

Abstract
Background
Vemurafenib, a selective inhibitor of BRAF kinase, is approved for the treatment of adult stage IIIc/IV BRAF V600 mutation-positive melanoma. We conducted a phase I, open-label, dose-escalation study in pediatric patients aged 12–17 years with this tumor type (NCT01519323).
Procedure
Patients received vemurafenib orally until disease progression. Dose escalation was conducted using a 3+3 design. Patients were monitored for dose-limiting toxicities (DLTs) during the first 28 days of treatment to determine the maximum tolerated dose (MTD). Safety/tolerability, tumor response, and pharmacokinetics were evaluated.
Results
Six patients were enrolled (720 mg twice daily [BID]; n=3]; 960 mg BID [n=3]). The study was terminated prematurely due to low enrollment. No DLTs were observed; thus, the MTD could not be determined. All patients experienced at least one adverse event (AE); the most common were diarrhea, headache, photosensitivity, rash, nausea, and fatigue. Three patients experienced serious AEs, one patient developed secondary cutaneous malignancies, and five patients died following disease progression. Mean steady-state plasma concentrations of vemurafenib following 720 mg and 960 mg BID dosing were similar or higher, respectively, than in adults. There were no objective responses. Median progression-free survival and overall survival were 4.4 months (95% confidence interval [CI]=2.7–5.2) and 8.1 months (95% CI=5.1–12.0), respectively.
Conclusions
A recommended and effective dose of vemurafenib for patients aged 12–17 years with metastatic or unresectable melanoma was not identified. Extremely low enrollment in this trial highlights the importance of considering the inclusion of adolescents with adult cancers in adult trials.
Keywords: vemurafenib, BRAF mutation, pediatric, melanoma, clinical trial, oncology
INTRODUCTION
The incidence of melanoma in individuals aged <20 years in the United States is 4.2 cases per 1 million and increasing.1 Although rare in young children, 73% of melanoma cases before the age of 20 years occur in adolescents aged 15–19 years, with melanoma accounting for 7% of all malignancies in this age group.2,3 Approximately 85–90% of pediatric melanoma patients in this age range present with localized disease that is amenable to surgical resection.4 A further 10% of pediatric patients have resectable disease with regional spread (stage IIIa/IIIb), and there are reports of treatment with high-dose interferon alfa 2b adjuvant therapy.5–7 Unresectable (stage IIIc) or metastatic (stage IV) melanoma is exceptionally rare in pediatric patients, and outcome is particularly poor with an estimated 5-year overall survival (OS) of 12–20%.8,9
Elucidation of oncogenic mutations in melanoma has led to the development of novel targeted therapies, which have improved survival in adults with advanced melanoma. Approximately 50% of adult melanomas carry a somatic BRAF mutation in codon V600, most commonly V600E, leading to constitutive BRAF activation.10–13 BRAF activation results in downstream mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase phosphorylation, and ultimately to the activation of transcription factors responsible for regulating cell cycle and protein synthesis. While not independently sufficient to promote oncogenesis, BRAF V600 mutations are a major regulator of melanoma cell proliferation.
Vemurafenib, a selective inhibitor of BRAF kinase, is indicated for use in adults with BRAF V600 mutation-positive stage IIIc/IV melanoma.14 The phase III BRAF Inhibitor in Melanoma 3 (BRIM-3) trial compared vemurafenib with dacarbazine in treatment-naïve adult patients with BRAF V600-mutant stage IIIc/IV melanoma.15 Compared with dacarbazine, vemurafenib significantly improved progression-free survival (PFS) and OS, and was associated with 63% and 74% relative reductions in the risk of death and in the risk of death or disease progression, respectively (P < 0.001 for both comparisons).15 Response rates were 48% for vemurafenib and 5% for dacarbazine.15 The most common adverse events (AEs) associated with vemurafenib during clinical development were arthralgia, rash, fatigue, alopecia, keratoacanthoma or squamous cell carcinoma (SCC), photosensitivity, nausea, and diarrhea.15–17
There is a lack of prospectively evaluated therapies in adolescents with metastatic melanoma. Given the poor outcomes observed in this population8 and the improved outcomes reported with vemurafenib in adult patients with metastatic melanoma,15,17 we conducted a clinical trial of vemurafenib in adolescent patients with BRAF V600 mutation-positive stage IIIc/IV melanoma.
METHODS
Study design
BRAF Inhibitor in Melanoma – Pediatric (BRIM-P; NCT01519323; NO25390) was a phase I, open-label, multicenter, single-arm, dose-escalation study of oral vemurafenib in patients aged 12–17 years. The study was sponsored by F. Hoffmann-La Roche Ltd and conducted at 26 sites in 10 countries between December 2011 and December 2015.
The protocol was approved by participating institutions’ review boards, and the trial was conducted in accordance with the principles of the Declaration of Helsinki and International Conference on Harmonization guidelines for Good Clinical Practice. All patients and/or parent or legal guardians provided written, informed consent, and patient safety was reviewed by an independent Data Safety Monitoring Board.
The study included a dose-escalation phase, using a 3 + 3 design18 (see supplementary materials), and a planned extension phase where additional patients would be recruited to further evaluate safety and efficacy. The starting dose was 720 mg twice daily (BID) for patients who weighed ≥45 kg; for patients who weighed <45 kg, the starting dose was 480 mg BID regardless of the dose-escalation cohort open at the time of enrollment. All patients were to receive oral vemurafenib at their assigned dose until the maximum tolerated dose (MTD) for the extension phase was determined.
