etoposide core can function as a sequence-specific topoisomerase II poison, we designed an initial OTI (OTI28) that utilized a DNA sequence spanning a strong drug-induced topoisomerase II cleavage hotspot within intron 6 of the PML gene (bases 1482–1489) (42). This cleavage hotspot is also present in the t(15;17) translocation of a patient with APL who had been treated with mitoxantrone (30). The interaction of an OTI that spanned the APL breakpoint sequence with topoisomerase II will be discussed in a later section.
The sequence of the central 30 nucleotides of the 50-base pair-oligonucleotide used for the initial experiments (bases 1461–1510) is shown in Figure 3A. When the unmodified oligonucleotide duplex (radiolabeled on its top/target strand) was treated with free etoposide in the presence of topoisomerase IIα, several strong DNA cleavage sites were observed (Figure 3). The cleavage site between bases 24–25 (1484–1485) is also a strong site for mitoxantrone and is believed to be the hotspot that results in ∼50% of the t(15;17) translocations observed in patients with therapy-related APLs following treatment with mitoxantrone (17,24,30,42,49).
Figure 3.

An oligonucleotide-linked etoposide core increases topoisomerase II-mediated DNA cleavage. (A) The central 30 base pairs of a double stranded 50-mer oligonucleotide sequence corresponding to bases 1471–1500 (top strand) of PML intron 6 is shown. The yellow box denotes the position of the tethered etoposide core and linker moieties on OTI28 or LIN28. Arrows indicate sites of DNA cleavage induced by free etoposide (blue) or OTI28 (yellow). (B) Comparison of DNA cleavage mediated by human topoisomerase IIα (hTIIα, left) and topoisomerase IIβ (hTIIβ, right) of the radiolabeled, unmodified PML top strand hybridized to an unmodified PML bottom strand in the presence of free etoposide or hybridized to OTI28 (bottom strand). For each gel, lane 1 contains the unmodified PML oligonucleotide. Lanes 2–5 contain the unmodified PML duplex treated with 0–500 μM free etoposide. Lanes 7 and 8 contain the unmodified PML top strand hybridized with OTI28. Lanes 10 and 11 contain the unmodified top strand duplexed with LIN28 (bottom strand oligonucleotide that contains the linker at position 28 with no attached etoposide core). Lanes 6, 9, and 12 contain reference (R) oligonucleotides that were 24, 23 and 19 bases in length. Gels are representative of at least three independent experiments. (C) Quantification of the relative levels of DNA cleavage mediated by topoisomerase IIα (left) and topoisomerase IIβ (right). DNA cleavage at each site was normalized to the cleavage observed at site 24–25 in reactions containing unmodified duplex in the absence of etoposide (lane 2). Cleavage results of the unmodified duplex in the presence of 500 μM free etoposide are shown in blue (lane 5) and those with an unmodified top strand hybridized to OTI28 are shown in yellow (lane 8). Error bars represent the standard error of the mean of an average of two to five independent experiments. Significance was determined by paired t-tests. P-values are indicated by asterisks (*P < 0.05; **P < 0.005; ***P < 0.0005).
To examine the effects of a linked drug on topoisomerase II-mediated DNA cleavage of this sequence, the active core of etoposide was covalently attached to cytosine 28 on the bottom strand (Figure 3A) through a flexible linker as described above. On the basis of modeling studies, the tethered drug moiety should be able to intercalate into the scissile bond between bases 24–25, stabilizing cleavage at that site (Figures 1 and 4A). As seen in Figure 3, this was the case when the unmodified bottom strand was replaced with the drug-linked oligonucleotide (OTI28, bottom strand). Topoisomerase IIα cleaved at this position (between bases 24–25 on the top strand) at levels that were even higher than those observed in the unmodified duplex in the presence of 500 μM free etoposide. High enzyme-mediated cleavage was also observed one base away (between bases 23 and 24 on the top strand), although at slightly lower levels than at site 24–25. Drug insertion at this position is also supported by modeling studies (Figure 4B).
Four additional points should be noted regarding the data shown for topoisomerase IIα (Figure 3B, C; left): first, no DNA cleavage was observed in the absence of topoisomerase IIα (lane 1), indicating that the tethered etoposide core does not induce cleavage via a spontaneous chemical reaction. Second, OTI28 did not induce topoisomerase IIα-mediated cleavage at any of the other strong cleavage sites induced by free etoposide (compare lanes 8 and 5). This finding suggests that the specificity of the etoposide core can be constrained by its covalent attachment to an oligonucleotide. Third, some minor sites of topoisomerase IIα-mediated DNA cleavage with OTI28 were not predicted by our modeling studies (Figures 1 and 4). These sites were not related to the conditions utilized to anneal the OTIs to their complementary strand. Similar DNA cleavage patterns were observed when annealing temperatures were varied by 30°C and cooling rates differed over an 8-fold range (Supplementary Figure S5). These findings suggest that the presence of the drug may subtly alter the structure of the duplex that affects its interaction with topoisomerase IIα. Lastly, no enzyme-mediated DNA cleavage was observed when the bottom strand was replaced with an oligonucleotide that contained the linker alone (i.e. a linker with no attached drug moiety; lane 11). This provides strong evidence that it is the presence of the etoposide core rather than the linker alone on the oligonucleotide that enhances topoisomerase IIα-mediated DNA cleavage.
Results similar to those with topoisomerase IIα also were observed with topoisomerase IIβ (Figure 3B, C; right). The same two strong sites of enzyme-mediated DNA cleavage were observed in a duplex containing OTI28. Once again, scission at both cleavage sites (24–25, 23–24) was higher than that seen in the unmodified duplex in the presence of 500 μM free etoposide (compare lanes 8 and 5). However, cleavage at 23–24 was similar to that seen at 24–25. Finally, as seen with the α isoform, topoisomerase IIβ did not cleave a duplex with a bottom strand that contained a linker but no tethered etoposide core at position 28 (lane 11).
