

Abstract
Background
Circular RNAs (circRNAs) have emerged as important regulators in carcinogenesis and metastasis. However, the knowledge of circRNAs in bladder cancer remains limited. This study aimed to investigate the role and mechanism of circRNAs in the development and progression of bladder cancer.
Material/Methods
Three pairs of bladder carcinomas (including high- and low-grade tumors) and adjacent normal tissues were collected from patients. The total RNAs were extracted from these samples and subjected to Clariom D microarray assays to detect the differentially expressed circRNAs and mRNAs. The mRNA targets for these circRNAs were predicted by miRanda in combination with stringent differential mRNA filters. The interaction network for the circRNA-mRNA pairs was generated by Cytoscape.
Results
Among the 1038 circRNAs detected by the Clariom D microarray assay, we identified 7 significantly differentially expressed circRNAs in the tumors (fold change >2, FDR <0.05). Principal component analysis of the differential circRNAs confirmed that the tumor samples were separated from the normal samples. Hierarchical clustering analyses on these RNAs and their predicted mRNA targets showed that the majority of differentially expressed circRNAs and mRNAs had been up-regulated in the bladder tumors. KEGG signaling pathway analysis has indicated that genes involved in cell proliferation, oncogenic transformation, and metastasis are potentially regulated by these circRNAs.
Conclusions
The current study provides a molecular basis for further investigating the mechanisms by which circRNAs regulate bladder cancer. The clinical significance of the identified circRNAs is highlighted by their potentials as diagnostic and prognostic biomarkers for bladder cancer patients.
MeSH Keywords: Gene Expression Profiling; RNA, Untranslated; Urinary Bladder Neoplasms
Background
Bladder cancer is ranked as one of the major genitourinary malignancies worldwide, with men being 4 times more likely than women to be diagnosed with this disease [1]. Urothelial carcinoma accounts for over 90% of all bladder cancer cases, and thus is the focus of bladder cancer research [2]. The economic burden of bladder cancer remains high and has been among the most costly malignancies for decades [3]. Despite great advances in understanding its molecular pathogenesis, detailed mechanisms involved in the initiation, progression, and metastasis of bladder cancer await elucidation. In addition, genetic alterations are the key to understanding the tumorigenesis of bladder cancer. Previous studies have laid out the mutational landscape for this cancer, highlighting genes of chromatin-remodeling and cell cycle pathways [4], along with the commonly known mutations in TP53 [5], HRAS [6], FGFR3 [7], PIK3CA [8], RB1 [9], KRAS [6], and TSC1 [10]. Many resources and tools have empowered in-depth studies on the mechanisms accounting for cancer progression [11], and regulatory non-coding RNAs (ncRNAs) have emerged as one such important mechanism to regulate carcinogenesis and metastasis [12]. However, knowledge of the regulatory ncRNAs and their roles in bladder cancer is still limited. Clinically, regulatory ncRNAs may serve as promising biomarkers and/or therapeutic targets for bladder cancer, and potentially offer significant benefits to patients. In the clinic, some strategies have been proposed to predict the prognosis of the bladder cancer patients, and correct stratification of patients into low-, intermediate-, and high-risk groups may assist with recommendation of proper adjuvant treatment [13,14]. A comprehensive analysis of the transcriptomes of early-stage urothelial carcinomas revealed distinct subclasses of non-muscle-invasive bladder cancer, which may improve prognostication and treatment selection [15]. Therefore, there exists a compelling reason to investigate the regulatory ncRNAs in bladder cancer.
