

Abstract
Previous studies have shown that increased levels of chemokine receptor CXCR7 are associated with the increased invasiveness of prostate cancer cells. We now show that CXCR7 expression is upregulated in VCaP and C4‐2B cells after enzalutamide (ENZ) treatment. ENZ treatment induced apoptosis (sub‐G1) in VCaP and C4‐2B cells, and this effect was further increased after combination treatment with ENZ and CCX771, a specific CXCR7 inhibitor. The levels of p‐EGFR (Y1068), p‐AKT (T308) and VEGFR2 were reduced after ENZ and CCX771 combination treatment compared to single agent treatment. In addition, significantly greater reductions in migration were shown after combination treatment compared to those of single agents or vehicle controls, and importantly, similar reductions in the levels of secreted VEGF were also demonstrated. Orthotopic VCaP xenograft growth and subcutaneous MDA133‐4 patient‐derived xenograft (PDX) tumor growth was reduced by single agent treatment, but significantly greater suppression was observed in the combination treatment group. Although overall microvessel densities in the tumor tissues were not different among the different treatment groups, a significant reduction in large blood vessels (>100 μm2) was observed in tumors following combination treatment. Apoptotic indices in tumor tissues were significantly increased following combination treatment compared with vehicle control‐treated tumor tissues. Our results demonstrate that significant tumor suppression mediated by ENZ and CXCR7 combination treatment may be due, in part, to reductions in proangiogenic signaling and in the formation of large blood vessels in prostate cancer tumors.
Keywords: CXCR7, mCRPC, CCX771, enzalutamide, drug resistance
Short abstract
What's new?
Despite promising initial responses to androgen deprivation therapy, advanced prostate cancer eventually progresses to metastatic castration‐resistant disease in the majority of men. This increased aggressiveness in tumor behavior is associated with elevated expression of chemokine receptor CXCR7. Here, in VCaP and C4‐2B prostate cancer cell lines, combined treatment with the androgen receptor signaling inhibitor enzalutamide (ENZ) and the CXCR7 inhibitor CCX771 was found to enhance apoptosis and suppress cell motility, invasion and proangiogenic signaling. Experiments in orthotopic VCaP xenograft and subcutaneous MDA133‐4 patient‐derived xenograft models corroborated observations in cells and demonstrated significant reductions in blood vessel formation.
Abbreviations
- ADT
androgen deprivation therapy
- AR
androgen receptor
- ATCC
American Type Culture Collection
- CCX771
CXCR7 inhibitor
- C‐PARP
cleaved Poly(ADP‐ribose) polymerase
- CXCL12/SDF1
C‐X‐C motif chemokine ligand 12/stromal cell‐derived factor 1
- CXCR4
C‐X‐C motif chemokine receptor 4
- CXCR7
C‐X‐C motif chemokine receptor 7
- DAB
3;3′‐diaminobenzidine
- DMSO
dimethyl sulfoxide
- ENZ
enzalutamide
- FACS
fluorescence‐activated cell sorting
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- HMGB1
high‐mobility group protein 1
- HUVEC
human umbilical vein endothelial cells
- mCRPC
metastatic castration‐resistant prostate cancer
- PCa
prostate cancer
- PDX
patient‐derived xenograft
- p‐EGFR
phosphor‐epidermal growth factor receptor
- PI
propidium iodide
- QRT‐PCR
quantitative real‐time polymerase chain reaction
- SCID
severe combined immunodeficiency
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- VEGF
vascular endothelial growth factor
- WB
Western blotting
Androgen deprivation therapy (ADT) remains the standard therapy for advanced prostate cancer (PCa). Approximately 80–90% of men with PCa initially respond to ADT; however, in nearly all cases, the tumors develop resistance to ADT and progress to metastatic castration‐resistant prostate cancer (mCRPC).1 Enzalutamide (ENZ), an androgen receptor (AR) signaling inhibitor, suppresses androgen signaling in PCa cells, in part through the regulation of gene expression.2 In clinical studies, ENZ was shown to prolong patient survival by 4.8 months.3, 4 This moderate increase in survival is likely due to the development of resistance that arises from aberrant regulation of AR target gene expression.5, 6 However, the mechanisms of resistance to AR inhibition in PCa are complex, involving adaptive responses and various genomic alterations that create conditions for cell selection. Ultimately, resistance to ENZ therapy develops in most patients; and this condition inexorably leads to malignant progression. The development of novel, more effective combination therapy strategies for mCRPC is clearly needed.
Various reports have suggested that inflammation plays an important role in the development and progression of PCa.7, 8 It is widely accepted that chemokines and the chemokine receptor networks are fundamental drivers of the complex interactions between inflammatory cells and the benign prostate and PCa cells. Specifically, it has been shown that the C‐X‐C motif chemokine ligand 12 (CXCL12/SDF1) is secreted by human prostate stromal fibroblasts and osteoblasts in the bone marrow and is a major paracrine factor that stimulates proliferation and induces invasiveness of prostate epithelial cells.9, 10, 11, 12 C‐X‐C motif chemokine receptor 7 (CXCR7), a G‐protein‐coupled receptor, was recently identified as a scavenger receptor for the chemokine SDF1 and is expressed in PCa. Although it is well‐accepted that the dominant SDF1 receptor is the C‐X‐C motif chemokine receptor 4 (CXCR4), emerging data has shown that CXCL12 binds to the CXCR7 receptor with high affinity.13, 14, 15 CXCR7 has been implicated in the increased survival, adhesiveness and invasiveness of PCa cells.16, 17, 18 CXCR7 expression is positively correlated with the PCa Gleason grade and is elevated in PCa metastatic lesions, including those present in both soft tissues and bone.19 Knockdown of CXCR7 led to reduced proliferative activities, whereas the overexpression of CXCR7 led to reduced apoptosis.19, 20 SDF1‐CXCR7 signaling was shown to contribute to PCa invasiveness through the regulation of CD44 and cadherin‐11.19 In addition, CXCR7 can activate AKT signaling and upregulate the secretion of interleukin‐8 (IL‐8) and vascular endothelial growth factor (VEGF), potentially contributing to tumor angiogenesis.19 Further studies demonstrated that IL‐8 treatment can upregulate CXCR7 and suggested that an IL‐8‐CXCR7 positive‐feedback loop contributes to malignant progression.19, 20 Remarkably, CXCR7 was reported to interact with epidermal growth receptor (EGFR) and stimulate increased levels of phospho‐EGFR (p‐EGFR) and phospho‐ERK1/2 (p‐ERK1/2).20
A recent publication demonstrated the functional interactions between AR and CXCR721; however, combination treatment of mCRPC with an inhibitor of AR and CXCR7 signaling has not been reported. In the present study, we report the significant effects of ENZ and CCX771, a novel specific CXCR7 inhibitor,22 in the suppression of cell survival, migration and angiogenesis in ENZ‐treated PCa cells. We further show that this combination treatment suppresses tumor growth in CRPC cell line‐based and patient‐derived xenograft (PDX) models.