Study participants
Pediatric patients (aged 12–17 years) with histologically confirmed, surgically-incurable and unresectable stage IIIc or IV melanoma that tested positive for the BRAF V600 mutation (Cobas® 4800 BRAF V600 Mutation Test, Roche Molecular Systems, Inc., Branchburg, NJ) were enrolled. Other key inclusion criteria included a Karnofsky Performance Score of >50 (patients aged ≥16 years) or a Lansky score of ≥60 (patients aged <16 years), a life expectancy >3 months and adequate hematologic, hepatic, and renal function. Patients with asymptomatic, previously treated, radiographically stable central nervous system (CNS) lesions were eligible if the patient had not required CNS-directed therapy for at least 3 months prior to starting treatment, and had not required glucocorticoids or anticonvulsants for at least 21 days prior to starting treatment. Patients were excluded if they had any of the following: active or untreated CNS lesions; a history of spinal cord compression or carcinomatous meningitis; prior treatment with selective/specific BRAF or MEK inhibitors or vemurafenib; were pregnant or lactating; a QTc ≥450 msec or a history of congenital long QT syndrome. Concomitant treatment with any other anticancer therapy was not permitted (see supplementary materials).
Objectives
The primary objective of the study was to estimate the MTD and identify the recommended dose of oral vemurafenib for pediatric patients aged 12–17 years with unresectable stage IIIc/IV BRAF mutation-positive melanoma. Secondary objectives were evaluations of the safety and tolerability, efficacy, and pharmacokinetic profile of vemurafenib in this population. Exploratory objectives included investigation into potential biomarkers.
Safety assessments
Patients were evaluated for dose-limiting toxicities (DLTs; see supplementary materials) during the first 28 days of study treatment. Safety and tolerability were assessed by monitoring the frequency and intensity of AEs throughout the study and until 28 days after the last dose of vemurafenib. Additionally, patients were monitored during study treatment and the subsequent 6-month follow-up period for the occurrence of new primary melanoma and SCC (cutaneous and non-cutaneous). AEs were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, version 4.0). Additional safety assessments were performed throughout the study until 28 days after the last dose of vemurafenib and included laboratory parameters (hematology, urinalysis, and clinical chemistry), performance status (Karnofsky or Lansky), vital signs, physical examinations, and electrocardiograms (until treatment completion only). Evaluations (dermatologic, physical, and radiologic) to identify cutaneous and non-cutaneous SCC and new primary melanoma were performed regularly during the study; suspicious lesions were biopsied on an as-needed basis, and evaluations were continued following the last dose of vemurafenib until loss to follow-up, death, withdrawal of consent, or study closure. Patients who developed an SCC or suspicious skin lesion could opt to continue or discontinue the trial based on consultation with the investigator.
Efficacy assessments
Tumor response was assessed by computed tomography (CT) scan or magnetic resonance imaging at baseline (reference measure), after 4 weeks (cycle 2), and then every 8 weeks (cycles 4–12) or 12 weeks (thereafter) until the completion of treatment. Findings were judged according to the Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1).
Pharmacokinetic assessments
Venous blood samples (2 ml) were collected pre-dose (morning), and at 2, 4, 8, 12, and 24 hr post-dose on days 1 and 22, and pre-dose and 2 hr post-dose every 4 weeks (cycles 2–4), 8 weeks (cycles 6–12), and 12 weeks (thereafter) until the end of treatment. All samples were collected following an 8-hr overnight fast, and patients continued to fast for 4 hr after the morning drug administration on days 1 and 22. Plasma concentrations of vemurafenib were determined using validated liquid chromatography-tandem mass spectrometry. The lower limit of quantitation for vemurafenib in human plasma was 0.025 μg/ml, with linearity demonstrable to 50 μg/ml (upper limit of quantitation), using a sample volume of 0.05 ml.
Statistical analyses
Target enrollment for the planned extension phase was 20 patients. The primary analysis variable was the MTD, defined as one dose level below the dose that induced a DLT in at least one of three patients following expansion of that dose level (i.e., two of six patients, or fewer if unacceptable toxicity prohibited further enrollment into a dose level). All safety findings were summarized using descriptive statistics. Secondary efficacy variables included PFS, OS, best overall response rate (BORR), and clinical benefit rate. PFS and OS were estimated using Kaplan-Meier methodology. Pharmacokinetic parameters were obtained by non-compartmental methods using WinNonlin software (version 6.4; Pharsight Corporation, Mountain View, CA). Data from samples collected on days 1 and 22 were used to obtain the maximum plasma concentration (Cmax) and area under the plasma concentration-time curve between time zero and 12 hr (AUC0–12); the accumulation ratio was calculated using AUC0–12 values (day 22:day 1). Vemurafenib concentrations and pharmacokinetic parameters are presented using descriptive statistics.