To further characterize the mechanism of enzyme-mediated DNA cleavage induced by the OTI, the effects of the tethered etoposide core on rates of DNA ligation mediated by topoisomerase IIα were compared to those of free etoposide on an unmodified duplex oligonucleotide. Similar inhibition of religation was observed for both the tethered and the untethered drug (Figure 5A). Thus, like free etoposide, the OTI increases levels of cleavage complexes by inhibiting ligation of the cut DNA.
Figure 5.

OTI28 inhibits DNA ligation and stabilizes cleavage complexes similarly to free etoposide. (A) Enzyme-mediated ligation of DNA. (B) Persistence of cleavage complexes. For both A and B, cleavage results of the unmodified PML duplex in the presence of 500 μM free etoposide are shown in blue and those with an unmodified PML top/target strand hybridized to OTI28 are shown in yellow. Error bars represent the standard deviation of at least three independent experiments.
In addition, cleavage complexes formed with OTI28 versus free etoposide persisted for similar lengths of time (Figure 5B) following 20-fold dilution of the reaction mixtures. Therefore, the OTI appears to stabilize cleavage complexes to a comparable extent as the free etoposide. These findings further suggest that the effects of tethered etoposide core within the cleavage complex produce similar effects to the free drug.
DNA lesions have the capacity to act as topoisomerase II poisons. Abasic sites are among the most effective DNA lesions at inducing topoisomerase II-mediated DNA cleavage (39,50,51). Thus, to provide further evidence that the effects of OTI28 were mediated by the tethered drug moiety as opposed to a distortion in the double helix, we synthesized an oligonucleotide that replaced the cytosine-linker-etoposide core construct at position 28 with a tetrahydrofuran abasic site analog (AP28). DNA cleavage results are shown in Figure 6. The abasic site induced lower levels of cleavage than OTI28, with the major site of DNA cleavage (position 19–20) being five bases away from that of OTI28 (24–25) (compare lanes 7 to 5). The differences in the cleavage patterns obtained with AP28 and OTI28 provide additional strong evidence that DNA cleavage induced by OTI28 is due to the actions of the tethered etoposide core by type II topoisomerases and not to a distortion or lesion in the helix at the site of drug attachment.
Figure 6.
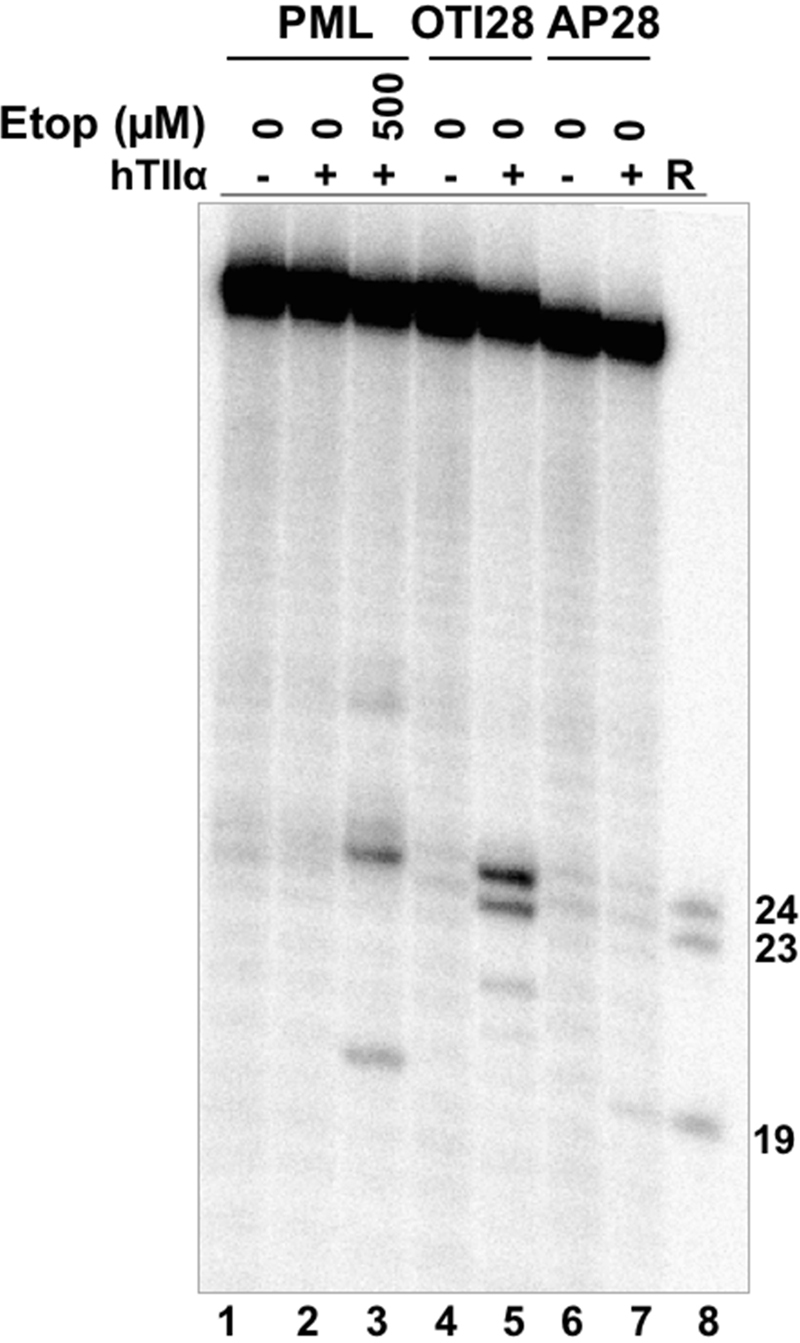
An oligonucleotide with an abasic site analog at position 28 generates a different DNA cleavage pattern than does OTI28. Lanes 1–3 contain a radiolabeled unmodified PML top/target strand hybridized to an unmodified PML bottom strand in the absence of enzyme, or in the presence of enzyme and 0–500 μM free etoposide. Lanes 4 and 5 contain a radiolabeled PML top strand hybridized with OTI28 (bottom strand). Lanes 6 and 7 contain a radiolabeled unmodified PML top strand hybridized to a bottom strand oligonucleotide containing an abasic site analog at position 28 (AP28). Lane 8 contains reference (R) oligonucleotides that are 24, 23 and 19 bases in length. The gel is representative of at least three independent experiments.