Material and Methods
Cell lines and reagents
Human PCa LNCaP (American Type Culture Collection [ATCC], 2005), VCaP (ATCC, 2010) and LNCaP C4‐2B (C4‐2B) (derived from LNCaP tumors grown in a castrated host23) cell lines were validated by short tandem repeat DNA fingerprinting with the AmpFℓSTR Identifier kit (Applied Biosystems) in MD Anderson's Cell Line Core Facility. CCX771 (CXCR7‐specific inhibitor), and CCX704 (control for CCX771, i.e., a close analog of CXCR7 without affinity for CXCR7) were provided by ChemoCentryx (Mountain View, CA). AMD3100 (CXCR4 inhibitor) and ENZ (MDV3100) were purchased from SelleckChem, and SDF1 was purchased from GeneScript.
In vitro ENZ and CCX771 treatment
VCaP and C4‐2B cells were pretreated with 1 µM ENZ or DMSO vehicle control for 24 hr. On the following day, additional drug combinations were added and incubated for 48 hr as follows: 100 ng/ml SDF1 + 20 µg/ml AMD3100, 100 ng/ml SDF1 + 1 µM CCX704, 100 ng/ml SDF1 + 800 nM CCX771 or no drugs. Cells were then collected and prepared for cDNA microarray, quantitative real‐time polymerase chain reaction (QRT‐PCR), Western blotting (WB), immunofluorescence staining, fluorescence‐activated cell sorting (FACS), wound‐healing assay, transwell migration assay or ELISA. IC50 and EC50 for AMD3100 and CCX771 were published in our previous study.24
Quantitative real‐time polymerase chain reaction
QRT‐PCR was carried out using the following conditions: initial denaturation for 10 min at 95°C, followed by 40 cycles of denaturation at 95°C for 3 sec, annealing at 60°C for 30 sec. The 2–ΔΔCt method was used to analyze the relative CXCR7 mRNA expression over the control group. Specific PCR primers for CXCR7 (forward:5′‐CACAGCACAGCCAGGAAGG‐3′; reverse:5′‐GTTCCCTGGCTCTGAGTAGTCGA‐3′), and for GAPDH (forward:5′‐AGCACCCCTGGCCAAGGTCA‐3′; reverse:5′‐GCAGTGGGGACACGGAAGGC‐3′) were used (ThermoFisher Scientific).
Western blotting analysis
Antibodies against EGFR, p‐EGFR (Tyr1068), AKT, p‐AKT (Thr308), cleaved PARP (C‐PARP) and GAPDH were purchased from Cell Signaling Technology, and those against CXCR7, and VEGFR2 from Abcam. Densitometry analysis for WB was performed using FIJI imaging software.25 Expression levels of p‐EGFR and p‐AKT (308) were normalized to total EGFR and AKT levels, and VEGFR2 to GAPDH. The value from each drug‐treated group was further normalized with the control. The control was set as 1.
Immunofluorescence
VCaP and C4‐2B cells were seeded on coverslips in 24‐well plates. Forty‐eight hours after ENZ (1 µM) or DMSO treatment the cells were rinsed with PBS, and fixed with acetone‐methanol (1:1) at 4°C for 8 min. After 20 min of blocking in Dako protein block (Dako), the cells were incubated with polyclonal CXCR7 antibody (GeneTex, cat#100027) for 2 hr, followed by incubation with an Alexa 488‐conjugated secondary antibody (Invitrogen) for 40 min. The specificity of immunofluorescence was validated by incubating some cells in PBS instead of primary antibody.
Fluorescence‐activated cell sorting analysis
Treated cells as described above were stained with propidium iodide (PI) and then analyzed on a FACS Canto II (BD Biosciences). Three independent experiments were performed in triplicate. The quantitative data were generated using FlowJo software (Tree Star).
Wound‐healing assay
C4‐2B cells were grown to 80% confluence in 6‐well plates, and a straight line was made in triplicate wells. The medium was removed, and the plates were washed with culture media to remove the floating cell debris. After being treated as described above, the cells were fixed and stained with crystal violet dye. Wound closure was measured using the MRI Wound Healing Tool macro for ImageJ (v1.50b).26 Three independent experiments were performed.
Transwell migration assay
BD Falcon™ (BD Biosciences) 24‐well cell culture inserts with an 8.0‐µm PET membrane were used for transwell migration assays. A total of 1 × 105 (VCaP) and 1 × 104 (C4‐2B) cells/well in 250 µl of culture media with 1% FBS were seeded into the cell culture insert in triplicate. The lower chamber was filled with 750 µl culture media with 10% FBS. After the treatments described above, the cells in the inner surface of the inserts were carefully removed, and the cells that had migrated onto the outer membrane of the inserts were fixed with Protocol® HEMA 3® stain (ThermoFisher Scientific). The number of cells that had migrated to the outer surface of the membrane were counted in nine random high‐power fields (HPFs) under a light microscope (Nikon Instruments). Three independent experiments were performed.
VEGF ELISA
Aliquots of supernatants from the cells were collected and levels of human VEGF were quantified according to the manufacturer's instruction for the ELISA kit (R&D systems). The cells were seeded at a concentration of 1.5 × 106 (VCaP) and 3 × 105 (C4‐2B) cells/well in six well‐plates in triplicate and treated as described above. Three independent experiments were performed. The sensitivity of the assay was <5.0 pg/ml.
Orthotopic VCaP xenografts
VCaP cells that were infected with lentivirus to stably express luciferase, that is, VCaP‐luc (3 × 106 cells), were injected into the dorsolateral prostate of 4‐ to 6‐week‐old athymic nude male mice (Taconic Farm, Hudson, NY) as reported previously.27 When the tumor volume achieved 50 mm3 according to the bioluminescence signal, which was usually 2 weeks after the injection, mice were randomly distributed into the different experimental groups to receive one of the following treatments for 35 days: ENZ (10 mg/kg, oral gavage, daily), CCX771 (25 mg/kg, subcutaneously, daily), ENZ + CCX771 combination, vehicle control for ENZ (1% carboxymethyl cellulose, 0.1%Tween‐80, 5% DMSO in water) or Captisol® (vehicle control for CCX771 provided by ChemoCentryx). The number of animals per treatment group ranged from 17 to 23. Tumor growth was monitored weekly by luminescence signal using the IVIS Lumina Imaging System (Perkin Elmer). At the end of the experimental period, all mice were euthanized, and their tumors were collected.
