

Abstract
Background
Atypical idiopathic pulmonary fibrosis (IPF) including multiple cysts or markedly atelectatic induration in upper lung predominance occasionally can confirm the diagnosis of IPF through a multidisciplinary discussion (MDD) between clinician, radiologist and, pathologist in clinical practice. The aim of this study was to clarify the differences in clinico-radiological characteristics and the efficacy of anti-fibrotic agents between atypical IPF and typical IPF.
Methods
We retrospectively evaluated the differences in clinico-radiological characteristics between patients with atypical IPF (n=44) and those with typical IPF (n=87) and examined efficacy of anti-fibrotic agents in atypical IPF. Atypical IPF was characterized by the presence of markedly atelectatic induration in upper lung predominance (pleuroparenchymal fibroelastosis; PPFE like lesion) with and without multiple thick-walled large cysts (TWLC), so-called macrocystic honeycombing (TWLC; >2.5 cm in diameter with 1–3 mm thickness) in addition to honeycombing in the bilateral lower lobes predominance.
Results
There was no difference in the baseline disease severity for IPF between both groups. The annual change value of fibrotic score and traction bronchiectasis (TBE) score, and decreased changes in forced vital capacity (FVC) during 6 months were significantly higher in atypical IPF than those in typical IPF. Survival time was significantly lower in patients with atypical IPF (MST: 33.4 vs. 47.9 months, P=0.03). The multivariate Cox regression model demonstrated that the prognostic predictors were presence of atypical IPF and increased Gender-Age-Physiology (GAP) staging. Moreover, the rate of decrease in FVC value 6 months after treatment with anti-fibrotic agents was significantly higher in atypical IPF than those in typical IPF (−11.8%±14.0% vs. −1.0%±12.7%; P=0.01).
Conclusions
This study demonstrated that the prognosis for atypical IPF was significantly worse than that for typical IPF. Future studies are required prospective analyses of efficacy of anti-fibrotic agents for patients with atypical IPF.
Keywords: Idiopathic pulmonary fibrosis (IPF), usual interstitial pneumonia (UIP), pleuroparenchymal fibroelastosis, anti-fibrotic agents, prognosis
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonias (IIPs) characterized radiologically and histologically by the features of usual interstitial pneumonia (UIP) (1). Although the median survival for patients with IPF is recognized 2.5–3.5 years after diagnosis (2), the clinical course of each patient is variable (3).
According to American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) guidelines published in 2011 (1), chest high-resolution computed tomography (HRCT) is an essential key tool of the diagnostic algorithm for IPF. A definite UIP pattern on chest HRCT includes subpleural/basal predominance, reticular abnormalities, honeycombing with or without traction bronchiectasis (TBE), and lack of features inconsistent with UIP. However, the identification of atypical IPF including multiple cysts or atelectatic induration occasionally can confirm the diagnosis of IPF through a multidisciplinary discussion (MDD) between clinician, radiologist and, pathologist in clinical practice.
To our knowledge, few reports are available on comparison of clinic-radiological characteristics and outcomes between patients with atypical and typical IPF. Herein, we compared the clinical features and outcomes between atypical and typical IPF divided based on chest HRCT images, as well as to elucidate prognostic factors and efficacy of anti-fibrotic agents.
Methods
Study population
The study cohort included patients enrolled at Toho University Omori Medical Center in Japan between April 2003 and March 2015. During the study period, 87 patients with typical IPF and 44 patients with atypical IPF were analyzed retrospectively. The diagnosis of typical IPF was made by a multidisciplinary clinico-radiologic-pathological review of the patient data in accordance with the 2011 ATS/ERS/JRS/ALAT consensus statement (1). In contrast, atypical IPF was characterized by the presence of markedly atelectatic induration in upper lung predominance (pleuroparenchymal fibroelastosis; PPFE like lesion) with and without multiple thick-walled large cysts (TWLC), so-called macrocystic honeycombing (TWLC; >2.5 cm in diameter with 1–3 mm thickness) in addition to honeycombing predominantly in both lower lobe (classic UIP features) (Figure 1).
Figure 1.

The representative cases of typical and atypical IPF. (A) Typical IPF; (B) PPFE like lesion; (C) mixed type: PPFE + TWLC. IPF, idiopathic pulmonary fibrosis; PPFE, pleuroparenchymal fibroelastosis; TWLC, thick-walled large cysts.