Finally, the ability of OTI28 to induce DNA cleavage mediated by an etoposide-resistant type II topoisomerase was assessed. The yeast topoisomerase II H1011Y mutant (52) was used for these experiments for two reasons. First, target-based resistance to topoisomerase II-targeted drugs is generally associated with the loss of one enzyme allele or the deletion of nuclear localization signals (53,54). Consequently, point mutations that impart etoposide resistance to human type II topoisomerases have not been well described or characterized. Second, yeast topoisomerase II is highly sensitive to etoposide, and the H1011Y mutant is the only type II enzyme that has been shown to be etoposide resistant due to a reduced affinity for the drug (55).
As seen in Figure 7, yeast topoisomerase II H1011Y is ∼4-fold resistant to the tethered etoposide core in OTI28 as compared to the wild-type enzyme. This result is consistent with the resistance of the mutant enzyme to free etoposide (52).
Figure 7.
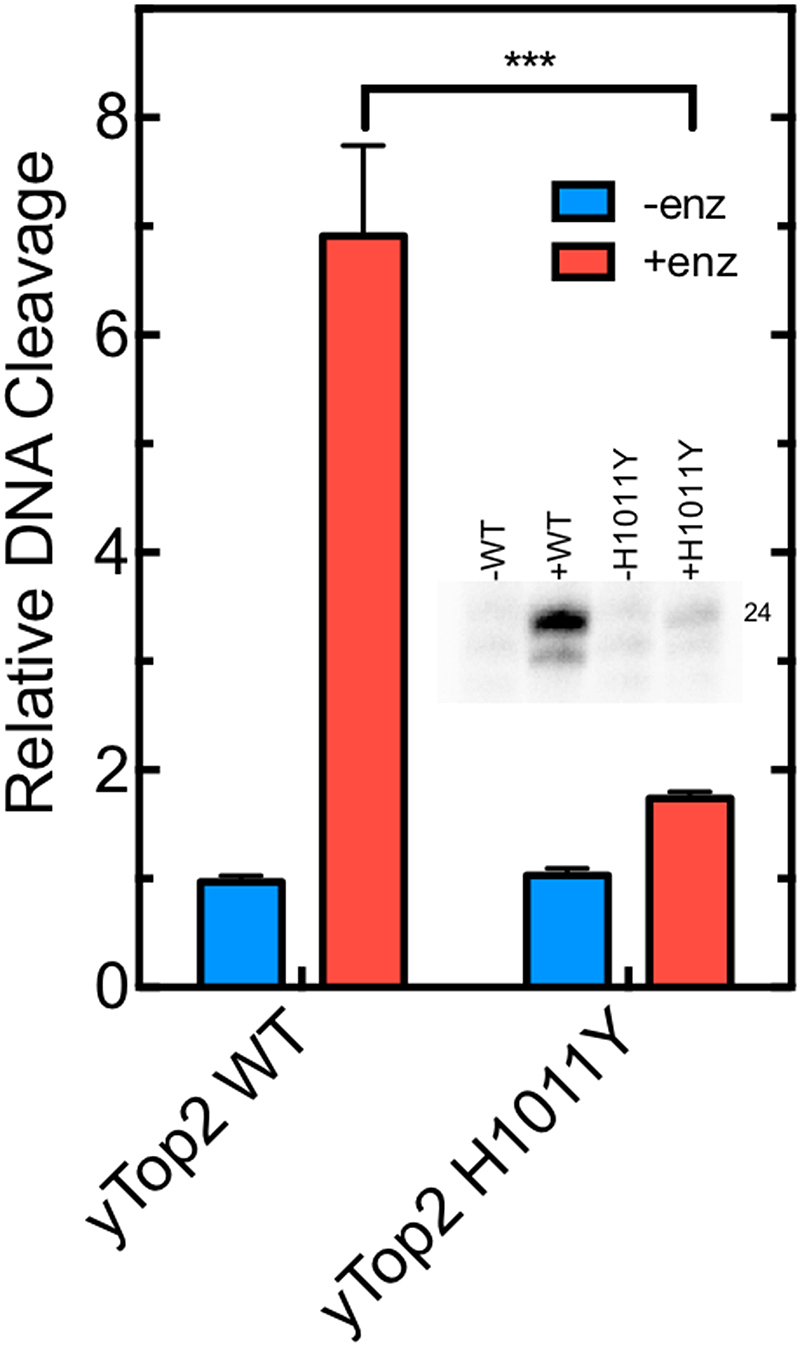
OTI28 induces lower levels of DNA cleavage mediated by an etoposide-resistant mutant yeast topoisomerase II (H1011Y) as compared to wild-type yeast topoisomerase II. Quantification of the relative levels of enzyme-mediated DNA cleavage at site 24–25 (indicated as the band labeled 24 in the inset) mediated by wild-type (yTop2WT) and H1011Y mutant (yTop2H1011Y) yeast topoisomerase II on an unmodified PML top strand hybridized to OTI28 (graph: +enz, red; inset: +WT, +H1011Y). DNA cleavage is normalized to background levels of DNA when no enzyme is present (graph: -enz, blue; inset: -WT, -H1011Y). Error bars represent the standard deviation of three independent experiments. Significance was determined by a paired t-test. P-values are indicated by asterisks (***P < 0.0005).
Taken together, the above experiments provide strong evidence that the DNA cleavage induced by OTI28 is due to the presence of the tethered etoposide core.
OTIs enhance topoisomerase II-mediated DNA cleavage in a sequence-dependent manner
As described above, the major site of topoisomerase II-mediated DNA cleavage on the top/target strand induced by OTI28 was located at a position two bases away and opposite the tethered etoposide core (site 24–25). To determine whether OTIs generate a predictable pattern of cleavage, we shifted the location of the tethered etoposide core to three additional locations along the sequence of the bottom oligonucleotide: positions 29 (OTI29), 33 (OTI33) and 23 (OTI23) (Figure 8A). These locations were chosen to see if it was possible to move the major site of DNA cleavage to the second-strongest site of scission induced by OTI28 (site 23–24), to the site of secondary DNA cleavage induced when free etoposide was added to an unmodified duplex (site 19–20), and to a site that was not cleaved in the presence of free etoposide (site 29–30), respectively.