Subcutaneous PDX MDA133‐4
MDA133‐4 tumors28 were established by subcutaneously implanting 0.125 cm3 tumor fragments into the left flanks of pre‐castrated severe combined immunodeficiency (SCID) mice (Charles River Laboratories). Tumors were allowed to grow until they reached a volume of 50 mm3. The experimental groups received treatment as described in the orthotopic prostate model above, but for 28 days. The number of animals per treatment group ranged from 9 to 13. Tumor growth was measured by calipers, and tumor volumes were calculated using the formula: length × width2/2.29 At the end of the experimental period, the mice were euthanized, and the tumors were collected. All animal experiments were conducted in accordance with accepted standards of humane animal care approved by MD Anderson Cancer Center IACUC.
Immunohistochemical analysis
CD34 and HMGB1 immunostaining was carried out on formalin‐fixed and paraffin‐embedded tissue sections from the MDA133‐4 and VCaP xenografts harvested at Day 28 and 35 respectively. Briefly, after tissue sections were deparaffinized and rehydrated through a graded alcohol series, they were microwaved in 0.01 mol/L citrate buffer at pH 6.0 for 10 min to retrieve antigen. After a 30‐min incubation in Dako protein block, tissue sections were incubated in rabbit monoclonal antibodies to CD34 (Abcam, cat#AB81289) or HMGB1 (Abcam, cat#AB79823,) for 90 min, followed by incubation in a HRP polymer conjugated secondary antibody (Dako, cat#K4061) for 40 min. The immunoreactions were visualized using DAB/H2O2. Specificity of the immunoreactions was verified by replacing the primary antibodies with PBS.
Vascular density was quantified on CD34 labeled tissue sections using an image‐analysis system (Eclipse 90i; Nikon Instruments). Tumor regions with the highest vascular density were first identified by low‐power (40×) screening. Vascular measurement, including both the counts of the vascular profiles and the size of the individual vascular profiles that were outlined by CD34 staining, was carried out in the “hot” region on five 200× measuring fields using Nikon NIS‐Elements version 3.0 software. To quantify necrotic lesions, cytoplasmic and nuclear HMGB1 immunostaining in tumors was identified and digitalized using the Nikon image analysis system. The ratio of the total cytoplasmic versus nuclear immunostaining intensity was used to determine the extent of necrotic lesions.30, 31
Apoptosis detection and quantification
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the ApopTag® Peroxidase in situ Apoptosis Detection Kit (EMD Millipore, cat#S7100) according to the manufacturer's instructions.
Statistical analysis
The experimental data was presented as the mean ± SE. Comparisons between the treatment groups were performed using Student's t test or the Mann‐Whitney rank test when it was appropriate. All p values were derived from two‐tailed analysis. A p value <0.05 was considered statistically significant for all analyses.
Results
Upregulation of CXCR7 expression in ENZ‐treated prostate cancer cells
To determine alterations in gene activity that may contribute to ENZ resistance, we conducted a series of cDNA microarray analyses of ENZ‐treated PCa cells. CXCR7 was identified as one of the upregulated genes in ENZ‐treated VCaP and LNCaP cells (microarray data are deposited in the Gene Expression Omnibus [http://www.ncbi.nlm.nih.gov/geo/], accession number GSE69249).27 Significantly increased CXCR7 mRNA levels were demonstrated by QRT‐PCR in both VCaP and C4‐2B cells after ENZ treatment (Fig. 1 a). WB analysis corroborated the increased expression of CXCR7 in ENZ‐treated VCaP and C4‐2B cells compared to the control cells (Fig. 1 b). Indirect immunofluorescence labelling showed an increased cytoplasmic staining for CXCR7 in ENZ‐treated VCaP and C4‐2B cells compared to the control (DMSO)‐treated cells (Fig. 1 c). Cytoplasmic immunofluorescence likely indicates internalization of CXCR7. This mediates active signaling, which was expected given that the membranous receptor CXCR7 may be internalized and mediate active signaling in the cytoplasm.32
Figure 1.
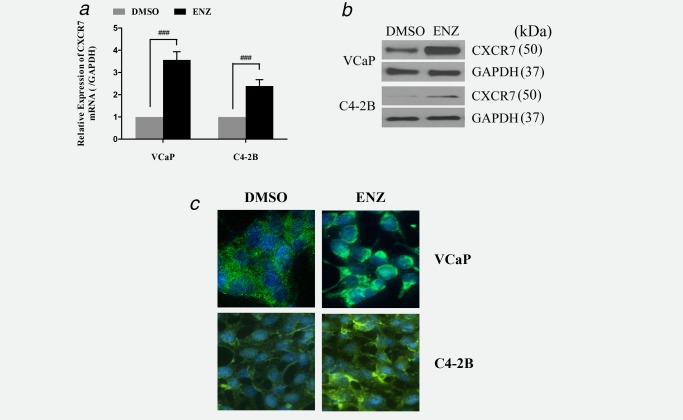
CXCR7 expression is upregulated by enzalutamide (ENZ) treatment in prostate cancer cells. (a) RT‐PCR was used to analyze the expression profile of CXCR7 mRNA in VCaP and C4‐2B cells after treatment with ENZ. (b) Western blotting and (c) immunofluorescence staining were used to analyze CXCR7 protein levels in these cells. The data are represented as the mean ±SE of 3 experiments performed in triplicate. ### p < 0.0001.
ENZ + CCX771 combination treatment leads to reduction in EGFR and AKT phosphorylation, reduced VEGFR2 signaling and induction of apoptosis in prostate cancer cells
ENZ treatment resulted in reductions in p‐EGFR and VEGFR2 compared to the control DMSO in VCaP and C4‐2B cells, although the reductions in C4‐2B cells were modest compared to those in VCaP cells (Fig. 2). Except for the notable reduction of VEGFR2 in C4‐2B, single agent CCX771 treatment did not reduce p‐EGFR or VEGFR2 expressions in either cell line compared to the control CCX704. Single agent AMD3100 treatment did not reduce expression of p‐EGFR, VEGFR2 and p‐AKT in either cell line (Fig. 2). ENZ and CCX771 combination treatment resulted in significant reductions of p‐EGFR, p‐AKT and VEGFR2 expression in both VCaP and C4‐2B cells (Fig. 2). ENZ and AMD3100 combination treatment resulted in reductions of p‐EGFR and VEGFR2 expression but not in p‐AKT in both cell lines, and these reductions were less than those of ENZ and CCX771 combination treatment (Fig. 2).