In the present study, we focused on identifying differentially expressed circular RNAs (circRNAs) in the tumors and revealing their roles in carcinogenesis and metastasis of bladder cancer. CircRNAs are a class of endogenous regulatory ncRNAs, produced by back-splicing circularization from precursor mRNAs of numerous eukaryotic genes [16]. This class of RNAs was initially identified by next-generation RNA sequencing (RNA-seq) and is generally transcribed at low levels, but their transcription is widespread among various cells and tissues [17–19]. The presence of circRNAs in biological samples like saliva and blood makes them convenient and potentially diagnostic or predictive biomarkers for cancer development, progression, and prognosis [16]. The tissue-specific and disease-associated expression patterns of circRNAs are suggestive of their roles [20]. Since some circRNAs are derived from genomic loci that are related to development of diseases, it is proposed that they may regulate physiological and pathological conditions as well [12]. Furthermore, it has been demonstrated that the levels of particular circRNAs are significantly altered during oncogenic proliferation [21]. CircRNAs may serve as sponges for microRNAs and lift inhibition on target mRNA expression, causing transcriptomic alterations in the cell [22]. For instance, a circular RNA – circHIPK3 – sponges the microRNA miR-558 to suppress the expression of heparanase, and negatively regulates the progression of bladder cancer [23]. As a result, it is conceivable that dysregulation of circRNAs can lead to de-regulated signaling pathways that are involved in several cellular processes such as deregulation of cellular proliferation, which is a hallmark of carcinogenesis. Therefore, in the present study, we hypothesize that a particular set of circRNAs are differentially expressed in the bladder tumors and contribute to the development and progression of bladder cancer.
To test this hypothesis, we systematically investigated the differentially expressed circRNAs in clinical samples of bladder tumors and adjacent normal tissues. Specifically, we extracted total RNAs from these samples and detected circRNAs and mRNAs by microarray assays. We applied integrative analysis to identify the differentially expressed circRNAs in the tumors. We further performed hierarchical clustering analysis on these RNAs and predicted their mRNA targets. Finally, we applied differential mRNA filters and deduced the signaling pathways for which the putative mRNA targets may be involved. We also validated the findings of this study by quantitative RT-PCR. Our study identifies circRNAs that have not been reported previously for bladder cancer. Most importantly, the pathway analysis demonstrates that genes involved in cell proliferation, oncogenic transformation, and metastasis are potentially subject to the regulations of these circRNAs. Our findings establish a molecular basis for delving into the detailed mechanisms by which circRNAs regulate the oncogenic process in bladder cancer. The clinical significance of the identified circRNAs needs to be established by systematic biomarker studies, for which circRNAs may be promising candidates for diagnostic and prognostic predictions.
Material and Methods
Clinical samples
From February to August 2017, 3 cases of bladder carcinoma (high-grade and low-grade) with resection specimens containing adjacent normal bladder tissue were collected at Beijing Luhe Hospital (Table 1). All samples were confirmed by histological examination prior to the microarray chip assay. The samples were processed according to a previously published method and stored at −80°C [24]. The collection protocol was approved by the Institutional Human Research Ethics Committee. All participating patients have read and signed the informed consent agreement.
Table 1.
The information of the bladder cancer patients recruited to the current study.
| ID | Gender | Age (years) | Pathology | TNM staging |
|---|---|---|---|---|
| 1 | Male | 76 | High grade urothelial carcinoma | T2N1M0 |
| 2 | Male | 72 | High grade urothelial carcinoma | T2N1M0 |
| 3 | Male | 49 | Low grade urothelial carcinoma | T2N0M0 |
Total RNA extraction
Total RNA was isolated with TRIzol (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. Extracted RNA was quantified spectrophotometrically with NanoDrop ND-1000 (Thermo Fisher Scientific, Wilmington, DE, USA). The quality of RNA was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Microarray hybridization
Isolated total RNA was amplified, labeled, and hybridized using the Clariom D platform following the manufacturer’s instructions (Affymetrix, Santa Clara, CA, USA). Briefly, the Affymetrix GeneChip WT Pico Kit was used for cDNA preparation and biotin labeling. cRNA was purified using an Affymetrix magnetic bead protocol. The Affymetrix GeneChip Hybridization, Wash, and Stain kit was used for array processing. Arrays were incubated for 16 h in an Affymetrix GeneChip 645 hybridization oven at 45°C with rotation at 60 rpm. The chips were subsequently scanned with an Affymetrix GeneChip Scanner 3000. Raw data were analyzed using Affymetrix Expression Console and Transcriptome Analysis Console software prior to downstream analysis. circRNAs with ≥2-fold-change (FC) and p<0.05 were selected as being significantly differentially expressed (Figure 1).
Figure 1.

Bioinformatics analysis to identify the differentially expressed circRNA-mRNA pairs. (A) To identify the differentially expressed circRNAs, RNAs displaying fold change greater than 2 (FC ≥2) with statistical significance (FDR [false discovery rate] ≤0.05) were selected. The putative circRNA-mRNA pairs were determined after application of multiple differential filters. (B) The predicted interaction network of the circRNA-mRNA pairs. FC – fold change; FDR – false discovery rate.