Figure 8.

Moving the position of the linked etoposide core along the bottom strand (OTI) sequence alters the topoisomerase II-mediated cleavage pattern of the top strand. (A) Sequences of the top/target and bottom PML strands are shown. The different-colored boxes indicate the position of the tethered etoposide core in each OTI (bottom strand), including OTI28 (yellow), OTI29 (orange), OTI33 (green), and OTI23 (purple). Arrows indicate the cleavage sites induced by each OTI (shown by corresponding colors). Large arrows indicate the major site of cleavage. T and B (first column) indicate top and bottom strands, respectively. (B) Comparison of DNA cleavage mediated by human topoisomerase IIα (hTIIα, left) and topoisomerase IIβ (hTIIβ, right) of the radiolabeled, unmodified PML top strand hybridized to an unmodified PML bottom strand in the presence of 0–500 μM free etoposide (lanes 2–3) or hybridized to the bottom strands OTI28 (lanes 5–6), OTI29 (lanes 8–9), OTI33 (lanes 11–12), or OTI23 (lanes 14–15). For each gel, lane 1 contains an unmodified PML duplex. Reference (R) oligonucleotides are 24, 23, 20 and 19 bases long. Gels are representative of at least three independent experiments.
In all three cases, strong topoisomerase IIα-mediated DNA cleavage induced by the new OTIs was observed two bases away opposite the drug location at the predicted sites described above (Figure 8B; left). As with OTI28, some of the new OTIs also induced DNA scission at sites that were not predicted by our modeling studies (Figures 1 and 4). However, these sites were all in the vicinity of the predicted sites of cleavage. Similar sites of DNA cleavage were obtained with these OTIs and topoisomerase IIβ (Figure 8B; right).
Two important conclusions can be drawn from these studies. First, OTIs can be used to induce DNA cleavage, at least in part, in a predictable manner. Second, sites of cleavage can be moved to sequences not normally associated with the actions of free etoposide (Figures 3A and 8A, B). Thus, the use of OTIs is not restricted to pre-existing etoposide-induced sites of cleavage, implying that it may be possible to design OTIs against almost any patient sequence associated with a cancer mutation or chromosomal breakpoint.
OTIs can be directed against a t(15;17) translocation breakpoint seen in a patient with APL
One of the goals in characterizing OTIs is to determine if it would be possible to target them to cancer-specific sequences present in malignant cells. Therefore, having established the proof of principle that OTIs can be used to direct the location of etoposide-induced topoisomerase II-mediated DNA cleavage, we designed an OTI against a t(15;17) chromosomal translocation (breakpoint at 1484–1485 in PML and 12034–12035 in RARA) in a patient with t-APL (Figure 9A) (30). This sequence was chosen because it encompasses the site of cleavage in the PML gene induced by OTI28 (Figure 3). Initial studies with the translocation sequence utilized a 50-base pair oligonucleotide that was comprised of bases 1461–1488 of intron 6 of PML (Figure 9A; blue) and 12039–12060 of intron 2 of RARA (Figure 9A; orange).
Figure 9.

OTIs designed against a patient-observed PML-RARA translocation increase DNA cleavage mediated by human type II topoisomerases. (A) Sequences of the top and bottom strands of each PML-RARA duplex are shown. The blue portion corresponds to the segment derived from the PML gene, and the orange portion corresponds to the segment derived from the RARA gene. The yellow box indicates the position of the tethered etoposide core on each OTI (bottom strand). The OTIs were 50, 30 or 20 bases in length (black lines below the diagram). Arrows indicate sites of DNA cleavage induced by free etoposide (blue) and the translocation OTIs (yellow). (B) Comparison of DNA cleavage mediated by human topoisomerase IIα (hTIIα, left) and topoisomerase IIβ (hTIIβ, right) of the radiolabeled, unmodified PML-RARA top/target strand hybridized to an unmodified PML-RARA bottom strand in the presence of free etoposide or of the radiolabeled PML-RARA top strand hybridized to a 50-mer, 30-mer or 20-mer PML-RARA OTI bottom strand. Lanes 1–5 contain the unmodified PML-RARA duplex in the absence of enzyme, or in the presence of enzyme and 0–500 μM free etoposide. Lanes 7 and 8 contain the unmodified PML-RARA top strand hybridized with the 50-mer OTI. Lanes 10 and 11 contain the unmodified top strand hybridized with the 30-mer OTI. Lanes 13 and 14 contain the unmodified top strand hybridized with the 20-mer OTI. Lanes 6, 10, 13, and 15 contain reference (R) oligonucleotides 24, 23, 20 and 19 bases in length. Gels are representative of at least three independent experiments. (C) Quantification of the relative levels of enzyme-mediated DNA cleavage. DNA cleavage at each site is normalized to the cleavage observed at site 24–25 in reactions containing an unmodified duplex in the absence of etoposide (lane 2). Results with unmodified PML–RARA duplex in the presence of 500 μM free etoposide (blue) or with unmodified top strand hybridized with 50-mer OTI (yellow), 30-mer OTI (orange), or 20-mer OTI (green) bottom strand are shown. Error bars represent the standard deviation of at least three independent experiments. Significance was determined by paired t-tests. P-values are indicated by asterisks (*P < 0.05; **P < 0.005; ***P < 0.0005).
First, the unmodified breakpoint oligonucleotide duplex was treated with free etoposide (0–500 μM) in the presence of topoisomerase IIα, and several cleavage sites were observed (Figure 9B, C; left). The strongest of these sites were observed at positions 24–25, 25–26 and 19–20 on the top strand (lane 5), in order of decreasing strength of cleavage.