Figure 2.
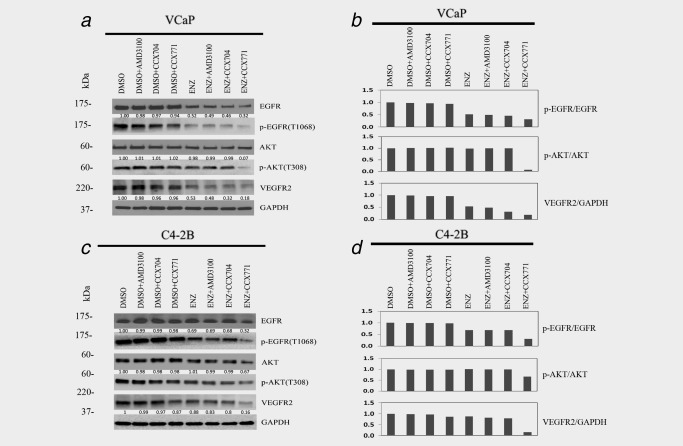
ENZ and CCX771 combination treatment suppresses EGFR, AKT and VEGFR2 signaling. Western blotting analysis revealed that CCX771 and ENZ markedly reduced phosphorylation levels of EGFR, AKT and VEGFR2 in VCaP (a) and C4‐2B (c) cells. Combination treatment (ENZ + CCX771) showed increased suppression of EGFR/AKT signaling compared to both single agents. The quantified ratio of the band intensity for the phosphorylated protein normalized to the total protein and DMSO control, and the VEGFR2 normalized to GAPDH and DMSO control for VCaP (b) and C4‐2B (d) cells.
Analysis of the apoptotic activity response by Western blot analysis of PARP cleavage showed modest inductions by CCX771 in VCaP cells and by ENZ and AMD3100 combination treatment in C4‐2B cells (Figs. 3 a and 3 b). However, substantially increased PARP cleavage was demonstrated by ENZ and CCX771 combination treatment in both cell lines (Figs. 3 a and 3 b). FACS/cell‐cycle analysis of sub‐G1 cells demonstrated that ENZ treatment increased the percentage of cells in the sub‐G1 phase for both VCaP and C4‐2B compared to control (DMSO) treatment (VCaP and C4‐2B: p < 0.05) (Figs. 3 c and 3 d). Single agent CCX771 also increased the percentage of sub‐G1 cells compared to the control treatment in both cell lines (VCaP and C4‐2B: p < 0.001) (Figs. 3 c and 3 d). ENZ and CCX771 combination treatment significantly increased the percentage of sub‐G1 cells compared to CCX771 treatment (VCaP and C4‐2B: p < 0.001) or ENZ treatment (VCaP and C4‐2B: p < 0.001). Single agent AMD3100 increased the percentage of sub‐G1 cells in VCaP (p < 0.001) (Fig. 3 c), but not C4‐2B cells (Fig. 3 d). ENZ and AMD3100 combination treatment significantly increased the percentage of sub‐G1 cells compared to ENZ or AMD3100 treatment alone in both VCaP and C4‐2B cells (VCaP and C4‐2B: p < 0.001). However, ENZ and CCX771 combination treatment increased the percentage of sub‐G1 cells to a greater extent than any single or combination treatment in both VCaP and C4‐2B cells (VCaP and C4‐2B: p < 0.001) (Figs. 3 c and 3 d). These results demonstrate that ENZ and CCX771 combination treatment targeting both AR and CXCR7 resulted in substantially increased apoptosis in these PCa models.
Figure 3.
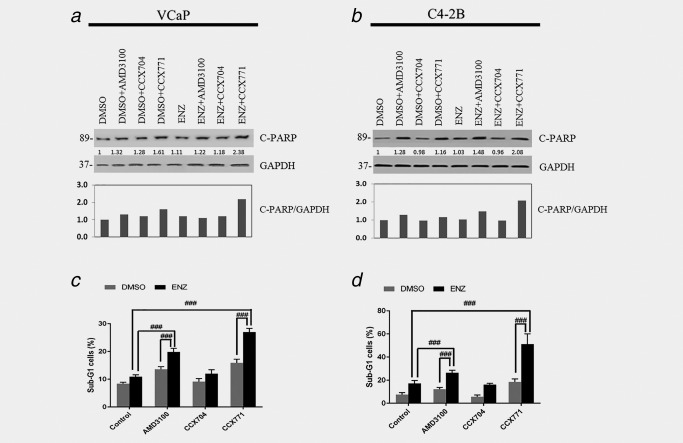
ENZ and CCX771 combination treatment induces apoptosis in prostate cancer cells. WB of C‐PARP showing increased apoptotic activity by ENZ+CCX771 combination treatment in VCaP (a, c) and C4‐2B cells (b, d). ENZ significantly increased the percentage of sub‐G1 cells compared to the controls in both VCaP and C4‐2B cells (p < 0.05, not indicated in the figure) and AMD3100 significantly increased sub‐G1 cells in VCaP (p < 0.001, not indicated in the figure), but not in C4‐2B cells. Combination treatments showed superior pro‐apoptotic activities compared to single agent ENZ, CCX771 or AMD3100 treatment. The data are represented as the mean ± SE of three experiments performed in triplicate. ### p < 0.001.
ENZ and CCX771 combination treatment suppresses migration and angiogenesis of prostate cancer cells
To analyze the specific biological effects of ENZ and CCX771 combination treatment that may result, in part, from the suppression of EGFR‐ or AKT‐mediated signaling, we analyzed the migration and drivers of angiogenesis, that is, VEGF secretion, in VCaP and C4‐2B PCa cells under the specified treatment conditions.
Transwell migration assay results showed the ENZ and CCX771 combination treatment significantly reduced the cell migration compared to single agent treatment with ENZ (p < 0.05) or CCX771 (p < 0.01) in VCaP cells (Figs. 4 a and 4 b). Similar reductions in migration induced by the combination treatment were observed in C4‐2B cells when compared to single agent treatment with ENZ (p < 0.01) or CCX771 (p < 0.001) (Figs. 4 c and 4 d). ENZ and AMD3100 combination treatment showed a significant reduction in migration compared to single agent ENZ, but not AMD3100, in C4‐2B cells (p < 0.05) (Figs. 4 c and 4 d).
Figure 4.