Acute exacerbation (AE) of IPF was diagnosed by criteria proposed by Collard et al. (4).
Gender-Age-Physiology (GAP) score was calculated by gender, age, forced vital capacity (FVC) % predicted and diffusion capacity (DLco) % predicted and patients divided to severity of staging such as stage I–III as previously described (5).
The study was approved by the local Ethics Committee of the Toho University Omori Medical Center (No. M16263).
Chest CT scan
A helical CT scanner (Aquilion 16, Toshiba, Tokyo, Japan) was applied. Thin-section CT scans were obtained at full inspiration and scanning protocol consisted of reconstruction of 1–2-mm collimation sections with a high-spatial-frequency algorithm at 1- or 2-cm intervals. CT images of the chest were photographed at window settings appropriate for the lung parenchyma [window level from −600 Hounsfield Units (HU); width from 1,600 HU] for all patients.
The lungs were divided into 6 lung zones (upper, middle, and lower zones in the bilateral lugs).The upper zone was defined as the area of the lung above the level of the tracheal carina, the lower zone as the area of the lung below the level of the inferior pulmonary vein, and the middle zone as the area of the lung between the upper and lower zones, respectively. The extent of lung involvement was evaluated visually and independently for each of the 6 lung zone. To examine interstitial fibrosis, fibrosis score was assigned: 0, none; 1, ground glass opacity without reticulation; 2, ground glass and fine reticular opacity; 3, reticular opacity and microcysts less than 3 mm in diameter; or 4, coarse reticular opacity and large cysts more than 3 mm in diameter (6). TBE was originally assigned with categorical severity score that considered the average degree of airway dilatation within areas of fibrosis; 0, none; 1, mild; 2, moderate; 3, severe (7). The fibrosis or TBE scores were assessed for each of the 6 lung zones and then summed. Extent of fibrosis (the overall % of lung involvement) was calculated by averaging the 6 lung zones and recorded to the nearest 5%. Extent of TBE was classified into 4 groups; 0, none; 1, existence of 1 segment; 2, existence of 2 segments; 3, existence of more than 2 segments. A consensus reading of the CT images was analyzed independently by 1 pulmonologist (K.S.) and 1 radiologist (K.M.).
Pulmonary function test (PFT)
Spirometry and the measurement of DLco were performed using a PFT system (Chestac-33, CHEST Co. Ltd., Tokyo, Japan). The diffusion capacity was measured by the single breath technique (8). The composite physiologic index (CPI) was calculated using the formula proposed by Wells (9).
Measurement of the estimated systolic pulmonary arterial pressure (esPAP)
The esPAP was calculated from measurements using transthoracic Doppler echocardiography with room air. The transtricuspid pressure gradient was calculated using the modified Bernoulli equation and was considered to be equal to the equal to the esPAP in the absence of right ventricular outflow obstruction: esPAP = transtricuspid pressure gradient + right atrial pressure.
Measurement of the levels of the serum markers
The serum level of Krebs von den Lungen (KL)-6 and surfactant protein (SP)-D were measured using commercially a KL-6 enzyme-linked immunosorbent assay (ELISA) kit (Eisai Co. Ltd., Tokyo, Japan) and a SP-D ELISA kit (Yamasa, Tokyo, Japan), respectively. The normal levels of serum KL-6 and SP-D were <500 U/mL and <110 ng/mL, respectively.
Statistical analysis
Data were expressed as mean ± standard deviation. Statistical analysis for continuous data between two groups was performed using the Wilcoxon rank sum test or the Student t-test, as appropriate. When categorical variables were compared, the Chi-square test and Fisher’s exact test was used, as appropriate. Prognostic significance of each parameter was analyzed by univariate and multivariate Cox proportional hazard regression analysis. The survival rate was calculated by the Kaplan-Meier method, and the log rank test was applied with the significance level set at <5%. P<0.05 was regarded as statistically significant. Statistical analysis was performed using JMP, version 10.0.0 (SAS Institute, Cary, NC, USA).
Results
Baseline clinical differences between patients with atypical and typical IPF
The ratio of males, smoking history, smoking index values, rate of dust exposure, and the incidence of lung cancer and AE were significantly higher in patients with typical IPF than in those with atypical IPF. There was no difference in the baseline disease severity (GAP staging) for IPF between both groups (stage I/II/III =18/20/6 vs. 40/35/12, P=0.84) and duration from the onset to initial visit (P=0.64) (Table 1). On the other hand, duration from initial visit to disease progression was shorter in patients with atypical IPF than in those with typical IPF (MST =12.1 vs. 40.4 months, P<0.0001) (Figure 2).
