

Abstract
Motivation
The concept of a ‘mechanism-based taxonomy of human disease’ is currently replacing the outdated paradigm of diseases classified by clinical appearance. We have tackled the paradigm of mechanism-based patient subgroup identification in the challenging area of research on neurodegenerative diseases.
Results
We have developed a knowledge base representing essential pathophysiology mechanisms of neurodegenerative diseases. Together with dedicated algorithms, this knowledge base forms the basis for a ‘mechanism-enrichment server’ that supports the mechanistic interpretation of multiscale, multimodal clinical data.
Availability and implementation
NeuroMMSig is available at http://neurommsig.scai.fraunhofer.de/
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The development of novel high throughput ‘omic’ technologies in the last decade has revealed new insight and progresses in areas of cancer, cardiovascular and metabolic disorders. The datasets coming from these technologies have led to the discovery of candidate biomarkers and potential drug targets. However, in other areas such as neurodegenerative diseases, this mechanistic understanding is either rather limited or almost absent.
Readouts in translational biomedicine are going beyond molecular level: they can span from genes and genetic variation information to imaging and organ-level (or even organism-level) data and markers. The definition of a disease as ‘dysregulated pathways’ may hold true for cancer, but is inappropriate for neurodegenerative diseases as pathways refer typically to rapid molecular processes and the alterations in neurodegenerative diseases are slow and multi-facetted. There is simply no such thing as a ‘degeno-gene’ (in analogy to the ‘onco-gene’). Supporting that, there have not been any described ‘cause-effect’ relationships that would explain the different pathological changes observed in patients with these disorders. When the effects of dysregulation can be easily observed—like in monogenic diseases—it is generally not so difficult to link the phenotype with the event that lead to it. This is likely to be attributed to a short and direct chain of causality (Hofmann-Apitius et al., 2015a). Hence, because the complexity of neurodegenerative diseases is enormous; it is crucial to integrate a wider spectrum of causal assertions into models that represent and organize the available mechanistic knowledge.
MSigDB (Subramanian et al., 2015) is the prototypic implementation of a system that allows for the identification of perturbed pathways. However, the output of ranking algorithms like GSEA is usually a list of associated canonical pathways that do not contain disease-specific information and multimodal data. In addition, canonical pathways are also biased towards cancer biology (Hofmann-Apitius et al., 2015b).
Adopting the fundamental principle of ‘running patterns in data against a knowledge base of established patterns’ (‘pathways’; ‘signatures’), we have developed a mechanism enrichment server and extended it towards multiscale and multimodal data. This is where the two ‘M’ of NeuroMMSig come from: Multimodal and Mechanistic. It is noteworthy that the difference between NeuroMMSig and other, conventional methods for pathway enrichment or functional gene annotation lies in the specificity of the disease context. Pathway enrichment is based upon canonical pathways, which are not disease specific. The multimodal mechanisms behind NeuroMMSig, however, are manually curated and contain detailed representations of multimodal pathophysiology in a well-defined disease context.
Here, we present NeuroMMSig, a web server for mechanism enrichment that allows submission of multiscale data from molecular to clinical level to return mechanisms that fit best the data. We have focused on neurodegenerative diseases, as we try to establish a ‘mechanism-based taxonomy of Alzheimer’s Disease (AD) and Parkinson’s Disease (PD)’. This is the core of the AETIONOMY project (www.aetionomy.eu) and in fact, NeuroMMSig (DB and Server) form the backbone of attempts at stratifying patient subgroups based on disease mechanisms.
2 Systems and methods
2.1 Categorization of NDD pathways from mechanism based models
Disease knowledge assembly models were built using Biological Expression Language (BEL) which integrate literature-derived ‘cause and effect’ relationships in the form of triples (Kodamullil et al., 2015). We have captured a representative subsample of the scientific knowledge on existing canonical pathways in AD and PD (Iyappan et al., 2016) which have been grouped into subgraphs.
2.2 Multimodal data integration, data sources and software
NeuroMMSig’s subgraphs have been enriched with multimodal data (e.g. imaging features, variant information and drugs). The methodology describing how the linking across different data scales was performed is provided in the Supplementary text. Moreover, we have developed an enrichment algorithm to rank the subgraphs based on the input.