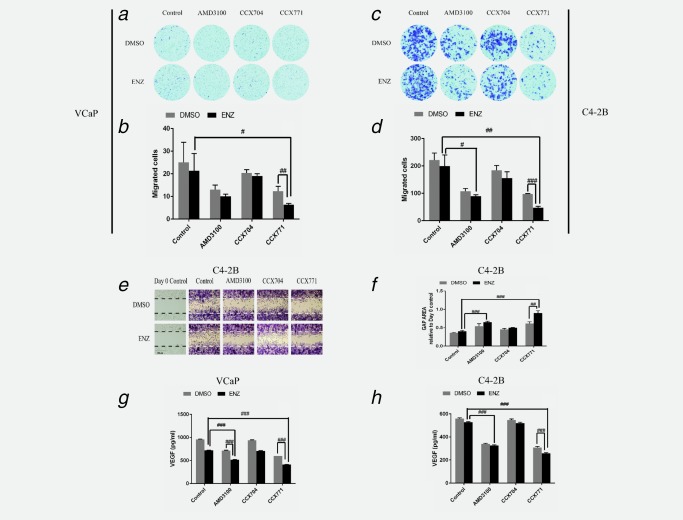
ENZ and CCX771 combination treatment suppresses migration of prostate cancer cells. (a–d) Transwell migration assays demonstrated that ENZ+CCX771 combination treatment led to reduced migration compared to single agent ENZ or CCX771 treatment in prostate cancer cells. (e, f) Wound‐healing assay results showed that ENZ + CCX771 combination treatment also demonstrated reduced migration compared to single agent ENZ or CCX771 treatment in C4‐2B cells. (g, h) VEGF secretion was significantly reduced by CCX771 or AMD3100 compared to the controls (p < 0.001, not indicated in the figure), and reduced to a greater extent by ENZ + CCX771 combination treatment compared to ENZ or CCX771 single agent treatment in prostate cancer cells. The data are represented as the mean ± SE of three experiments performed in triplicate. ### p < 0.001, ## p < 0.01, # p < 0.05.
As VCaP cells tend to aggregate and form clumps, analyzing the migration using wound healing assays would not have yielded accurate results33, 34; therefore, only C4‐2B cells were used for cell migration assays. Wound‐healing migration assay results showed that CCX771 treatment led to significant reductions in C4‐2B cell migration compared to CCX704 (p < 0.01) treatments. We observed a significantly greater effect of ENZ and CCX771 combination treatment compared to ENZ (p < 0.001) or CCX771 (p < 0.01) single agent treatment (Figs. 4 e and 4 f). ENZ and AMD3100 combination treatment showed significant differences (p < 0.001) compared to ENZ alone; however, a significant difference was not observed between ENZ and AMD3100 combination treatment and AMD3100 alone.
VEGF has been shown to play pivotal roles in the regulation of normal and pathogenesis‐related angiogenesis. Among the three VEGF receptors (VEGFR1, 2 and 3), VEGFR2 is generally accepted as the major mediator of VEGF‐driven responses in tumor progression.35, 36 ENZ treatment alone led to reduced VEGF secretion compared to control treatment in VCaP cells (p < 0.001), but not in C4‐2B cells. Additionally, CCX771 or AMD3100 single agent treatment resulted in significant reductions in VEGF secretion in both cell lines (CCX771 and AMD3100: p < 0.001) (Figs. 4 g and 4 h). In the present study, WB analysis verified that VEGFR2 was markedly reduced by ENZ and CCX771 combination treatment compared to single ENZ or CCX771 treatment in both VCaP and C4‐2B cells (Fig. 2). ELISA assay results showed that ENZ and CCX771 combination treatment significantly reduced the secretion of VEGF compared to single ENZ or CCX771 treatment (VCaP and C4‐2B: p < 0.001) (Figs. 4 g and 4 h). ENZ and AMD3100 combination treatment showed significant reduction in VEGF secretion compared to ENZ alone (VCaP and C4‐2B: p < 0.001), and compared to AMD3100 alone in VCaP cells (p < 0.001); however, this combination treatment did not show a significant reduction of VEGF secretion compared to AMD3100 treatment in C4‐2B cells. To determine whether ENZ and CCX771 combination treatment could suppress paracrine‐mediated angiogenic activities, we performed tubule formation assays as previously described37 in human umbilical vein endothelial cells (HUVECs) (Supporting Information Figure 1) and found that incubation in conditioned media from VCaP or C4‐2B cells that had been treated with ENZ and CCX771 resulted in reduced HUVEC tubule formation compared to the control or ENZ‐treated cells (p < 0.05) (Supporting Information Figure 1).
ENZ and CCX771 combination treatment inhibits growth of prostate tumor xenografts
To further test our hypothesis that ENZ combination treatment with CCX771 is a promising therapy approach for CRPC, we used two different AR‐positive, androgen‐dependent CRPC models: VCaP orthotopic prostate xenografts and the PDX MDA133‐4 subcutaneous model. We treated mice bearing VCaP orthotopic tumors with ENZ, CCX771 or a combination of ENZ and CCX771 and monitored their tumor progression (Fig. 5). The results showed that each single agent treatment significantly reduced tumor growth (wet weight) (ENZ: p < 0.01, CCX771: p < 0.05) compared to vehicle control (combined Captisol® group and ENZ vehicle groups). ENZ and CCX771 combination treatment further reduced the wet tumor weights compared to single agent ENZ treatment (p < 0.05), or CCX771 treatment (p < 0.05) (Fig. 5 c).
Figure 5.
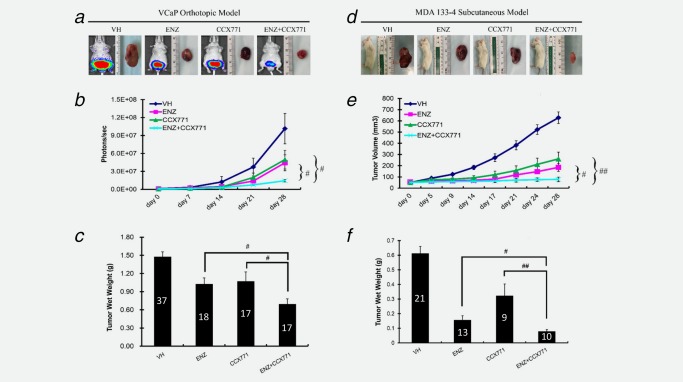
ENZ and CCX771 combination treatment inhibits the growth of prostate tumor xenografts. VCaP orthotopic xenografts and MDA133‐4 PDX subcutaneous models were both treated with vehicle control (presented data are combination of ENZ vehicle and CCX771 vehicle, Captisol), ENZ, CCX771 and ENZ+CCX771 for 28 days. (a–c) VCaP xenografts. (a) Representative bioluminescent images of mice with tumors from each treatment group. (b) Weekly tumor growth measurements by IVIS. (c) Tumor wet weight at the end of 35 days of treatment. (d–f) MDA133‐4 PDX subcutaneous xenografts. (d) Representative bioluminescent images of mice with tumors from each treatment group. (e) Weekly tumor volume measurements by calipers. (f) Tumor wet weight at the end of 28 days of treatment. The data are represented as the mean ± SE. ## p < 0.01, # p < 0.05. The number of animals used for each treatment group are depicted on bar graphs (c) and (f).