Table 1. Comparison of demographic characteristics between atypical and typical IPF.
| Variable | Atypical IPF (n=44) | Typical IPF (n=87) | P value |
|---|---|---|---|
| Age (years) | 72.6±7.3 | 71.8±7.7 | 0.55 |
| Sex (male/female) | 28/16 | 70/17 | 0.04 |
| Smoking history (current/ever/never) | 6/20/18 | 16/57/14 | 0.01 |
| Smoking index# | 404±630 | 771±573 | 0.001 |
| Dust exposure (%) | 2 (4.6) | 27 (31.0) | 0.0006 |
| mMRC score (0/I/II/III/IV) | 4/18/19/3/0 | 9/26/34/14/4 | 0.27 |
| Severity of IPF (GAP stage: I/II/III) | 18/20/6 | 40/35/12 | 0.84 |
| AE (%) | 5 (11.4) | 24 (27.6) | 0.03 |
| Primary lung cancer (%) | 0 (0.0) | 9 (10.3) | 0.03 |
| Histological UIP diagnosis (%) | 9 (20.5) | 25 (28.7) | 0.31 |
| Duration from the onset to initial visit (days) | 602±756 | 534±624 | 0.64 |
Data are presented as mean ± SD. #Smoking index, number of cigarettes consumed per day multiplied by years of smoking. IPF, idiopathic pulmonary fibrosis; mMRC, modified Medical Research Council; GAP, gender, age, and lung physiology; UIP, usual interstitial pneumonia; SD, standard deviation.
Figure 2.

Cumulative incidence of disease progression in the patients with typical IPF and atypical IPF. The prevalence of disease progression at a 5-year in patients with typical IPF (75.9%) was lower than in those with atypical IPF (100%) (MST: 12.1 vs. 22.6 months, P<0.0001). IPF, idiopathic pulmonary fibrosis.
Baseline values of %FVC, %FEV1, and serum value of KL-6 in patients with atypical IPF were significantly lower than those in patients with typical IPF, whereas CPI was significantly higher for atypical IPF patients. Moreover, stratified analysis for %FVC demonstrated that the change of FVC decline in atypical IPF patients with %FVC ≥70% was significantly greater than that in typical IPF patients, whereas, there were no significant differences between patients with atypical and typical IPF with %FVC <60% and 60%≤ %FVC <70% in the change of FVC decline during 6 months (Figure 3). The change in decline of FVC and %DLco during 6 months was significantly greater in atypical IPF patients, respectively (−0.23±0.34 vs. −0.11±0.18 L, P=0.01; −10.9±16.7 vs. −3.5±11.0 L, P=0.008) (Table 2).
Figure 3.
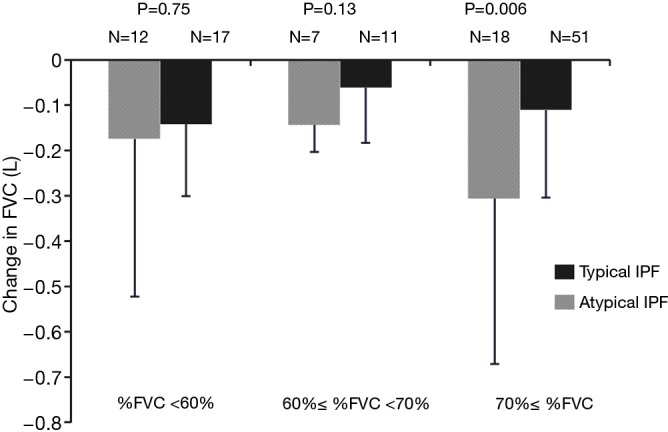
Comparison of stratification of change in FVC between typical IPF and atypical IPF. The change in FVC decline in atypical IPF patients with %FVC ≥70% was significantly greater than that in typical IPF patients, whereas, there were no significant differences between patients with atypical and typical IPF with %FVC <60% and 60%≤ %FVC <70% in the change of FVC decline during 6 months. FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis.