Exploratory biomarker assessments
Whole blood samples for biomarker assessments were collected at the start of treatment. Tumor tissue samples from BRAF mutation screening were retained for exploratory molecular analyses; SCC or suspicious lesion tissue samples were also collected. Mutations (single nucleotide variations [SNVs], small insertion/deletions [indels], structural variations, including inter- and intra-chromosomal rearrangements, and copy number variations (CNV)) and mutation load were assessed in tumor tissue using targeted genomic profiling methods (see supplementary materials).19–21
RESULTS
Patients
A total of six patients, all weighing ≥45 kg, were enrolled into two dose cohorts. Three patients received vemurafenib at a dose of 720 mg BID, and three patients subsequently received vemurafenib at a dose of 960 mg BID. Baseline characteristics and prior therapies are presented in Table 1. Patients in the 720 mg BID dose cohort received 76, 83, and 138 days of treatment, respectively; patients in the 960 mg BID dose cohort received 130, 144, and 291 days of treatment, respectively. All patients were enrolled between January 2013 and August 2014; study enrollment was discontinued in December 2015 due to the inability to recruit eligible patients.
TABLE 1.
Baseline characteristics, prior therapy and disease burden
| Patient | Sex | Age, years |
Weight, kg |
BMI, kg/m2 |
Race | Histologic subtype |
Surgical excisiona |
Systemic therapya |
Target lesion sitesb |
Non- target lesion sitesb |
Vemurafenib dose, mg BID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 17 | 52.4 | 17.3 | White | Nodular | Yes | None | Liver, lymph node | Lung, skin | 720 |
| 2 | Male | 15 | 138.5 | 40.0 | White | Nodular | Yes | IFN | Lung | Lung, sacrum | 720 |
| 3 | Male | 16 | 64.7 | 19.7 | White | Superficial spreading | Yes | None | Lymph node | Lymph node | 720 |
| 4 | Female | 16 | 59.5 | 21.9 | White | NA | Yes | TMZ | Brain | Brain | 960 |
| 5 | Female | 15 | 86.4 | 34.1 | Black or AA | NA | No | None | Skin, lymph node, soft tissue | Soft tissue | 960 |
| 6 | Male | 16 | 49.2 | 18.3 | White | Superficial spreading | Yes | None | Liver | Bone, lung, liver | 960 |
AA, African-American; BID, twice daily; IFN, interferon; NA, not available; TMZ, temozolomide.
Prior to study entry.
At study entry.
Safety and tolerability
All six patients received at least one dose of vemurafenib and were included in the safety population. An MTD could not be determined, because no DLTs were observed in the small number of patients enrolled.
All patients experienced at least one AE. The most common AEs (reported in ≥3 patients) were diarrhea and headache (each occurring in four [66.7%] patients), and photosensitivity, rash, nausea, and fatigue (each occurring in three [50%] patients). The majority of AEs were grade 1 or 2 in severity, and four patients experienced grade ≥3 AEs (Table 2). There were no AEs leading to discontinuation or dose reduction of vemurafenib. Two patients had an AE (nephrolithiasis, n = 1; skin infection, n = 1) that required vemurafenib to be temporarily withheld.
TABLE 2.
AEs of grade ≥3 intensity occurring in any patient
| AE (MedDRA preferred term) | Vemurafenib 720 mg BID (n = 3) |
Vemurafenib 960 mg BID (n = 3) |
All patients (n = 6) |
Vemurafenib treatment-relatedb |
|---|---|---|---|---|
| Any AE | 2 (66.7) | 2 (66.7) | 4 (66.7) | |
| Neck pain | 0 | 1 (33.3) | 1 (16.7) | No |
| Spinal pain | 1 (33.3) | 0 | 1 (16.7) | No |
| BCC | 0 | 1 (33.3)a | 1 (16.7) | Yes |
| Intracranial tumor hemorrhage | 0 | 1 (33.3) | 1 (16.7) | No |
| SCC | 0 | 1 (33.3) | 1 (16.7) | Yes |
| Photosensitivity | 1 (33.3) | 0 | 1 (16.7) | Yes |
| Maculopapular rash | 0 | 1 (33.3) | 1 (16.7) | No |
| Diarrhea | 0 | 1 (33.3) | 1 (16.7) | No |
| Scrotal abscess | 0 | 1 (33.3) | 1 (16.7) | Yes |
| Lymphocyte count decreased | 0 | 1 (33.3) | 1 (16.7) | No |
| Hypokalemia | 0 | 1 (33.3) | 1 (16.7) | No |
| Headache | 0 | 1 (33.3) | 1 (16.7) | No |
AE, adverse event; BCC, basal cell carcinoma; BID, twice daily; MedDRA, Medical Dictionary for Regulatory Activities; SCC, squamous cell carcinoma.
One case report with two lesions.
As determined by the investigator.
Data are expressed as number of patients (%).
Serious AEs were reported in three patients during the study (Table 3). Patient 6, a 16-year-old white male, developed a cutaneous SCC that was considered related to vemurafenib, and the patient concomitantly experienced a cutaneous basal cell carcinoma (BCC; two lesions reported). The patient underwent excision of the SCC and BCC, and no further therapy for these lesions was administered. No patients had a prolongation of QTc >500 msec.
TABLE 3.