Single agent treatment of mice with subcutaneous MDA133‐4 PDX showed similar tumor suppressive effects as the VCaP xenograft regarding the tumor volume (ENZ, Day 28: p < 0.05 and CCX771, Day 28: p < 0.01) (Figs. 5 d and 5 e) and the tumor wet weight (ENZ: p < 0.05 and CCX771: p < 0.01) (Fig. 5 f). ENZ and CCX771 combination treatment further suppressed tumor growth (tumor volume: p < 0.05 vs. ENZ, p < 0.01 vs. CCX771, and wet weight: p < 0.05 vs. ENZ, p < 0.01 vs. CCX771) compared to single agent treatment.
ENZ and CCX771 combination treatment effects on angiogenesis and tumor cell death in CRPC xenografts
We used CD34 immunostaining (Supporting Information Figure 2) to analyze and compare the microvessel density (MVD) in tumor tissue samples from each treatment group and the vehicle control group from the experiments described above. MVD was not significantly different between the treatment and vehicle control groups in either the VCaP or MDA133‐4 model (Figs. 6 a and 6 b). However, the percentage of microvessels with dimensions larger than 100 μm2 was significantly lower in the ENZ and CCX771 combination treatment groups compared to the vehicle control in both the VCaP and MDA133‐4 model (both p < 0.05) (Figs. 6 c and 6 d). Apoptotic bodies in the tumors that were labeled by TUNEL staining (Supporting Information Figure 3) were analyzed. The results showed an increased apoptotic index in response to single agent treatment, that is, ENZ or CCX771 (both p < 0.01) in the VCaP model (Fig. 6 e). Although a trend toward an increased apoptotic index in response to single treatments was observed in the 133–4 model, the differences were not significant. However, apoptotic indices in the ENZ and CCX771 combination treatment groups were significantly increased compared to the vehicle (VCaP: p < 0.001 and MDA133‐4: p < 0.05) (Figs. 6 e and 6 f). As the ENZ‐CCX771 treatment induced a significant reduction in large vessel density, we expected that necrosis could also be induced in these tumors. To address this possibility, we performed immunostaining with an antibody to HMGB1, a necrosis marker,38 in the xenograft models (Supporting Information Figure 4). In the MDA133‐4 model, ENZ and CCX771 combination treatment induced significantly higher necrotic activity compared with vehicle and ENZ (both p < 0.001). Although there was an apparent trend toward increased necrotic activities by a single agent and ENZ and CCX771 combination treatment, these effects were not statistically significant in the VCaP model (Figs. 6 g). However, single agent CCX771 treatment resulted in a significant increase (p < 0.01) in necrotic lesions in the MDA133‐4 model (Fig. 6 h).
Figure 6.
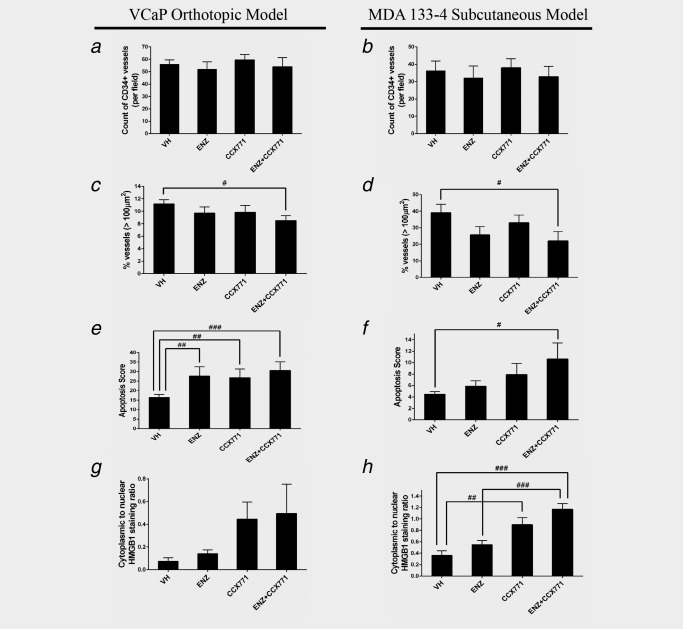
ENZ and CCX771 combination treatment inhibits angiogenesis, and induces apoptosis and necrosis in prostate tumor xenografts. (a–d) Vasculature labeled by CD34 immunostaining was analyzed by image analysis and microvessel density (MVD) was determined in both the VCaP orthotopic xenografts and MDA133‐4 PDX subcutaneous models. Although the differences in overall MVD between any treatment group and the vehicle control group did not reach statistical significance (a, b), the percentage of large vascular profiles (>100 μm2) was significantly lower in the ENZ + CCX771 combination treatment group compared to the vehicle control in both VCaP and MDA133‐4 xenografts (c, d). (e, f) Apoptotic cells were detected by TUNEL staining. ENZ + CCX771 combination treatment induced a significant increase in the apoptotic score in both VCaP and MDA133‐4 xenografts. (g, h) Necrotic lesions were analyzed by HMGB1 immunostaining and represented as the ratio of cytoplasmic to nuclear staining. Compared with the vehicle control, both ENZ + CCX771 and CCX771 treatment induced significant increases in necrotic lesions in the MDA133‐4 PDX xenograft. The data are represented as the mean ± SE. ### p < 0.001, ## p < 0.01, # p < 0.05.