Quantitative RT-PCR (qRT-PCR)
FastQuant RT Super Mix (Tiangen, Beijing, China) was used to synthesize cDNA templates. Quantitative RT-PCR was performed by SYBR Green in 20 μl of reactions containing 2 μl of cDNA, 0.5 μM of forward primers, and 0.5 μM of reverse primers. PCR was set at 95°C for 10 min for pre-denaturation, followed by 40 cycles of [95°C for 10 s and 60°C for 60 s]. RPS18 was used as a reference. All PCRs were performed in triplicate. The relative expression level of circRNA was calculated by 2−ΔΔCt method.
Bioinformatics and statistical analysis
Differentially expressed circRNAs-targeted miRNAs pairs were predicted using miRanda software combined with stringent differential mRNA filters (Figure 1). The sponge microRNA targets of the differentially expressed circRNAs were predicted by use of Miranda software on miRbase (http://www.mirbase.org). The potential circRNA-mRNA pair prediction was performed by integrating the microRNA-mRNA targets data from www.microrna.org. Candidate circRNA-mRNA pairs were further subject to a differential mRNA expression filter using data from the same microarray assay. Principal component analysis was performed for the differentially expressed circRNAs and mRNAs data obtained from the microarray assays. Volcano plots and hierarchical clustering analyses were also performed for the differentially expressed circRNAs and the mRNAs detected by the microarray to gain statistical and biological insights. circRNAs identified by the profiling data were subjected to gene ontology (GO) and KEGG pathway analyses based on their correlated mRNAs using Gene Ontology (http://www.geneo-ngoloty.org/). The interaction network was predicted and generated by Cytoscape.
Statistical tests were dependent on data distributions: parametric tests were applied for normally distributed data (means, t test, and Pearson correlation), and non-parametric tests were applied for non-normally distributed data (medians, Kruskal-Wallis rank sum test, and Spearman’s rank correlation). Statistical analysis was performed with R and R-packages (version 3.4.1).
Results
Discovery of the differentially expressed circRNAs and their potential mRNA targets in bladder tumors
From the Clariom D microarray assay, we detected hybridization signals for 1038 circRNAs, among which 527 (50.7%) had been annotated previously, while the rest remained to be further characterized and had not been named yet. To identify the differentially expressed circRNAs, we selected those displaying fold change greater than 2 (FC >2) with statistical significance (FDR [false discovery rate] <0.05), and determined that 7 circRNAs were either up- or down-regulated in the tumors (hsa_circ_0000172, chr5_158368701_158368987, chrX_131412035_131417060, chr15_61512925_61513309, chr22_28943661_28946139, hsa_circ_0002495, and chr9_74522734_74523039) (Figure 1). To identify the mRNA targets of these circRNAs, we first predicted the sponge targets of microRNA for these circRNAs based on Miranda software predictions on miRbase. After integrating the microRNA-mRNA targets data from www.microrna.org, we determined the potential circRNA-mRNA pairs. These candidate circRNA-mRNA pairs were further subjected to a differential mRNA expression filter using data from the same microarray assay. Finally, we identified 2 differentially expressed circRNAs with 89 predicted target mRNAs (Figure 1).
Robustness of the microarray-based strategy for discovering tumor-specific circRNAs
To confirm the validity of the microarray experiment, we evaluated whether the identified differentially expressed circRNAs can distinguish between samples from tumors and adjacent normal tissues. We performed principal component analysis (PCA) on the differential circRNAs retrieved by the microarray data and confirmed that the tumor and normal samples were divided into distinctive groups (Figure 2A). Similarly, the PCA plot of the 1221 differentially expressed mRNAs showed that the tumor samples were also separated from the normal samples (Figure 2B). Therefore, the differentially expressed circRNAs and mRNAs could distinguish between the tumorous and normal tissues, strongly supporting the robustness of the microarray-based strategy for discovering tumor-specific circRNAs for bladder cancer.
Figure 2.