Serious AEs
| Patient | AE (MedDRA preferred term) |
Vemurafenib dose, mg BID | Onset, study day | Duration, days | Gradea | Vemurafenib treatment-relatedb |
|---|---|---|---|---|---|---|
| 1 | Spinal pain | 720 | 160 | 19 | 3 | No |
|
| ||||||
| Nephrolithiasis | 960 | 32 | 5 | 2 | No | |
| 4 | Adrenal insufficiency | 171 | 7 | 2 | No | |
| Intracranial tumor hemorrhagec | 304 | 5 | 5 | No | ||
|
| ||||||
| Maculopapular rash | 960 | 9 | 105 | 3 | No | |
| 6 | BCC | 85 | 12 | 3 | Yes | |
| SCC | 85 | 12 | 3 | Yes | ||
AE, adverse event; BCC, basal cell carcinoma; BID, twice daily; MedDRA, Medical Dictionary for Regulatory Activities; SCC, squamous cell carcinoma.
Maximum observed CTCAE grade.
As determined by the investigator.
Occurred 10 days after the patient had discontinued study drug due to PD (confirmed by brain CT).
All six patients discontinued study treatment following disease progression. Five of the six patients died subsequent to disease progression, and four patient deaths were attributed to progressive disease (PD). One death occurred due to an AE of intracranial tumor hemorrhage in a patient with pre-existing intracranial disease (Patient 4). The AE occurred 10 days after the patient had discontinued the study drug due to PD (confirmed by brain CT). In the opinion of the investigator, the event of intracranial tumor hemorrhage was not related to the study drug.
Efficacy
All patients were assessed for tumor response during the dose-escalation phase and were included in the efficacy population. The BORR was 0% (95% CI = 0–45.93) as no patient had a confirmed response. One patient had an unconfirmed partial response (observed at only one time point; Supplementary Fig. S1). Four patients had a BORR status of stable disease for >6 weeks; therefore, the clinical benefit rate was 66.7% (Table 4). Median PFS was 4.4 months (95% CI = 2.7–5.2), and all six patients subsequently experienced disease progression. Median OS was 8.1 months (95% CI = 5.1–12.0), and one patient was alive at the time of study closure 16.9 months after study drug initiation. No patients were enrolled in the efficacy extension phase.
TABLE 4.
Best overall response, time to progression, and overall survival in pediatric patients receiving vemurafenib 720 mg or 960 mg BID
| Vemurafenib dose, mg BID | Best overall response | Time to progression, months | Patient status | Overall survival, months |
|---|---|---|---|---|
| 720 | SD | 4.5 | Died | 6.1 |
| 720 | PD | 2.7 | Died | 12.0 |
| 720 | PD | 2.5 | Died | 3.1 |
| 960 | SD | 9.7 | Died | 10.1 |
| 960 | SD | 5.2 | Alive | 16.9a |
| 960 | SDb | 4.3 | Died | 5.1 |
BID, twice daily; PD, progressive disease; SD, stable disease.
Censored observation.
Patient had an unconfirmed partial response (observed at only one time point).
Pharmacokinetics
All patients had at least one post-dose blood sample and were included in the pharmacokinetic population. Individual steady-state plasma concentration-time profiles (day 22) are presented by vemurafenib dose in Figure 1. Steady-state concentration was relatively constant over the dosing interval in both dose cohorts following oral administration of 720 mg BID or 960 mg BID vemurafenib. Mean Cmax (% coefficient of variation [CV]) following single doses of vemurafenib 720 mg and 960 mg BID (day 1) were 2.593 μg/ml (72%) and 7.897 μg/ml (52%), respectively; corresponding values at steady state (day 22) were 50.367 μg/ml (26%) and 93.367 μg/ml (18%), respectively. Mean AUC0–12 (CV) following single doses of vemurafenib 720 mg and 960 mg BID (day 1) were 19.510 μg∙hr/ml (76%) and 67.967 μg∙hr/ml (58%), respectively; corresponding values at steady state (day 22) were 498.333 μg∙hr/ml (28%) and 982.333 μg∙hr/ml (23%), respectively. Inter-patient variability in vemurafenib exposure was greater on day 1 (CV: 52–76%) compared with day 22 (i.e., steady state; CV: 18–28%). Substantial accumulation (day 22:day 1) was observed following multiple oral doses of vemurafenib (720 mg and 960 mg BID); individual accumulation ratios (AUC, day 22:day 1) ranged from approximately 11 to 67.
FIGURE 1.
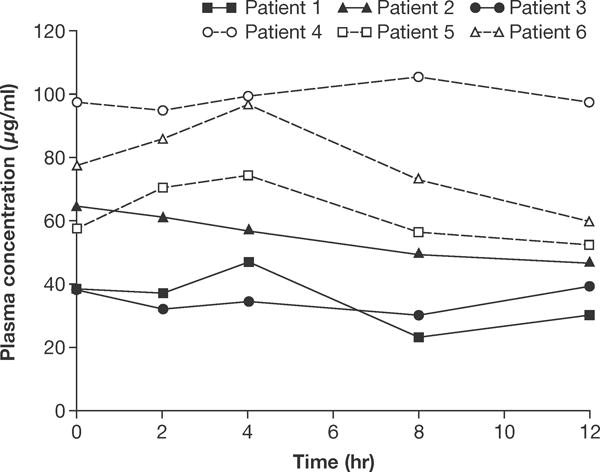
Individual steady-state (day 22) plasma concentration-time profiles following treatment with vemurafenib 720 mg BID (patients 1–3) or 960 mg BID (patients 4–6). BID, twice daily.