Discussion
ENZ inhibits AR signaling and downregulates multiple AR‐regulated pro‐oncogenic signaling pathways, yet it also de‐represses specific signaling pathways that may promote malignant progression.5, 27, 39, 40, 41 Although multiple PCa cell lines derived from metastases, including LNCaP, C4‐2B (LNCaP‐derived highly metastatic cells), DU145 and PC‐3, have been reported to co‐express CXCR4 and CXCR7,42, 43 CXCR4 was not one of the top regulated genes identified in our microarray analysis of ENZ‐treated PCa cells. Gene expression, IHC and WB analyses supported the microarray results and demonstrated substantially upregulated CXCR7 in VCaP and C4‐2B cells in response to ENZ treatment (Fig. 1). Inhibition of CXCR7 using the novel CXCR7‐specific inhibitor CCX771 significantly increased apoptotic activities in these models, and this effect was potentiated by ENZ treatment in vitro (Figs. 3 a and 3 b), and confirmed in our in vivo experiments (Figs. 6 e and 6 f). CXCR4‐CXCR7 may form homodimers or heterodimers, and these activities likely play an important role in the modulation of downstream signaling.44 We used the CXCR4 inhibitor AMD3100 in our study to compare its effects with CCX771 and found that CCX771 had a superior effect compared to the inhibition of CXCR4 by the CXCR4 selective inhibitor AMD3100 in our model. Although SDF1‐CXCR4 signaling has been widely characterized in multiple malignancies, SDF1‐CXCR7 signaling, which is less studied, may play an important role in metastatic PCa. As ENZ treatment led to significantly upregulated CXCR7, it is conceivable that ENZ‐stimulated CXCR7 signaling may play a role in ENZ resistance through homodimerization or heterodimerization with CXCR445 and that targeting AR together with CXCR7 would counteract the development of ENZ resistance and generate significant therapeutic effects in mCRPC. Although testing this hypothesis is beyond the scope of this report, it is worthy of consideration in future studies.
We demonstrated that the protein expression of p‐EGFR (Y1068), p‐AKT (T308) and VEGFR2 were modestly reduced after ENZ treatment, but the combination of ENZ and CCX771 resulted in further significant reduction of these proteins in both VCaP and C4‐2B models (Fig. 2). The migration capacity of PCa cells was measured using transwell and wound healing migration assays. The results showed similar reductions in the migration of the cells after AMD3100, CCX771 or ENZ and AMD3100 combination treatment. However, statistically significant reductions in migration were demonstrated by ENZ and CCX771 combination treatment compared to single agent treatments (Fig. 4). We used two PCa xenograft models, which reflect the diversity of CRPC, to corroborate the in vitro results with regard to ENZ and CCX771 combination therapeutic activities (Fig. 5). As we observed relatively modest effects by AMD3100 and/or ENZ and AMD3100 combination treatment, these treatments were not tested in the xenograft models.
Previous studies have shown that AR signaling regulates VEGF expression in PCa cells.46 In addition to PCa cells, CXCR7 is also highly expressed in tumor‐associated endothelial cells and plays an important role in angiogenesis.12, 47 Our results show that ENZ and CCX771 combination treatment results in substantial reduction in VEGF secretion (Figs. 4 g and 4 h) in VCaP and C4‐2B cells. Therefore, it is conceivable that suppression of VEGF‐VEGFR signaling by ENZ and CCX771 combination treatment in PCa cells leads to the suppression of autocrine and paracrine‐mediated angiogenic activities. Our results show that incubation with conditioned media from VCaP or C4‐2B cells treated with ENZ and CCX771 significantly reduced HUVEC tubule formation compared to the control or ENZ‐treated cells, supporting the paracrine concept (Supporting Information Figure 1). Previous studies have shown that CCX771 directly inhibits tumor‐associated endothelial cells.12, 47 Our immunohistochemical analysis using the CD34 antibody did not show significant differences in overall vascular density between ENZ, CCX771 or ENZ and CCX771 combination treatment groups and the control group in the VCaP or MDA133‐4 xenograft tissues harvested at 5 (VCaP) or 4 weeks (MDA133‐4) after treatment. However, further analysis showed that ENZ and CCX771 combination treatment resulted in significant reductions in the large vessel fraction (vessels a profile area >100 µm2) of the tumor‐associated vasculature (Figs. 6 c and 6 d). Our results support the notion that the CXCR7‐specific inhibitor CCX771 may cooperate with ENZ to generate a stronger and more durable anti‐angiogenic effect that mainly involves larger vessels. These results are consistent with those from a recent report which showed that both genetically silenced CXCR7 expression or pharmacological inhibition of CXCR7 by CCX771 mainly exerted an effect on larger size capillaries.48 We found it interesting, and potentially important, that ENZ further suppressed VEGF secretion in combination with AMD3100 in VCaP cells, but not in C4‐2B cells, whereas ENZ further suppressed VEGF secretion in combination with CCX771 in both VCaP and C4‐2B cells (Figs. 4 g and 4 h). These results likely reflect the increased androgen sensitivity of VCaP cells compared to C4‐2B cells. Our results also indicate that in PCa cells that are relatively ENZ‐resistant, such as C4‐2B cells, AR and CXCR7‐mediated stimulation of VEGF secretion may drive angiogenesis and indirectly stimulate PCa cell growth. Therefore, ENZ treatment may have particular therapeutic impact for some ADT‐refractory PCa patients in combination with CXCR7 inhibition.
In summary, in this study we demonstrated that ENZ treatment enhanced the downregulation of p‐EGFR, p‐AKT, VEGFR2 and secreted VEGF when combined with a CXCR7‐specific inhibitor, CCX771, in VCaP and C4‐2B PCa cells. These effects were observed to a lesser extent for ENZ and AMD3100 combination treatment compared to single agent treatment. However, in general, the ENZ and CCX771 combination was more effective in the suppression of these signaling activities. In addition, we showed that ENZ and CCX771 combination treatment was more effective in suppressing migration compared to single agent treatment than ENZ and AMD3100 combination treatment. Importantly, ENZ and CCX771 combination treatment showed significantly greater growth suppression activities in PCa cell line xenograft and PDX models of CRPC, and an analysis of tumor tissues showed a significant reduction in the large tumor‐associated vessels in response to this combination treatment. However, this was not the case with single agent treatment. We suggest that in vivo suppression of tumor growth is related to the suppression of autocrine and paracrine VEGF‐VEGFR2 signaling by ENZ and CCX771 combination treatment.
Supporting information
Supporting Fig1
Supporting Fig2
Supporting Fig3
Supporting Fig4
Conflict of interest: Drs. Zhang, Campbell and Schall are full‐time employees of ChemoCentryx, Inc. The other authors declare no conflicts of interest.