Second, the unmodified target strand was hybridized with a complementary 50-base OTI in which the tethered etoposide core was located at the equivalent position in the PML gene as OTI29 (Figure 9A). As predicted, this OTI induced strong cleavage at position 23–24 (lane 8), at a site two bases away and opposite the tethered drug. Strong cleavage was also observed at sites 20–21 and 24–25 (lane 8).
Human type II topoisomerases display a DNA length-dependence for scission. The enzyme cleaves a 50-mer duplex oligonucleotide considerably better than it does a 40-mer, and little cleavage is seen with oligonucleotides shorter than a 30-mer (43). However, when one of the two strands is maintained at a 50-base length, levels of enzyme-mediated DNA cleavage remain relatively high until the length of the complementary strand is reduced to ∼20 bases (43). Therefore, to see if shorter OTIs could still be used to induce high levels of topoisomerase IIα-mediated DNA cleavage in the translocation sequence, we tested a 30-mer version of the breakpoint OTI in which 10 bases were removed from each terminus (Figure 9A). High levels of cleavage were maintained at sites 23–24 and 19–20; however, cleavage disappeared from site 24–25 (Figure 9B, C; lane 12). As predicted, when the breakpoint OTI was further reduced in size to a 20-mer version, DNA cleavage levels dropped precipitously (Figure 9B, C; lane 14).
In all respects, results obtained with topoisomerase IIβ and the chromosomal translocation model oligonucleotides (with either a free or a tethered etoposide core) were similar to those obtained with topoisomerase IIα (Figure 9B, C; right).
Finally, to determine whether the breakpoint OTIs displayed specificity for the translocation over the parental PML and RARA sequences, the 50-mer, 30-mer and 20-mer breakpoint OTIs were hybridized to 50-mer top strands of each parental gene. Very low levels of DNA cleavage were generated by either topoisomerase IIα (Figure 10A) or topoisomerase IIβ (Figure 10B) in the PML oligonucleotide (lanes 8, 10 and 12) and no cleavage was observed in the RARA oligonucleotide with any of the breakpoint OTIs (lanes 18, 20, 22). This finding suggests that OTIs could potentially be directed to cancer-specific breakpoints without generating significant levels of DNA cleavage in the wild-type parental sequences.
Figure 10.

OTIs that incorporate an APL patient-derived PML-RARA translocation sequence do not increase DNA cleavage mediated by human type II topoisomerases when they are hybridized with the parental PML or RARA sequences. Comparison of DNA cleavage mediated by human topoisomerase IIα (hTIIα) (A) and topoisomerase IIβ (hTIIβ) (B) of the radiolabeled top strand of an unmodified PML-RARA duplex in the presence of free etoposide, a radiolabeled, unmodified PML or RARA top strand hybridized to a PML-RARA bottom strand in the presence of free etoposide, or a radiolabeled, unmodified PML or RARA top strand hybridized to a 50-mer OTI, a 30-mer OTI, or a 20-mer PML-RARA OTI bottom strand. For each gel, lanes 1–3 contain unmodified PML-RARA top/target strand hybridized to the unmodified 50-mer PML-RARA bottom strand, in the absence of enzyme, or in the presence of enzyme and 0–500 μM etoposide. Lanes 4–6 contain unmodified parental PML top/target strand hybridized to the unmodified 50-mer PML-RARA bottom strand, in the absence of enzyme, or in the presence of enzyme and 0–500 μM etoposide. Lanes 7–12 contain the unmodified parental PML top strand hybridized to the 50-mer, 30-mer or 20-mer PML-RARA OTI bottom strand in the absence or presence of enzyme. Lanes 14–16 contain unmodified parental RARA top strand hybridized to the 50-mer PML-RARA bottom strand, in the absence of enzyme, or in the presence of enzyme and 0–500 μM etoposide. Lanes 17–22 contain the unmodified parental RARA top strand hybridized to the 50-mer, 30-mer, or 20-mer PML-RARA OTI bottom strand in the absence or presence of enzyme. Lanes 13 and 23 contain a combination of reference (R) oligonucleotides 24, 23, 20 and 19 bases in length. Gels are representative of at least three independent experiments.
DISCUSSION
Etoposide and other topoisomerase II-targeted drugs are important therapeutics for the treatment of cancer (1,2,15,17,22,56). Unfortunately, the induction of topoisomerase II-mediated DNA cleavage throughout the genome, coupled with the lack of specificity toward cancer cells, limits the safe usage of these drugs. Among the most insidious of toxicities related with treatment with topoisomerase II poisons is the development of secondary leukemias (17,23,49,57). Indeed, as many as 3% of patients treated with etoposide or doxorubicin develop AMLs with translocations at chromosomal band 11q23 (58,59) or treated with mitoxantrone develop APLs with t(15;17) translocations (42,60).
One approach to providing anticancer agents with reduced toxicities is to develop drugs that specifically inhibit the activity of driver oncoproteins. An example is imatinib, which targets the TK domain of the bcr-abl fusion tyrosine kinase that is generated by the chromosomal translocation in chronic myelogenous leukemia (61). An alternative approach would be to target the chromosomal translocation itself by specifically cleaving the DNA in the vicinity of the translocation site. This could lead to disruption of the oncogene or could interfere with its transcription. If either occurred, it ultimately could rob the cell of the oncoprotein that drives malignant tumor growth.
Previous attempts to impart specificity to topoisomerase-targeted drugs were based on the attachment of either camptothecin (a topoisomerase I poison) (62) or etoposide derivatives (63) to the ends of triplex-forming oligonucleotides. The attached camptothecin induced topoisomerase I-mediated DNA scission at sites that were consistent with the location of the linked drug (62). However, cleavage appeared to be constrained to pre-existing cleavage sites (62). In contrast, the attachment of etoposide to the end of a triplex-forming oligonucleotide induced DNA cleavage at locations that were approximately 10 base pairs away from sites that could have come into physical contact with the drug based on the length of the drug and linkers employed (63). Thus, the basis for etoposide-induced DNA cleavage that resulted from the triplex-forming oligonucleotides could not be determined (63).