Table 2. Comparison of PFTs, serum markers, esPAP, and 6MWT among patients with atypical and typical IPF.
| Variable | Atypical IPF (n=44) | Typical IPF (n=87) | P value |
|---|---|---|---|
| FVC % predicted | 67.0±15.2 | 77.6±18.3 | 0.002 |
| FEV1% predicted | 85.5±24.6 | 94.8±21.3 | 0.03 |
| TLC % predicted | 72.1±13.8 | 76.0±16.5 | 0.18 |
| DLco % predicted | 58.9±18.9 | 54.8±17.9 | 0.27 |
| CPI | 63.3±16.6 | 52.2±15.2 | 0.0002 |
| ÄFVC/6 mo, L | −0.23±0.34 | −0.11±0.18 | 0.01 |
| Ä%DLco/6 mo, % | −10.9±16.7 | −3.5±11.0 | 0.008 |
| KL-6, U/mL | 883±496 | 1,190±716 | 0.01 |
| SP-D, ng/mL | 279±179 | 267±183 | 0.72 |
| ÄKL-6/6 mo, U/mL | −78±440 | −40±547 | 0.69 |
| ÄSP-D/6 mo, ng/mL | 25±119 | −13±130 | 0.11 |
| esPAP, mmHg | 33.7±9.8 | 30.7±7.8 | 0.22 |
| ÄesPAP/6 mo, mmHg | 5.6±9.1 | 4.0±10.6 | 0.46 |
| 6MWD, m | 335±126 | 313±138 | 0.39 |
| Lowest SpO2, % | 89.3±4.8 | 88.3±5.0 | 0.32 |
Data are presented as mean ± SD. IPF, idiopathic pulmonary fibrosis; CPI, composite physiologic index; KL-6, Kreb von den Lungen-6; SP-D, surfactant protein D; esPAP, estimated systolic pulmonary arterial pressure; 6MWD, 6-minute walking distance; SD, standard deviation.
Comparison of chest CT images between patients with atypical and typical IPF
There were no significant differences between patients with atypical and typical IPF in the baseline chest CT images. However, the annual change value of fibrosis score, extent of fibrosis, severity of TBE, and extent of TBE were significantly greater in patients with atypical IPF (Table 3).
Table 3. Comparison of chest CT images among patients with atypical and typical IPF.
| Variable | Atypical IPF (n=44) | Typical IPF (n=87) | P value |
|---|---|---|---|
| Fibrosis score | 13.7±4.0 | 15.1±3.4 | 0.08 |
| Extent of fibrosis | 25.7±11.2 | 26.8±10.1 | 0.64 |
| Severity of BE (none/mild/moderate/severe) | 0/16/26/2 | 2/44/40/1 | 0.57 |
| Extent of BE | 1.8±0.8 | 1.5±0.8 | 0.09 |
| ÄFibrosis score/12 mo | 4.5±2.8 | 2.0±2.0 | <0.0001 |
| ÄExtent of fibrosis/12 mo | 8.6±5.9 | 5.5±3.9 | 0.002 |
| ÄSeverity of BE/12 mo | 0.8±0.5 | 0.3±0.6 | 0.0001 |
| ÄExtent of BE/12 mo | 0.7±0.7 | 0.3±0.4 | 0.0002 |
Data are presented as mean ± SD. IPF, idiopathic pulmonary fibrosis; BE, bronchiectasis; SD, standard deviation.
Overall survival and prognostic significance of patients with atypical IPF
Duration of survival was significantly shorter in patients with atypical IPF (MST: 33.4 vs. 47.9 months, P=0.03) (Figure 4). In particular, atypical IPF complicating PPFE like lesion with multiple TWLC had worst prognosis (MST: Typical IPF/PPFE like lesion/mixed type =47.9/59.5/20.6 months; P=0.0004) (Figure 5). With regard to prognostic factors for entire IPF patients for survival, the multivariate Cox regression model demonstrated that the prognostic predictors were presence of atypical IPF (HR =2.07, 95% CI: 1.087–3.897, P=0.03) and increased GAP staging (Table 4). Regarding the most unfavorable prognostic predictors for patients with atypical IPF, mixed type with PPFE like lesion and multiple TWLC (HR =2.77, 95% CI: 1.535–4.8205, P=0.001) and increased GAP staging were significant factors in multivariate Cox proportional hazard regression analyses (Table 5). Also, overall survival was poorer in atypical IPF patients than in typical IPF patients with %FVC ≥70% (MST: 37.7 vs. 63.2 months, P=0.055). With regard to prognostic factors for IPF patients with %FVC ≥70%, the multivariate Cox proportional hazard regression model revealed males (HR =3.19, 95% CI: 1.03–14.07, P=0.04), atypical IPF with mixed type (HR =6.73, 95% CI: 2.04–19.69, P=0.003), and %DLco (HR =0.96, 95% CI: 0.94–0.99, P=0.003).