References
- 1. Schrijvers D. Androgen‐independent prostate cancer. Recent Results Cancer Res 2007;175:239–49. [DOI] [PubMed] [Google Scholar]
- 2. Guerrero J, Alfaro IE, Gomez F, et al. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration‐resistant prostate cancer. Prostate 2013;73:1291–305. [DOI] [PubMed] [Google Scholar]
- 3. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–97. [DOI] [PubMed] [Google Scholar]
- 5. Karantanos T, Evans CP, Tombal B, et al. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol 2015;67:470–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013;32:5501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sfanos KS, Hempel HA, De Marzo AM. The role of inflammation in prostate cancer. Adv Exp Med Biol 2014;816:153–81. [DOI] [PubMed] [Google Scholar]
- 8. Nelson WG, De Marzo AM, DeWeese TL, et al. The role of inflammation in the pathogenesis of prostate cancer. J Urol 2004;172:S6–11; discussion S–2. [DOI] [PubMed] [Google Scholar]
- 9. Begley L, Monteleon C, Shah RB, et al. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell 2005;4:291–8. [DOI] [PubMed] [Google Scholar]
- 10. Begley LA, MacDonald JW, Day ML, et al. CXCL12 activates a robust transcriptional response in human prostate epithelial cells. J Biol Chem 2007;282:26767–74. [DOI] [PubMed] [Google Scholar]
- 11. Sun YX, Pedersen EA, Shiozawa Y, et al. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF‐1/CXCL12. Clin Exp Metastasis 2008;25:765–76. [DOI] [PubMed] [Google Scholar]
- 12. Sun X, Cheng G, Hao M, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev 2010;29:709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darash‐Yahana M, Pikarsky E, Abramovitch R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J 2004;18:1240–2. [DOI] [PubMed] [Google Scholar]
- 14. Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF‐1 and I‐TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 2006;203:2201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bachelerie F, Graham GJ, Locati M, et al. New nomenclature for atypical chemokine receptors. Nat Immunol 2014;15:207–8. [DOI] [PubMed] [Google Scholar]
- 16. Sun YX, Wang J, Shelburne CE, et al. Expression of CXCR4 and CXCL12 (SDF‐1) in human prostate cancers (PCa) in vivo. J Cell Biochem 2003;89:462–73. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, Li G, Stanco A, et al. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron 2011;69:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kallifatidis G, Munoz D, Singh RK, et al. Beta‐arrestin‐2 counters CXCR7‐mediated EGFR transactivation and proliferation. Mol Cancer Res 2016;14:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF‐1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 2005;280:35760–6. [DOI] [PubMed] [Google Scholar]
- 20. Crump MP, Gong JH, Loetscher P, et al. Solution structure and basis for functional activity of stromal cell‐derived factor‐1; dissociation of CXCR4 activation from binding and inhibition of HIV‐1. EMBO J 1997;16:6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsiao JJ, Ng BH, Smits MM, et al. Androgen receptor and chemokine receptors 4 and 7 form a signaling axis to regulate CXCL12‐dependent cellular motility. BMC Cancer 2015;15:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cruz‐Orengo L, Holman DW, Dorsey D, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med 2011;208:327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thalmann GN, Sikes RA, Wu TT, et al. LNCaP progression model of human prostate cancer: androgen‐independence and osseous metastasis. Prostate 2000;44:91–103. [DOI] [PubMed] [Google Scholar]
- 24. Zabel BA, Wang Y, Lewen S, et al. Elucidation of CXCR7‐mediated signaling events and inhibition of CXCR4‐mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol 2009;183:3204–11. [DOI] [PubMed] [Google Scholar]
- 25. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L, Karanika S, Yang G, et al. Androgen receptor inhibitor‐induced “BRCAness” and PARP inhibition are synthetically lethal for castration‐resistant prostate cancer. Sci Signal 2017;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee YC, Cheng CJ, Bilen MA, et al. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res 2011;71:5194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989;24:148–54. [DOI] [PubMed] [Google Scholar]
- 30. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–5. [DOI] [PubMed] [Google Scholar]
- 31. Ge X, Antoine DJ, Lu Y, et al. High mobility group box‐1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J Biol Chem 2014;289:22672–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu B, Xu D, Deng X, et al. CXCL12 enhances human neural progenitor cell survival through a CXCR7‐ and CXCR4‐mediated endocytotic signaling pathway. Stem Cells 2012;30:2571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harma V, Virtanen J, Makela R, et al. A comprehensive panel of three‐dimensional models for studies of prostate cancer growth, invasion and drug responses. PLoS One 2010;5:e10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vainio P, Lehtinen L, Mirtti T, et al. Phospholipase PLA2G7, associated with aggressive prostate cancer, promotes prostate cancer cell migration and invasion and is inhibited by statins. Oncotarget 2011;2:1176–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberts E, Cossigny DA, Quan GM. The role of vascular endothelial growth factor in metastatic prostate cancer to the skeleton. Prostate Cancer 2013;2013:418340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo S, Colbert LS, Fuller M, et al. Vascular endothelial growth factor receptor‐2 in breast cancer. Biochim Biophys Acta 2010;1806:108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tahir SA, Yang G, Goltsov AA, et al. Tumor cell‐secreted caveolin‐1 has proangiogenic activities in prostate cancer. Cancer Res 2008;68:731–9. [DOI] [PubMed] [Google Scholar]
- 38. Raucci A, Palumbo R, Bianchi ME. HMGB1: a signal of necrosis. Autoimmunity 2007;40:285–9. [DOI] [PubMed] [Google Scholar]
- 39. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell 2011;19:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine‐specific demethylase 1. Cancer Cell 2011;20:457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thompson TC, Li L. New targets for resistant prostate cancer. Oncotarget 2014;5:8816–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J, Shiozawa Y, Wang J, et al. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF‐1 in prostate cancer. J Biol Chem 2008;283:4283–94. [DOI] [PubMed] [Google Scholar]
- 43. Kiss DL, Windus LC, Avery VM. Chemokine receptor expression on integrin‐mediated stellate projections of prostate cancer cells in 3D culture. Cytokine 2013;64:122–30. [DOI] [PubMed] [Google Scholar]
- 44. Sánchez‐Martín L, Sánchez‐Mateos P, Cabañas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med 2013;19:12–22. [DOI] [PubMed] [Google Scholar]
- 45. Decaillot FM, Kazmi MA, Lin Y, et al. CXCR7/CXCR4 heterodimer constitutively recruits beta‐arrestin to enhance cell migration. J Biol Chem 2011;286:32188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eisermann K, Broderick CJ, Bazarov A, et al. Androgen up‐regulates vascular endothelial growth factor expression in prostate cancer cells via an Sp1 binding site. Mol Cancer 2013;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamada K, Maishi N, Akiyama K, et al. CXCL12‐CXCR7 axis is important for tumor endothelial cell angiogenic property. Int J Cancer 2015;137:2825–36. [DOI] [PubMed] [Google Scholar]
- 48. Wani N, Nasser MW, Ahirwar DK, et al. C‐X‐C motif chemokine 12/C‐X‐C chemokine receptor type 7 signaling regulates breast cancer growth and metastasis by modulating the tumor microenvironment. Breast Cancer Res 2014;16:R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Fig1
Supporting Fig2
Supporting Fig3
Supporting Fig4