Although the present study also utilized drug-linked oligonucleotides to enhance the specificity of etoposide-induced DNA cleavage, we took the approach of designing sequences that would form duplex (rather than triplex) structures at the proposed sites of cleavage. To this end, a series of oligonucleotides that contained a linked etoposide core (OTIs) was synthesized using a copper-catalyzed click chemistry scheme. OTIs were designed to take advantage of the following: 1. Etoposide acts by a known mechanism in which it enhances DNA cleavage mediated by type II topoisomerases (2,7,22). 2. The type II enzymes are validated targets for chemotherapeutic agents (2,15,16,22). 3. Levels of type II topoisomerases are generally higher in malignant cells, which generates higher levels of DNA strand breaks in treated cells (64,65). 4. Cancer cells often have high metabolic rates, which results in more replication and transcription complexes that convert topoisomerase II-DNA cleavage complexes into permanent double-strand breaks (2,10,15).
Results indicate that the tethered etoposide core in OTIs acts against type II topoisomerases in a manner similar to that of the free drug, but with a specificity (at least for the major sites of cleavage) that is tied to its site of linkage on the oligonucleotide. Furthermore, the linked drug can induce enzyme-mediated DNA cleavage at sites of action of the free drug or at sites at which free drug does not act. This finding suggests that it might be possible to develop OTIs against a broad range of translocation-related sequences. Results further suggest that OTIs can be designed to have high activity against cancer-related breakpoints but display limited activity against the parental genes.
It is notable that in some cases minor cleavage was observed at positions that were not predicted from modeling studies (Figures 1, 4 and Supplementary Figures S3 and S4). Although the basis for cleavage at these minor sites is not obvious, it is possible that they are induced by distortions in the oligonucleotide caused by the presence of the OTI. Alternatively, an OTI attached to the transport helix could act in trans to induce cleavage within the gate helix (Figure 1A). However, this seems unlikely, as cleavage patterns for these minor sites do not coincide with those observed in the presence of free etoposide (compare Figures 3–8).
In order for OTIs to function in cells and block the expression of driver oncogenes, they would have to hybridize to their target sequence. It is envisioned that this could potentially take place when the two strands of the double helix are separated during replication or transcription. In vitro experiments suggest that single-stranded oligonucleotides as short as 24-mers can invade the double helix and form a D-loop (66). Alternatively, invasion can be greatly enhanced by the presence of recombination proteins (67,68). Experiments are currently underway to assess the ability of OTIs to invade the double helix under a variety of conditions.
The development of oligonucleotide-based therapeutics has been challenging (69). However, improved chemistry and targeted delivery systems, together with the recent FDA approval of new RNA therapeutics (69), hold promise for the potential development of oligonucleotide-based agents. Future studies will be directed toward determining the effects of the OTIs in normal and cancer cells.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Rachel E. Ashley and Elizabeth G. Gibson for critical reading of the manuscript, Ben Andrews for early discussions about chemistry, Steve Jackson for help with chiral separations, Mark Edbrooke for support and advice on oligonucleotide delivery, Neil McDonald for support in early stages of the project, and Andrew Leach for support and advice.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [GM033944 and GM126363 to N.O.]; NIH [GM008554 to L.I.L.]. Funding for open access charge: NIH grant GM126363.
Conflict of interest statement. None declared.
REFERENCES
- 1. Deweese J.E., Osheroff M.A., Osheroff N.. DNA topology and topoisomerases: teaching a “knotty” subject. Biochem. Mol. Biol. Educ. 2008; 37:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deweese J.E., Osheroff N.. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res. 2009; 37:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nitiss J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 2009; 9:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vos S.M., Tretter E.M., Schmidt B.H., Berger J.M.. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell. Biol. 2011; 12:827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S.H., Chan N.-L., Hsieh T.-S.. New mechanistic and functional insights into DNA topoisomerases. Ann. Rev. Biochem. 2013; 82:139–170. [DOI] [PubMed] [Google Scholar]
- 6. Pommier Y., Sun Y., Huang S.-y.N., Nitiss J.L.. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016; 17:703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Osheroff N. Effect of antineoplastic agents on the DNA cleavage/religation reaction of eukaryotic topoisomerase II: inhibition of DNA religation by etoposide. Biochemistry. 1989; 28:6157–6160. [DOI] [PubMed] [Google Scholar]
- 8. Bromberg K.D., Burgin A.B., Osheroff N.. A two-drug model for etoposide action against human topoisomerase IIα. J. Biol. Chem. 2003; 278:7406–7412. [DOI] [PubMed] [Google Scholar]
- 9. Deweese J.E., Osheroff N.. The use of divalent metal ions by type II topoisomerases. Metallomics. 2010; 2:450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu X., Davenport J.W., Urtishak K.A., Carillo M.L., Gosai S.J., Kolaris C.P., Byl J.A.W., Rappaport E.F., Osheroff N., Gregory B.D. et al. . Genome-wide TOP2A DNA cleavage is biased toward translocated and highly transcribed loci. Genome Res. 2017; 27:1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ju B.G., Rosenfeld M.G.. A breaking strategy for topoisomerase IIβ/PARP-1-dependent regulated transcription. Cell Cycle. 2006; 5:2557–2560. [DOI] [PubMed] [Google Scholar]
- 12. Ju B.G., Lunyak V.V., Perissi V., Garcia-Bassets I., Rose D.W., Glass C.K., Rosenfeld M.G.. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science. 2006; 312:1798–1802. [DOI] [PubMed] [Google Scholar]