3 Implementation
3.1 NeuroMMSig server
NeuroMMSig is available at http://neurommsig.scai.fraunhofer.de/. A user interface offers a simple, yet comprehensive menu (Fig. 1A). Input fields allow users to submit multimodal data (e.g. genes, SNPs, imaging features). Users can also set the enrichment algorithm parameters and define the operators of the query. After data submission, a ranked list of subgraphs is displayed to the user (Fig. 1B). Here, associated information to the submitted data is shown as icons in a user friendly table: drug-gene interactions, known regulating miRNA and co-expressed networks. Moreover, when the user selects one or multiple subgraphs and clicks on ‘Visualize Network’, NeuroMMSig displays the graph representing the selection where the user can investigate how the disruption of the network occurs (Fig. 1C). For that reason, NeuroMMSig offers multiple functionalities enabling graph mining and reasoning over the graphs (e.g. graph algorithms, search and exporting options, knowledge provenance and Sankey diagram representations for pathway analysis).
Fig. 1.
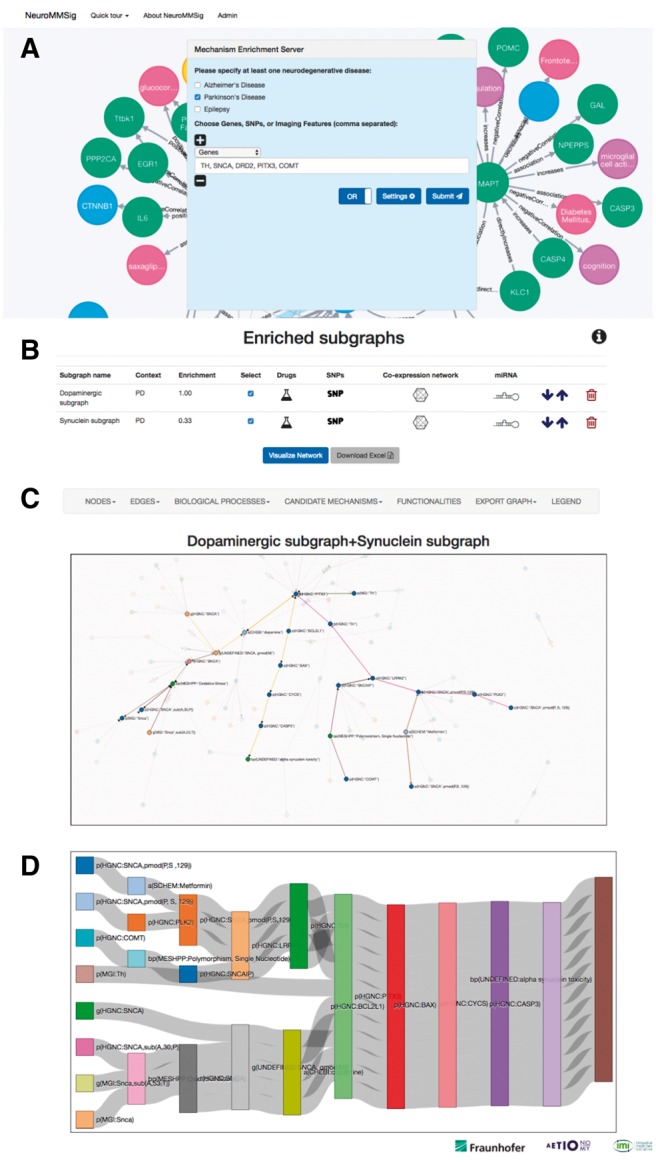
The web interface for NeuroMMSig. (A) Home page. Users can select the disease context and submit their data. In the navbar, links are provided to ‘Introduction’ and ‘How to use’ pages. (B) Example of the results provided by ranking algorithm with the most enriched subgraphs. (C) By clicking ‘Visualize Network’, the users can access the interactive network visualization with multiple functionalities. (D) Sankey diagram of causal relationships comprising the proposed mechanism
3.2 Application scenario
The five most relevant genes associated with ‘Dopamine signaling pathway’ in PD according to SCAIView [http://academia.scaiview.com/academia/] (Supplementary text) were used as an input (Fig. 1A). Two subgraphs were retrieved from NeuroMMSig (‘Dopaminergic subgraph’ and ‘Synuclein subgraph’) and they were selected for further analysis (Fig. 1B). Using the query tools, the two main hub nodes SNCA and Parkinson’s disease were removed from the network in order to avoid most of the paths going through them, which biases the retrieval of best candidate mechanisms. By choosing a process of interest such as ‘alpha synuclein toxicity’, the server proposes candidate mechanisms in which the data-mapped-nodes may perturb normal physiology (Fig. 1C and D).