Figure 4.

The Kaplan-Meier survival curve in patients with typical IPF (solid line) (n=87) and atypical IPF (dashed line) (n=44). Survival time was significantly shorter in those with atypical IPF than in those with typical IPF (MST: 33.4 vs. 47.9 months, P=0.03). IPF, idiopathic pulmonary fibrosis.
Figure 5.

Comparison of survival curves between 3 groups; atypical IPF with PPFE like lesion (dotted line) (n=20), atypical IPF with mixed type (PPFE + TWLC) (bold solid line) (n=24), and typical IPF (solid line) (n=87). The MST in each group was 59.5, 20.6, and 47.9 months, respectively. Survival in atypical IPF patients with mixed type of PPFE and TWLC had significantly worse than that in the other 3 groups (P=0.0004). IPF, idiopathic pulmonary fibrosis; PPFE, pleuroparenchymal fibroelastosis; TWLC, thick-walled large cysts.
Table 4. Prognostic factors of patients with entire IPF for survival (n=131)—The Multivariate Cox Regression Model.
| Variable | HR | 95% CI | P value |
|---|---|---|---|
| Atypical IPF | 2.073 | 1.087–3.897 | 0.03 |
| GAP severity (stage I vs. III) | 4.599 | 1.262–17.168 | 0.02 |
| GAP severity (stage II vs. III) | 4.522 | 1.725–11.711 | 0.003 |
| mMRC | 1.272 | 0.929–1.753 | 0.13 |
| Fibrosis score | 0.928 | 0.819–1.052 | 0.24 |
| Extent of fibrosis | 1.012 | 0.971–1.055 | 0.58 |
| BE severity | 1.626 | 0.828–3.145 | 0.16 |
| Extent of BE | 1.264 | 0.836–1.945 | 0.27 |
| SpO2 <90% | 1.046 | 0.595–1.876 | 0.88 |
| FVC % predicted | 0.995 | 0.969–1.023 | 0.74 |
| DLco % predicted | 0.988 | 0.967–1.008 | 0.24 |
IPF, idiopathic pulmonary fibrosis; GAP, gender, age, and lung physiology; mMRC, modified Medical Research Council; BE, bronchiectasis.
Table 5. Prognostic factors of patients with atypical IPF for survival (n=44)—The Multivariate Cox Regression Model.
| Variable | HR | 95% CI | P value |
|---|---|---|---|
| PPFE + TWLC | 2.773 | 1.535–4.8205 | 0.001 |
| %FVC | 0.986 | 0.964–1.007 | 0.178 |
| %DLco | 0.985 | 0.967–1.001 | 0.069 |
| GAP severity (stage I vs. III) | 4.664 | 1.419–15.386 | 0.011 |
| GAP severity (stage II vs. III) | 4.011 | 1.742–9.064 | 0.001 |
PPFE, pleuroparenchymal fibroelastosis; TWLC, thick-walled large cysts; GAP, gender, age, and lung physiology.
Treatments for patients with atypical and typical IPF
Forty two of 44 atypical IPF patients and 77 of 87 typical IPF patients received medication, respectively. The rate of administration of pirfenidone in patients with atypical IPF was significantly higher than that in patients with typical IPF (59.1% vs. 39.1%, P=0.04) (Table 6).
Table 6. Treatments for atypical and typical IPF.
| Variable, n (%) | Atypical IPF (n=44) | Typical IPF (n=87) | P value |
|---|---|---|---|
| Pirfenidone | 26 (59.1) | 34 (39.1) | 0.04 |
| Nintedanib | 2 (4.6) | 4 (4.6) | 1.00 |
| Inhaled N-acetylcysteine monotherapy | 8 (18.2) | 24 (27.6) | 0.29 |
| Prednisolone | 4 (9.1) | 3 (3.5) | 0.22 |
| Inhaled N-acetylcysteine + prednisolone | 1 (2.3) | 8 (9.2) | 0.27 |
| Prednisolone + cyclosporine | 1 (2.3) | 4 (4.6) | 0.66 |
| None | 2 (4.6) | 10 (11.5) | 0.34 |
IPF, idiopathic pulmonary fibrosis.