- 13. Yang X., Li W., Prescott E.D., Burden S.J., Wang J.C.. DNA topoisomerase IIβ and neural development. Science. 2000; 287:131–134. [DOI] [PubMed] [Google Scholar]
- 14. Lyu Y.L., Wang J.C.. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIβ. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:7123–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009; 9:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013; 8:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pendleton M., Lindsey R.H. Jr, Felix C.A., Grimwade D., Osheroff N.. Topoisomerase II and leukemia. Ann. N.Y. Acad. Sci. 2014; 1310:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu C.C., Li T.K., Farh L., Lin L.Y., Lin T.S., Yu Y.J., Yen T.J., Chiang C.W., Chan N.L.. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011; 333:459–462. [DOI] [PubMed] [Google Scholar]
- 19. Woessner R.D., Mattern M.R., Mirabelli C.K., Johnson R.K., Drake F.H.. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991; 2:209–214. [PubMed] [Google Scholar]
- 20. Puigvert J.C., Sanjiv K., Helleday T.. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016; 283:232–245. [DOI] [PubMed] [Google Scholar]
- 21. Murai J. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int. J. Clin. Oncol. 2017; 22:619–628. [DOI] [PubMed] [Google Scholar]
- 22. Baldwin E.L., Osheroff N.. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents. 2005; 5:363–372. [DOI] [PubMed] [Google Scholar]
- 23. Felix C.A., Kolaris C.P., Osheroff N.. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst.). 2006; 5:1093–1108. [DOI] [PubMed] [Google Scholar]
- 24. Joannides M., Mays A.N., Mistry A.R., Hasan S.K., Reiter A., Wiemels J.L., Felix C.A., Coco F.L., Osheroff N., Solomon E. et al. . Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2011; 3:e2011045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cowell I.G., Austin C.A.. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health. 2012; 9:2075–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cowell I.G., Sondka Z., Smith K., Lee K.C., Manville C.M., Sidorczuk-Lesthuruge M., Rance H.A., Padget K., Jackson G.H., Adachi N. et al. . Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowley J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973; 243:290–293. [DOI] [PubMed] [Google Scholar]
- 28. Barreca A., Lasorsa E., Riera L., Machiorlatti R., Piva R., Ponzoni M., Kwee I., Bertoni F., Piccaluga P.P., Pileri S.A. et al. . Anaplastic lymphoma kinase in human cancer. J. Mol. Endocrinol. 2011; 47:R11–R23. [DOI] [PubMed] [Google Scholar]
- 29. Stenman G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head Neck Pathol. 2013; 7(Suppl. 1):S12–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hasan S.K., Mays A.N., Ottone T., Ledda A., La Nasa G., Cattaneo C., Borlenghi E., Melillo L., Montefusco E., Cervera J. et al. . Molecular analysis of t(15;17) genomic breakpoints in secondary acute promyelocytic leukemia arising after treatment of multiple sclerosis. Blood. 2008; 112:3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P. Tools for ligand validation in Coot. Acta. Crystallogr. D. Struct. Biol. 2017; 73:203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chemical Computing Group ULC 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A,2R7, Vol. 2013.08. 2017.
- 33. Wang Y.-R., Chen S.-F., Wu C.-C., Liao Y.-W., Lin T.-S., Liu K.-T., Chen Y.-S., Li T.-K., Chien T.-C., Chan N.-L.. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017; 45:10861–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan P.F., Srikannathasan V., Huang J., Cui H., Fosberry A.P., Gu M., Hann M.M., Hibbs M., Homes P., Ingraham K. et al. . Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015; 6:10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt B.H., Burgin A.B., Deweese J.E., Osheroff N., Berger J.M.. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature. 2010; 465:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Worland S.T., Wang J.C.. Inducible overexpression, purification, and active site mapping of DNA topoisomerase II from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1989; 264:4412–4416. [PubMed] [Google Scholar]
- 37. Wasserman R.A., Austin C.A., Fisher L.M., Wang J.C.. Use of yeast in the study of anticancer drugs targeting DNA topoisomerases: expression of a functional recombinant human DNA topoisomerase IIα in yeast. Cancer Res. 1993; 53:3591–3596. [PubMed] [Google Scholar]
- 38. Elsea S.H., Hsiung Y., Nitiss J.L., Osheroff N.. A yeast type II topoisomerase selected for resistance to quinolones. Mutation of histidine 1012 to tyrosine confers resistance to nonintercalative drugs but hypersensitivity to ellipticine. J. Biol. Chem. 1995; 270:1913–1920. [DOI] [PubMed] [Google Scholar]
- 39. Kingma P.S., Greider C.A., Osheroff N.. Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry. 1997; 36:5934–5939. [DOI] [PubMed] [Google Scholar]
- 40. Byl J.A., Cline S.D., Utsugi T., Kobunai T., Yamada Y., Osheroff N.. DNA topoisomerase II as the target for the anticancer drug TOP-53: mechanistic basis for drug action. Biochemistry. 2001; 40:712–718. [DOI] [PubMed] [Google Scholar]
- 41. Liu Q., Wang J.C.. Identification of active site residues in the “GyrA” half of yeast DNA topoisomerase II. J. Biol. Chem. 1998; 273:20252–20260. [DOI] [PubMed] [Google Scholar]
- 42. Mistry A.R., Felix C.A., Whitmarsh R.J., Mason A., Reiter A., Cassinat B., Parry A., Walz C., Wiemels J.L., Segal M.R. et al. . DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N. Engl. J. Med. 2005; 352:1529–1538. [DOI] [PubMed] [Google Scholar]
- 43. Deweese J.E., Burgin A.B., Osheroff N.. Using 3′-bridging phosphorothiolates to isolate the forward DNA cleavage reaction of human topoisomerase IIα. Biochemistry. 2008; 47:4129–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Byl J.A., Fortune J.M., Burden D.A., Nitiss J.L., Utsugi T., Yamada Y., Osheroff N.. DNA topoisomerases as targets for the anticancer drug TAS-103: Primary cellular target and DNA cleavage enhancement. Biochemistry. 1999; 38:15573–15579. [DOI] [PubMed] [Google Scholar]