4 Discussion
Harmonization of heterogeneous and multiscale datasets is yet a tremendous challenge in the field of neurodegeneration. The gap between molecular and clinical data is too wide to establish stable and meaningful assertions between imaging features and genes, for instance. Thus, integration of different data scales is a necessary step to shed some light on the mechanisms underlying neurodegenerative diseases.
The modeling approach chosen in NeuroMMSig is capable of explaining causal and correlative relationships among different entities namely genes, proteins or biological processes in the context of neurological disorders (Kodamullil et al., 2015). These relationships reveal the upstream and downstream regulators of each node in the network and how they are activating/inhibiting their neighboring nodes. Thus, navigating through the network it is possible to identify the root or primarily cause of a dysfunctional gene or protein which eventually contributes to the disorder.
The inventory of mechanisms specific for neurodegenerative diseases, which forms the basis of NeuroMMSig, is composed of small cause-and-effect models encoded in OpenBEL. Evidences for the BEL-encoded mechanisms come from the scientific literature, from experimental data analysis and from clinical readouts such as imaging biomarkers. Furthermore, both AD and PD models incorporate genetic and epigenetic information, which might, for instance, indicate and partially explain the effect of a particular SNP in a mechanism (Khanam et al., 2015; Naz et al., 2016). The presented work also serves as a comparison tool between different diseases. Thus, it allows to systematically identify shared-mechanisms between them. Combining all together, the BEL-encoded mechanisms contain pathophysiology information at highest resolution, with highly curated evidences spanning from the genetics and epigenetics layer via cell-type specific information to clinical phenotypes and biomarkers. Hence, NeuroMMSig overcomes some of challenges that pathway analysis methods currently have, as indicated by Khatri et al. (2012).
Supplementary Material
Acknowledgements
We thank Andrej Konotopez, Sumit Madan, André Gemünd and Charles Tapley Hoyt for technical assistance and valuable advices. We would also like to acknowledge Apurva Gopisetty and Anka Güldenpfennig for their support curating the models. Finally, we thank Shweta Bagewadi and Sepehr Golriz Khatami for their inputs to towards metadata inclusions.
Funding
This work was supported by the European Union/European Federation of Pharmaceutical Industries and Associations (EFPIA) Innovative Medicines Initiative Joint Undertaking under AETIONOMY [grant number 115568], resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution.
Conflict of Interest: none declared.
References
- Hofmann-Apitius M. et al. (2015a) Bioinformatics mining and modeling methods for the identification of disease mechanisms in neurodegenerative disorders. Int. J. Mol. Sci., 16, 29179–29206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann-Apitius M. et al. (2015b) Towards the taxonomy of human disease. Nat. Rev. Drug Discov., 14, 75. [DOI] [PubMed] [Google Scholar]
- Iyappan A. et al. (2016) Towards a pathway inventory of the human brain for modeling disease mechanisms underlying neurodegeneration. J. Alzheimer's Dis., 52, 1343–1360. [DOI] [PubMed] [Google Scholar]
- Khanam I.A. et al. (2015) Computational modelling approaches on epigenetic factors in neurodegenerative and autoimmune diseases and their mechanistic analysis. J. Immunol. Res., 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P. et al. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol, 8, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodamullil A.T. et al. (2015) Computable cause-and-effect models of healthy and Alzheimer's disease states and their mechanistic differential analysis. Alzheimer's Dement., 11, 1329–1339. [DOI] [PubMed] [Google Scholar]
- Naz M. et al. (2016) Reasoning over genetic variance information in cause-and-effect models of neurodegenerative diseases. Brief. Bioinf., 17, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. (2015) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.