Tumor whole exome sequencing
Whole exome sequencing (WES) was performed in melanoma samples and paired normal blood samples from all six patients. The most prevalent SNVs (Supplementary Fig. S2), and SNVs detected in mitogen-activated protein kinase (MAPK) signaling pathway genes (Fig. 2) and known to be relevant to melanoma,22–25 are described here. Eighty-five percent of SNVs were cytidine to thymidine (C > T) or guanine to adenine (G > A) transitions. Supplementary Table S1 shows all coding and non-coding variants (SNVs and indels) as detected by WES in each sample. Figure 2 shows genes affected by copy number gains and losses in each sample. No CNV changes were seen in melanoma-associated genes (Supplementary Table S2). In five of six patients, WES confirmed the presence of a BRAF V600E mutation, as originally detected using the Cobas® 4800 BRAF V600 mutation test. No BRAF V600E mutation could be detected in one patient, likely due to low quality of formalin-fixed, paraffin-embedded DNA and poor next-generation sequencing coverage (Supplementary Table S3).
FIGURE 2.
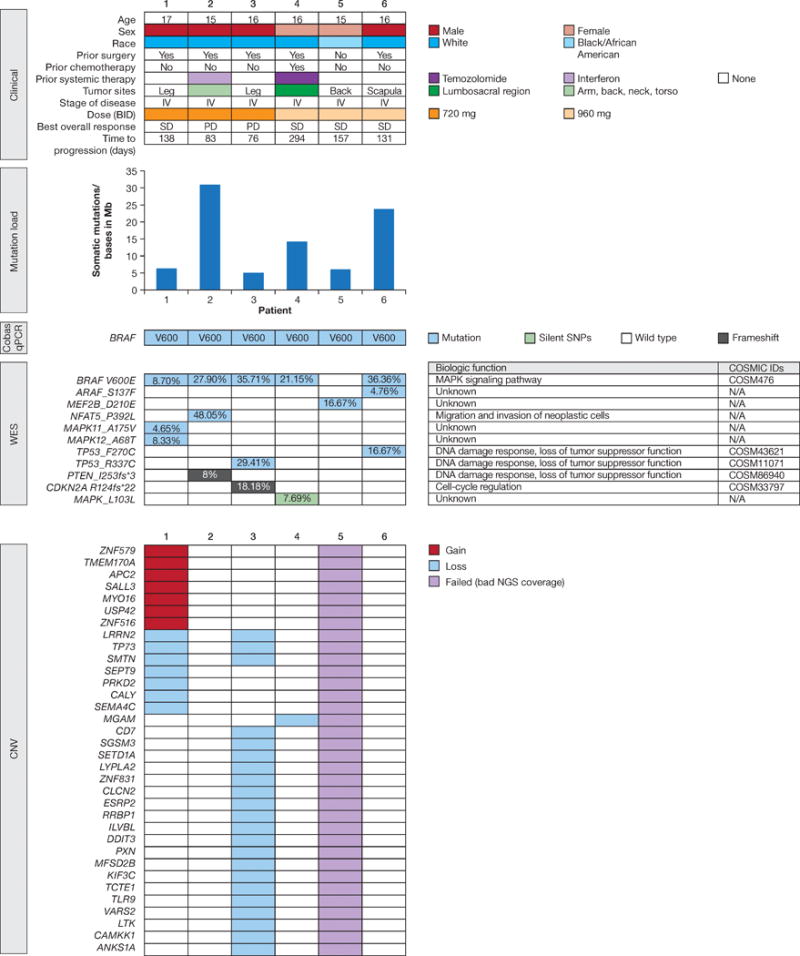
The genomic data for six pediatric melanomas analyzed by cobas® 4800 BRAF V600 Mutation Test and WES. The mutation load plot displays the number of somatic mutations per Mb. The SNVs shown here are related to the MAPK pathway. The CNV plot lists genes that either have a loss (blue) or a gain (red), or failed samples (grey).
CNV, copy number variation; MAPK, mitogen-activated protein kinase; SNV, single nucleotide variation; WES, whole exome sequencing.
The median observed mutation load in the coding regions was approximately 10.27 mutations/Mb (range, 4.83–31.26; Fig. 2). Mutation load, calculated from the WES data, did not show a correlation with time to progression.
DISCUSSION
The tolerability of vemurafenib in the small number of adolescent patients treated during this study was consistent with that observed in adults. The MTD of vemurafenib in pediatric and adolescent patients could not be determined as no DLTs were observed in the six treated patients (720 mg BID, n = 3; 960 mg BID, n = 3). The absence of DLTs should be interpreted cautiously given the small sample size. Secondary cutaneous malignancies (SCC and BCC) were observed in one patient, consistent with previous findings in adults.15–17
The pharmacokinetic characteristics of vemurafenib in adolescent patients were generally consistent with those observed in adults.26 As in adults, substantial accumulation of vemurafenib was observed following multiple BID doses in both dose cohorts. Inter-individual variability in vemurafenib exposure was greater following a single dose than at steady state, and steady-state exposure was relatively constant over the dosing interval. Steady-state vemurafenib plasma exposures in adolescent patients were found to be similar or higher than those observed in adults.26 For example, the mean steady-state Cmax following vemurafenib 720 mg BID was similar in adolescents (~50 μg/ml) to that observed in adults (~53 μg/ml); in contrast, the mean steady-state plasma concentration following vemurafenib 960 mg BID appeared greater in adolescents (~93 μg/mL) than in adults (~61 μg/mL).26
Based on the limited data available in this study, a dose recommendation cannot be made for patients with metastatic or unresectable melanoma <18 years of age. Given the established adult MTD of 960 mg BID, and the similar or higher exposure to vemurafenib in adolescents versus adults, further prospective evaluation of vemurafenib doses above 960 mg BID in patients aged <18 years and weighing ≥45 kg is not warranted. This study provides no information about dosing in patients aged <18 years who weigh <45 kg.