- 45. Bromberg K.D., Velez-Cruz R., Burgin A.B., Osheroff N.. DNA ligation catalyzed by human topoisomerase IIα. Biochemistry. 2004; 43:13416–13423. [DOI] [PubMed] [Google Scholar]
- 46. Bandele O.J., Osheroff N.. The efficacy of topoisomerase II-targeted anticancer agents reflects the persistence of drug-induced cleavage complexes in cells. Biochemistry. 2008; 47:11900–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilstermann A.M., Bender R.P., Godfrey M., Choi S., Anklin C., Berkowitz D.B., Osheroff N., Graves D.E.. Topoisomerase II - drug interaction domains: Identification of substituents on etoposide that interact with the enzyme. Biochemistry. 2007; 46:8217–8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bender R.P., Jablonksy M.J., Shadid M., Romaine I., Dunlap N., Anklin C., Graves D.E., Osheroff N.. Substituents on etoposide that interact with human topoisomerase IIα in the binary enzyme-drug complex: Contributions to etoposide binding and activity. Biochemistry. 2008; 47:4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joannides M., Grimwade D.. Molecular biology of therapy-related leukaemias. Clin. Transl. Oncol. 2010; 12:8–14. [DOI] [PubMed] [Google Scholar]
- 50. Kingma P.S., Osheroff N.. The response of eukaryotic topoisomerases to DNA damage. Biochim. Biophys. Acta. 1998; 1400:223–232. [DOI] [PubMed] [Google Scholar]
- 51. Sabourin M., Osheroff N.. Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res. 2000; 28:1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elsea S.H., Hsiung Y., Nitiss J.L., Osheroff N.. A yeast type II topoisomerase selected for resistance to quinolones. Mutation of histidine 1012 to tyrosine confers resistance to nonintercalative drugs but hypersensitivity to ellipticine. J. Biol. Chem. 1995; 270:1913–1920. [DOI] [PubMed] [Google Scholar]
- 53. Matsumoto Y., Takano H., Kunishio K., Nagao S., Fojo T.. Incidence of mutation and deletion in topoisomerase IIα mRNA of etoposide and mAMSA-resistant cell lines. Jpn. J. Cancer Res. 2001; 92:1133–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanagasabai R., Serdar L., Karmahapatra S., Kientz C.A., Ellis J., Ritke M.K., Elton T.S., Yalowich J.C.. Alternative RNA processing of topoisomerase IIα in etoposide-resistant human leukemia K562 cells: intron retention results in a novel C-terminal truncated 90-kDa isoform. J. Pharmacol. Exp. Ther. 2017; 360:152–163. [DOI] [PubMed] [Google Scholar]
- 55. Kingma P.S., Burden D.A., Osheroff N.. Binding of etoposide to topoisomerase II in the absence of DNA: decreased affinity as a mechanism of drug resistance. Biochemistry. 1999; 38:3457–3461. [DOI] [PubMed] [Google Scholar]
- 56. Bender R.P., Osheroff N.. Dai W. Checkpoint Responses in Cancer Therapy. 2008; 1st ednTotowa: Humana Press; 57–91. [Google Scholar]
- 57. Mays A.N., Osheroff N., Xiao Y., Wiemels J.L., Felix C.A., Byl J.A., Saravanamuttu K., Peniket A., Corser R., Chang C. et al. . Evidence for direct involvement of epirubicin in the formation of chromosomal translocations in t(15;17) therapy-related acute promyelocytic leukemia. Blood. 2010; 115:326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ratain M.J., Kaminer L.S., Bitran J.D., Larson R.A., Le Beau M.M., Skosey C., Purl S., Hoffman P.C., Wade J., Vardiman J.W. et al. . Acute nonlymphocytic leukemia following etoposide and cisplatin combination chemotherapy for advanced non-small-cell carcinoma of the lung. Blood. 1987; 70:1412–1417. [PubMed] [Google Scholar]
- 59. DeVore R., Whitlock J., Hainsworth J.D., Johnson D.H.. Therapy-related acute nonlymphocytic leukemia with monocytic features and rearrangement of chromosome 11q. Ann. Intern. Med. 1989; 110:740–742. [DOI] [PubMed] [Google Scholar]
- 60. Ramkumar B., Chadha M.K., Barcos M., Sait S.N., Heyman M.R., Baer M.R.. Acute promyelocytic leukemia after mitoxantrone therapy for multiple sclerosis. Cancer Genet. Cytogenet. 2008; 182:126–129. [DOI] [PubMed] [Google Scholar]
- 61. Druker B.J., Tamura S., Buchdunger E., Ohno S., Segal G.M., Fanning S., Zimmermann J., Lydon N.B.. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996; 2:561–566. [DOI] [PubMed] [Google Scholar]
- 62. Arimondo P.B., Thomas C.J., Oussedik K., Baldeyrou B., Mahieu C., Halby L., Guianvarc’h D., Lansiaux A., Hecht S.M., Bailly C. et al. . Exploring the cellular activity of camptothecin-triple-helix-forming oligonucleotide conjugates. Mol. Cell Biol. 2006; 26:324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Duca M., Guianvarc’h D., Oussedik K., Halby L., Garbesi A., Dauzonne D., Monneret C., Osheroff N., Giovannangeli C., Arimondo P.B.. Molecular basis of the targeting of topoisomerase II-mediated DNA cleavage by VP16 derivatives conjugated to triplex-forming oligonucleotides. Nucleic Acids Res. 2006; 34:1900–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davies S.M., Robson C.N., Davies S.L., Hickson I.D.. Nuclear topoisomerase II levels correlate with the sensitivity of mammalian cells to intercalating agents and epipodophyllotoxins. J. Biol. Chem. 1988; 263:17724–17729. [PubMed] [Google Scholar]
- 65. Coutts J., Plumb J.A., Brown R., Keith W.N.. Expression of topoisomerase IIα and β in an adenocarcinoma cell line carrying amplified topoisomerase IIα and retinoic acid receptor alpha genes. Br. J. Cancer. 1993; 68:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Necasova I., Janouskova E., Klumpler T., Hofr C.. Basic domain of telomere guardian TRF2 reduces D-loop unwinding whereas Rap1 restores it. Nucleic Acids Res. 2017; 45:12170–12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li X., Stith C.M., Burgers P.M., Heyer W.D.. PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol. Cell. 2009; 36:704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bower B.D., Griffith J.D.. TRF1 and TRF2 differentially modulate Rad51-mediated telomeric and nontelomeric displacement loop formation in vitro. Biochemistry. 2014; 53:5485–5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kaczmarek J.C., Kowalski P.S., Anderson D.G.. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017; 9:60–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.