Forty two patients with IPF were introduced anti-fibrotic agents during over 6 months (atypical IPF/typical IPF =20/22 cases). Eighteen of 20 patients with atypical IPF and 21 of 22 patients with typical IPF were treated with pirfenidone, and each remaining patient received nintedanib, respectively. The rate of decrease in FVC value 6 months after treatment with anti-fibrotic agents was significantly higher in atypical IPF than those in typical IPF (−11.8%±14.0% vs. −1.0%±12.7%; P=0.01) (Figure 6).
Figure 6.
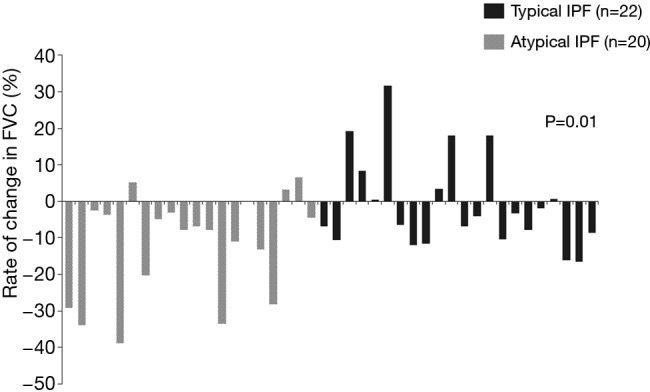
Relative rate of change in FVC after treatments with anti-fibrotic agents (atypical IPF/typical IPF =22/20 cases) for 6 months. The rate of decrease in FVC value 6 months after treatment with anti-fibrotic agents was significantly higher in atypical IPF than those in typical IPF (−11.8%±14.0% vs. −1.0%±12.7%; P=0.01). FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis.
Causes of death
The most frequent causes of death were pneumonia and chronic deterioration in patients with atypical IPF [each 10 of 27 deaths (37.0%)], and AE in patients with typical IPF [20 of 58 deaths (34.4%)], respectively. There were no significant differences in causes of death between patients with atypical and typical IPF.
Discussion
This is the first report that demonstrated differences of clinical features and outcomes in 2 subtypes of IPF divided distinctly atypical and typical pattern on chest HRCT.
IPF is a distinct clinical entity, associated with unexplained chronic progressive and fatal interstitial lung disease characterized by radiological and histological appearances consistent with UIP (1). The reasons for PPFE like lesion and/or the existence of multiple TWLC in the setting of IPF patients are still unclear. In our study, given that the ratio of males, smoking history, rate of dust exposure, and the incidence of lung cancer and AE were significantly lower in patients with atypical IPF than in those with typical IPF, atypical IPF may be treated as other disorders such as lung dominant connective tissue disease (CTD) or chronic hypersensitivity pneumonitis (CHP). However, secondary causes of pulmonary fibrosis were excluded to the extent possible, based on evaluation of clinical manifestation of CTD or clinical audit of relevant exposure history, detection of serum precipitins or autoantibody, lymphocytosis in bronchoalveolar lavage fluid, and/or characteristic histologic abnormalities such as granulomas, peribronchiolitis, and so on.
As reported by Sverzellati et al. (10), 34 of 55 biopsy-proven IPF patients had chest CT images that were regarded as having alternative diagnosis such as nonspecific interstitial pneumonia (NSIP), CHP, and sarcoidosis. In this study, there was no difference in survival between typical and atypical findings of UIP. In contrast, Silva et al. (11). reported that in 5 of 18 patients with initial findings suggestive of NSIP, the follow-up chest CT scans developed more suggestive of UIP. Indeed, survival differences between patients with typical and atypical UIP on chest HRCT patterns may reflect the lead-time bias of early or late diagnosis against IPF. In the present study, patients with atypical IPF have a worse prognosis than those with typical IPF. Although some atypical IPF in our study could be considered as advanced typical IPF, there was no difference in the baseline GAP staging for IPF and duration from the onset to initial visit between both groups. Furthermore, stratified analysis for %FVC demonstrated that the change of FVC decline in atypical IPF patients with %FVC ≥70%, who had relatively mild disease severity, was significantly greater than that in atypical IPF patients, whereas, there was no significant difference between patients with moderate-to-severe atypical and typical IPF in the change of FVC decline during 6 months. Thus, we want to emphasize that atypical IPF doesn’t always represent advanced IPF.