Unlike adult patients with BRAF V600-mutated metastatic melanoma, in whom objective responses to vemurafenib have been observed in 48–53% of cases in phase II and III trials,15,17 no objective responses were observed in adolescents with this tumor type. This cannot be attributed to drug resistance induced by prior chemotherapeutic intervention, as only two patients received systemic therapy prior to study enrollment. Moreover, the patient with the longest time to disease progression was the only one to receive cytotoxic chemotherapy prior to enrollment. Furthermore, drug exposure was equal to or higher than that observed in adults, and compliance with study drug administration was high. Finally, prolonged periods of study drug withholding due to toxicity or noncompliance did not occur. Thus, a clinical rationale to explain the apparent inferior outcomes in adolescents relative to adults has not been identified. Given the small sample size, the possibility that the difference may have occurred due to chance cannot be excluded.
Exploratory biomarker analysis detected no previously characterized genetic aberrations associated with resistance to vemurafenib therapy that would explain the lack of drug response (including loss of phosphatase and tensin homolog [PTEN]27 or neurofibromatosis-1,28 BRAF copy number amplification,29 CDK4 mutations and cyclin D1 amplifications,30 and NRAS or MEK mutations31).
Consistent with previous findings, our results suggest that the pediatric melanoma samples examined in the present study are heterogeneous, with mutations relevant to melanoma oncogenesis occurring in a mutually exclusive manner.32 The high proportion of C > T and G > A transitions are consistent with damage by ultraviolet radiation.33–37 The median mutation load in this pediatric cohort was also consistent with that observed previously in pediatric and adult samples.32,38
The primary limitations of this study were its low enrollment and consequent termination, for which there are several potential reasons. Foremost, metastatic melanoma requiring systemic therapy is exceedingly rare in adolescents.2 While the annual incidence of melanoma is approximately 4/1,000,000 pediatric patients,1 metastatic cancer at presentation comprises less than 3% of disease.8 Additionally, high cure rates of patients with regional disease treated with surgery and high-dose interferon alfa 2b limit the number of patients who recur with metastatic disease later on.5–7 Treatment of this adult cancer in adolescents occurs in both pediatric and adult oncology facilities, potentially limiting adequate patient concentration in, and referral to, tertiary study sites.3,39–41 Additionally, the majority of patients with melanoma who were considered for enrollment were BRAF V600 mutation negative, were not within the eligible age range, or had a disease stage that was too low. Finally, the standard-of-care treatment for metastatic melanoma has evolved considerably since the study began. Approval of six new medications (in addition to vemurafenib) for adult patients with unresectable or metastatic melanoma (cobimetinib, pembrolizumab, nivolumab, trametinib, dabrafenib, and ipilimumab) in the United States and European Union since the time of study initiation resulted in the potential availability of alternate therapeutic approaches for pediatric patients eligible for enrollment in this trial. Therefore, other clinical trials or off-label usage by treating physicians may have reduced enrollment.
Our experience highlights the need to identify appropriate solutions for conducting early phase trials in patients <18 years of age with cancers that are more typically seen in adults. Given that similar tolerability, pharmacokinetics, and recommended doses are typically observed in adolescents (relative to adults) in the evaluation of most drugs,37 the inclusion of adolescents with adult cancers is warranted in early phase trials to facilitate therapeutic development in younger patients. This holds particularly true considering that even a large adolescent multinational study may not recruit sufficient patients and may ultimately provide therapy considered obsolete relative to a rapidly evolving adult therapeutic landscape. Novel collaborative strategies among adult and pediatric centers and study sponsors are needed to facilitate referral of rare adolescent patients to tertiary centers recruiting on such trials. When dedicated phase I pediatric studies are warranted, mechanism-of-action based enrollment (as opposed to disease-specific enrollment) should be seriously considered so as not to risk successful completion of pharmacokinetic and tolerability objectives or render development of new drugs unfeasible in other potential pediatric indications. The experience of this trial, to which few patients were accrued despite extensive recruitment efforts, highlights the necessity of a multi-stakeholder approach among academics, regulators, industry sponsors, and advocacy groups to ensure broad pediatric development strategies, proper molecular prioritization, and minimized risk of low-enrolling or non-feasible trials.42
In conclusion, a recommended dose of vemurafenib in patients with BRAF V600-mutated metastatic or unresectable melanoma aged <18 years and weighing ≥45 kg was not identified in this study. No new safety signals were observed. No objective tumor responses were observed in this limited pediatric population. Based on limited data from six adolescent patients in two dose groups, the pharmacokinetic characteristics of vemurafenib appeared similar to those in adult patients. Inclusion of adolescents in adult trials and mechanism-of-action approaches in pediatric trials are critical potential mitigation strategies to ensure the timely evaluation of adolescents with rare diseases and successful completion of pharmacology and tolerability objectives in early phase trials in pediatric-aged patients.
Supplementary Material
SUPPLEMENTARY MATERIAL
SUPPLEMENTARY METHODS
SUPPLEMENTARY FIGURE S1 Best overall response in pediatric patients receiving vemurafenib 720 mg or 960 mg BID. *Progressive disease assessment was due to a new lesion(s). No post-baseline target lesion measurements were reported for patient 3.
SUPPLEMENTARY FIGURE S2 Landscape of the most prevalent coding mutations across six pediatric melanoma samples. Shaded boxes depict missense somatic mutations detected in the related gene per patient. In cases where multiple mutations per gene were found in a sample, only one mutation is shown.
SUPPLEMENTARY TABLE S1 All coding and non-coding variants (SNVs and indels) as detected by WES in each sample.
SUPPLEMENTARY TABLE S2 Genes affected by copy number gains and losses in each sample.
SUPPLEMENTARY TABLE S3 WES coverage and mutation load report from six pediatric melanoma samples. The sample from patient 5 had poor next-generation sequencing coverage due to poor formalin-fixed, paraffin-embedded DNA quality.
Acknowledgments
Medical writing assistance was provided by David Evans, PhD, and Emma McConnell, PhD, of Gardiner-Caldwell Communications, and was funded by F. Hoffmann-La Roche Ltd. The authors would like to thank Carina Mueller, of Metronomia Clinical Research GmbH, for biostatistics support and critical review of the manuscript, and Magalie Hilton, of F. Hoffmann-La Roche Ltd, for biostatistics support. IJD declares that this research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. JCC is supported by National Health Service funding to the National Institute for Health Research Biomedical Research Centre of the Royal Marsden Hospital.
Abbreviation Definition
- AE
Adverse event
- AUC0–12
Area under the concentration time curve during one dose interval (12 hours)
- BCC
Basal cell carcinoma
- BID
Twice daily
- BORR
Best overall response rate
- BRIM-3
BRAF Inhibitor in Melanoma 3
- BRIM-P
BRAF Inhibitor in Melanoma - Pediatric
- CI
Confidence interval
- Cmax
Maximum plasma concentration
- CNS
Central nervous system
- CNV
Copy number variation
- CT
Computed tomography
- CTCAE
Common Terminology Criteria for Adverse Events
- CV
Coefficient of variation
- DLT
Dose-limiting toxicity
- MAPK
Mitogen-activated protein kinase
- MedDRA
Medical Dictionary for Regulatory Activities
- MEK
Mitogen-activated protein kinase kinase
- MTD
Maximum tolerated dose
- NCI
National Cancer Institute
- NGS
Next-generation sequencing
- OS
Overall survival
- PD
Progressive disease
- PFS
Progression-free survival
- PTEN
Phosphatase and tensin homolog
- qPCR
Quantitative polymerase chain reaction
- RECIST
Response Evaluation Criteria in Solid Tumors
- SCC
Squamous cell carcinoma
- SNV
Single nucleotide variation
- WES
Whole exome sequencing
Footnotes
CONFLICT OF INTEREST
JCC reports consulting/advisory roles for F. Hoffmann-La Roche Ltd and Merck, and travel, accommodation, or expenses from F. Hoffmann-La Roche Ltd. IJD reports consulting roles for Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Eisai, and Pfizer. MC reports consulting/advisory roles for F. Hoffmann-La Roche Ltd. WZ and NR report current employment and stocks/shares at F. Hoffmann-La Roche Ltd. YJC, JP, MDT, SS, and NWRT report current employment at Genentech and stocks/shares in F. Hoffmann-La Roche Ltd. All other authors have no conflicts of interest to report.
References
- 1.Ries LAG, Smith MA, Gurney JG, et al. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995. Bethesda, MD: National Cancer Insitute; 1999. [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, et al. Lyon, France: International Agency for Research on Cancer; 2013. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No.11 [Internet] http://globocan.iarc.fr. Accessed July 18, 2016. [Google Scholar]
- 3.Herzog C, Pappo A, Bondy M, Bleyer A, Kirkwood J. Malignant melanoma. In: Bleyer A, O’Leary M, Barr R, Ries LAG, editors. Cancer Epidemiology in Older Adolescents and Young Adults 15 to 19 Years of Age, Including SEER Incidence and Survival: 1975–2000. Bethesda, MD: National Cancer Institute; 2006. pp. 53–63. [Google Scholar]
- 4.Strouse JJ, Fears TR, Tucker MA, et al. Pediatric melanoma: Risk factor and survival analysis of the surveillance, epidemiology and end results database. J Clin Oncol. 2005;23:4735–4741. doi: 10.1200/JCO.2005.02.899. [DOI] [PubMed] [Google Scholar]
- 5.Navid F, Furman WL, Fleming M, et al. The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer. 2005;103:780–787. doi: 10.1002/cncr.20860. [DOI] [PubMed] [Google Scholar]
- 6.Chao MM, Schwartz JL, Wechsler DS, et al. High-risk surgically resected pediatric melanoma and adjuvant interferon therapy. Pediatr Blood Cancer. 2005;44:441–448. doi: 10.1002/pbc.20168. [DOI] [PubMed] [Google Scholar]
- 7.Shah NC, Gerstle JT, Stuart M, et al. Use of sentinel lymph node biopsy and high-dose interferon in pediatric patients with high-risk melanoma: The Hospital for Sick Children experience. J Pediatr Hematol Oncol. 2006;28:496–500. doi: 10.1097/01.mph.0000212973.28996.e4. [DOI] [PubMed] [Google Scholar]
- 8.Lange JR, Palis BE, Chang DC, et al. Melanoma in children and teenagers: An analysis of patients from the National Cancer Data Base. J Clin Oncol. 2007;25:1363–1368. doi: 10.1200/JCO.2006.08.8310. [DOI] [PubMed] [Google Scholar]
- 9.Pappo AS. Melanoma in children and adolescents. Eur J Cancer. 2003;39:2651–2661. doi: 10.1016/j.ejca.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Beeram M, Patnaik A, Rowinsky EK. Raf: A strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–6790. doi: 10.1200/JCO.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 11.COSMIC. Catalogue of Somatic Mutations in Cancer. 2010 http://cancer.sanger.ac.uk/cosmic. Accessed March 27, 2017.