Recently, Oda et al. (12) reported that 9 of 11 patients consistent with radiologic criteria for PPFE were histologically diagnosed as PPFE with UIP pattern. Finally, as patients with PPFE with UIP pattern showed a trend toward poor prognosis, the authors emphasized that PPFE with UIP pattern is a disease entity distinct from IPF. Also in our study, atypical IPF with PPFE like lesion may represent a unique disorder entity. However, some patients with atypical IPF may be diagnosed as having IPF and treated with anti-fibrotic agents in real-world clinical practice, because such patients have also similar disease behavior of IPF.
Risk factors for mortality are known as follows; severity of dyspnea (13), change in FVC decline (14-16), lower FVC (17) or DLco (18), lower 6-minute walking distance (19), the presence of desaturation on 6-minute walking test (20), extent of fibrosis on chest HRCT (21,22), association with emphysema (23,24) and pulmonary arterial hypertension (25-27), change in body mass index decline (28), and so on. Currently, useful clinical score such as the GAP score also has been recognized worldwide (5). In the present study, prognostic factors were presence of atypical IPF and increased GAP staging in patients with IPF. Moreover, atypical IPF with mixed type had poorer prognosis. We suppose that this is why rate of staging by GAP in atypical IPF patients with mixed type were significantly higher GAP staging than that in those with PPFE like lesion (PPFE vs. mixed type; stage I/II/III =55%/45%/0% vs. 29%/46%/25.0%, P=0.03), and the levels of %DLco was significantly lower for atypical IPF patients with mixed type (PPFE/mixed type =69.8%/50.0%, P=0.005).
This study has some limitations. Firstly, this was a retrospective study at a single center and number of patients was small. Therefore, our results may not be representative of the entire atypical IPF population. Secondly, the atypical IPF we describe may correspond to unclassifiable pulmonary fibrosis. However, we believe that further stratification of patients, who had atypical imaging features on chest HRCT such as PPFE like lesion or multiple TWLC in addition to honeycombing in the bilateral lower lobes predominance, will be useful in assessing the outcome and efficacy of anti-fibrotic therapy. Thirdly, atypical IPF with mixed type may just go through the stage of atypical IPF with PPFE like lesion. At present, the mechanism remains unclear. Finally, most of atypical IPF patients were not able to diagnose pathologically. Indeed, atypical IPF patients with ground glass opacities, peribronchovascular distribution, micronodules, and so on should be diagnosed based on histological examination under surgical lung biopsy. However, we believe that surgical lung biopsy is rarely performed because most patients have severe impairment of pulmonary functions and/or honeycombing predominantly in the lower lobes on chest HRCT images. Basically, we aimed to diagnose not pathologically but clinico-radiologically.
In conclusion, this study demonstrated that the prognosis for atypical IPF was significantly worse than that for typical IPF. Future studies are required prospective analyses of efficacy of anti-fibrotic agents for patients with atypical IPF.
Acknowledgements
This work was supported by a grant-in-aid for diffuse lung diseases from the Japanese Ministry of Health, Labor and Welfare.