- 12.Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 13.Torres-Cabala CA, Curry JL. Genetics of Melanoma. New York, NY: Springer Science and Business Media; 2016. [Google Scholar]
- 14.EMA. Zelboraf: summary of product characteristics. 2016 http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002409/WC500124444.pdf Accessed March 27, 2017.
- 15.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storer BE. Design and analysis of phase I clinical trials. Biometrics. 1989;45:925–937. [PubMed] [Google Scholar]
- 19.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013 arXiv:1303.3997v2. [Google Scholar]
- 20.Mose LE, Wilkerson MD, Hayes DN, et al. ABRA: Improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30:2813–2815. doi: 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 23.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 24.Thuerauf DJ, Arnold ND, Zechner D, et al. p38 Mitogen-activated protein kinase mediates the transcriptional induction of the atrial natriuretic factor gene through a serum response element. A potential role for the transcription factor ATF6. J Biol Chem. 1998;273:20636–20643. doi: 10.1074/jbc.273.32.20636. [DOI] [PubMed] [Google Scholar]
- 25.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 26.Grippo JF, Zhang W, Heinzmann D, et al. A phase I, randomized, open-label study of the multiple-dose pharmacokinetics of vemurafenib in patients with BRAF V600E mutation-positive metastatic melanoma. Cancer Chemother Pharmacol. 2014;73:103–111. doi: 10.1007/s00280-013-2324-5. [DOI] [PubMed] [Google Scholar]
- 27.Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436) Clin Cancer Res. 2013;19:4868–4878. doi: 10.1158/1078-0432.CCR-13-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-scale RNA interference screen implciates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smalley KS, Lioni M, Dalla Palma M, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–1774. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 32.Lu C, Zhang J, Nagahawatte P, et al. The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol. 2015;135:816–823. doi: 10.1038/jid.2014.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 35.Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: An overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571:43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Leiter U, Garbe C. Epidemiology of melanoma and nonmelanoma skin cancer–the role of sunlight. Adv Exp Med Biol. 2008;624:89–103. doi: 10.1007/978-0-387-77574-6_8. [DOI] [PubMed] [Google Scholar]
- 37.Momper JD, Mulugeta Y, Green DJ, et al. Adolescent dosing and labeling since the Food and Drug Administration Amendments Act of 2007. JAMA Pediatr. 2013;167:926–932. doi: 10.1001/jamapediatrics.2013.465. [DOI] [PubMed] [Google Scholar]
- 38.Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrari A, Bisogno G, Cecchetto G, et al. Cutaneous melanoma in children and adolescents: The Italian rare tumors in pediatric age project experience. J Pediatr. 2014;164:376–382. doi: 10.1016/j.jpeds.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Neier M, Pappo A, Navid F. Management of melanomas in children and young adults. J Pediatr Hematol Oncol. 2012;34(Suppl. 2):S51–54. doi: 10.1097/MPH.0b013e31824e3852. [DOI] [PubMed] [Google Scholar]
- 41.Austin MT, Xing Y, Hayes-Jordan AA, et al. Melanoma incidence rises for children and adolescents: An epidemiologic review of pediatric melanoma in the United States. J Pediatr Surg. 2013;48:2207–2213. doi: 10.1016/j.jpedsurg.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Vassal G, Rousseau R, Blanc P, et al. Creating a unique, multi-stakeholder Paediatric Oncology Platform to improve drug development for children and adolescents with cancer. Eur J Cancer. 2015;51:218–224. doi: 10.1016/j.ejca.2014.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL
SUPPLEMENTARY METHODS
SUPPLEMENTARY FIGURE S1 Best overall response in pediatric patients receiving vemurafenib 720 mg or 960 mg BID. *Progressive disease assessment was due to a new lesion(s). No post-baseline target lesion measurements were reported for patient 3.
SUPPLEMENTARY FIGURE S2 Landscape of the most prevalent coding mutations across six pediatric melanoma samples. Shaded boxes depict missense somatic mutations detected in the related gene per patient. In cases where multiple mutations per gene were found in a sample, only one mutation is shown.
SUPPLEMENTARY TABLE S1 All coding and non-coding variants (SNVs and indels) as detected by WES in each sample.
SUPPLEMENTARY TABLE S2 Genes affected by copy number gains and losses in each sample.
SUPPLEMENTARY TABLE S3 WES coverage and mutation load report from six pediatric melanoma samples. The sample from patient 5 had poor next-generation sequencing coverage due to poor formalin-fixed, paraffin-embedded DNA quality.