Ethical Statement: The study was approved by the local Ethics Committee of the Toho University Omori Medical Center (No. M16263).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824. 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011;378:1949-61. 10.1016/S0140-6736(11)60052-4 [DOI] [PubMed] [Google Scholar]
- 3.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:285-92. 10.1513/pats.200601-005TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med 2016;194:265-75. 10.1164/rccm.201604-0801CI [DOI] [PubMed] [Google Scholar]
- 5.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156:684-91. 10.7326/0003-4819-156-10-201205150-00004 [DOI] [PubMed] [Google Scholar]
- 6.Sumikawa H, Johkoh T, Colby TV, et al. Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med 2008;177:433-9. 10.1164/rccm.200611-1696OC [DOI] [PubMed] [Google Scholar]
- 7.Jacob J, Bartholmai BJ, Rajagopalan S, et al. Automated quantitative computed tomography versus visual computed tomography scoring in idiopathic pulmonary fibrosis: Validation against pulmonary function. J Thorac Imaging 2016;31:304-11. 10.1097/RTI.0000000000000220 [DOI] [PubMed] [Google Scholar]
- 8.ATS committee on proficiency standards for clinical pulmonary function laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002;166:111-7. 10.1164/ajrccm.166.1.at1102 [DOI] [PubMed] [Google Scholar]
- 9.Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003;167:962-9. 10.1164/rccm.2111053 [DOI] [PubMed] [Google Scholar]
- 10.Sverzellati N, Wells AU, Tomassetti S, et al. Biopsy-proved idiopathic pulmonary fibrosis: Spectrum of nondiagnostic thin-section CT diagnoses. Radiology 2010;254:957-64. 10.1148/radiol.0990898 [DOI] [PubMed] [Google Scholar]
- 11.Silva CI, Müller NL, Hansell DM, et al. Nonspecific interstitial pneumonia and idiopathic pulmonary fibrosis: changes in pattern and distribution of disease over time. Radiology 2008;247:251-9. 10.1148/radiol.2471070369 [DOI] [PubMed] [Google Scholar]
- 12.Oda T, Ogura T, Kitamura H, et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest 2014;146:1248-55. 10.1378/chest.13-2866 [DOI] [PubMed] [Google Scholar]
- 13.Papiris SA, Daniil ZD, Malagari K, et al. The Medical Research Council dyspnea scale in the estimation of disease severity in idiopathic pulmonary fibrosis. Respir Med 2005;99:755-61. 10.1016/j.rmed.2004.10.018 [DOI] [PubMed] [Google Scholar]
- 14.Zappala CJ, Latsi PI, Nicholson AG, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J 2010;35:830-6. 10.1183/09031936.00155108 [DOI] [PubMed] [Google Scholar]
- 15.Collard HR, King TE, Jr, Bartelson BB, et al. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003;168:538-42. 10.1164/rccm.200211-1311OC [DOI] [PubMed] [Google Scholar]
- 16.Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:543-8. 10.1164/rccm.200209-1112OC [DOI] [PubMed] [Google Scholar]
- 17.du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011;184:1382-9. 10.1164/rccm.201105-0840OC [DOI] [PubMed] [Google Scholar]
- 18.Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003;168:531-7. 10.1164/rccm.200210-1245OC [DOI] [PubMed] [Google Scholar]
- 19.Lederer DJ, Arcasoy SM, Wilt JS, et al. Six-minute-walk distance predicts waiting list survival in Idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:659-64. 10.1164/rccm.200604-520OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lama VN, Flaherty KR, Toews GB, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:1084-90. 10.1164/rccm.200302-219OC [DOI] [PubMed] [Google Scholar]
- 21.Best AC, Meng J, Lynch AM, et al. Idiopathic pulmonary fibrosis: physiologic tests, quantitative CT indexes, and CT visual scores as predictors of mortality. Radiology 2008;246:935-40. 10.1148/radiol.2463062200 [DOI] [PubMed] [Google Scholar]
- 22.Mura M, Ferretti A, Ferro O, et al. Functional predictors of exertional dyspnea, 6-min walking distance and HRCT fibrosis score in idiopathic pulmonary fibrosis. Respiration 2006;73:495-502. 10.1159/000089656 [DOI] [PubMed] [Google Scholar]
- 23.Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur Respir J 2005;26:586-93. 10.1183/09031936.05.00021005 [DOI] [PubMed] [Google Scholar]
- 24.Sugino K, Ishida F, Kikuchi N, et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology 2014;19:239-45. 10.1111/resp.12207 [DOI] [PubMed] [Google Scholar]
- 25.Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006;129:746-52. 10.1378/chest.129.3.746 [DOI] [PubMed] [Google Scholar]
- 26.Hamada K, Nagai S, Tanaka S, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 2007;131:650-6. 10.1378/chest.06-1466 [DOI] [PubMed] [Google Scholar]
- 27.Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: Decreased survival associated with severe pulmonary arterial hypertension. Chest 2009;136:10-5. 10.1378/chest.08-2306 [DOI] [PubMed] [Google Scholar]
- 28.Kishaba T, Nagano H, Nei Y, et al. Body mass index-percent forced vital capacity-respiratory hospitalization: New staging for idiopathic pulmonary fibrosis patients. J Thorac Dis 2016;8:3596-604. 10.21037/jtd.2016.12.49 [DOI] [PMC free article] [PubMed] [Google Scholar]