The principle component analysis on the microarray data. (A) Principal component analysis (PCA) was performed on the differentially expressed circRNAs retrieved by the microarray data. The tumor samples are separated from the normal samples. (B) PCA plot of the 1221 differentially expressed mRNAs (FDR ≤0.05, FC ≥2). FC: fold change; FDR: false discovery rate.
A majority of circRNAs and coding mRNAs were up-regulated in the bladder tumors
To gain biological insights from the differentially expressed circRNAs, we further performed hierarchical clustering analyses on circRNAs and mRNAs detected by the microarray. The clustering analysis on the significantly differentially expressed 7 circRNAs (FDR <0.05, Figure 3A) showed that most circRNAs (hsa_circ_0000172, chr5_158368701_158368987, chrX_131412035_131417060, chr15_61512925_61513309, chr22_28943661_28946139, and hsa_circ_0002495) were up-regulated in the tumors, with only 1 circRNA (chr9_74522734_74523039) being down-regulated in these samples (Figure 3B). Some of these circRNAs are novel and not yet named, and we tentatively annotate them here using the format of “chromosome_start position_end position”. Concomitantly, clustering on the coding mRNAs detected by the same microarray showed a similar pattern: most of the coding mRNAs were up-regulated in the tumors (Figure 3C, 3D). Therefore, a vast majority of signaling pathways may have been aberrantly up-regulated in bladder tumors (see Discussion).
Figure 3.

Volcano plots and hierarchical clustering analysis of the differentially expressed circRNAs and mRNAs. (A) The volcano plot of the differentially expressed circRNAs (FDR <0.05). The 7 differentially expressed circRNAs are labeled in the plot. (B) Hierarchical clustering analysis of the differentially expressed circRNAs. The identities of the 7 significantly differentially expressed circRNAs are shown on the right of each row. Some circRNAs are unnamed and tentatively annotated here using the format “chromosome_start position_end position”. T – tumor samples, N – normal samples. (C) The volcano plot of all the differentially expressed coding mRNAs from the microarray (FDR <0.01). (D) Hierarchical clustering analysis of the differentially expressed coding mRNAs. T – tumor samples; N – normal samples; FC – fold change; FDR – false discovery rate.
To further confirm the findings from the microarray and the clustering results on the differentially expressed circRNAs, we evaluated the relative expressions of 5 out of the 7 circRNAs by quantitative RT-PCR (qRT-PCR). Significantly, the qRT-PCR results were consistent with the microarray findings: Has_circ_0000172, Has_circ_0002495, Chr22: 28943661, and Chr5: 158368701 were up-regulated in tumor, while Chr9: 74522734 was down-regulated (Figure 4).
Figure 4.
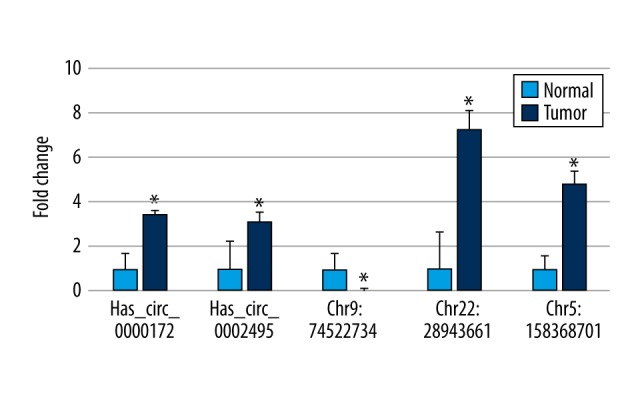
Validation of the differentially expressed circRNAs. To confirm the findings from the microarray analysis, the relative expression of circRNAs was measured by qRT-PCR. Five out of the 7 significantly differentially expressed circRNAs were measured. Note that some circRNAs have not yet been named yet, and are tentatively named here using the “chromosome: start_position” format. * p<0.05 vs. normal. qRT-PCR – quantitative reverse transcriptase-polymerase chain reaction.
The differentially expressed circRNAs may positively regulate a vast array of signaling pathways in bladder cancer
To demonstrate the biological functions of the differentially expressed circRNAs for bladder cancer, we performed KEGG signaling pathway analysis on the filtered predicted mRNA targets of the differential circRNAs (Figure 5). Presumably, these mRNAs were enriched for signaling pathways involved in cell proliferation and oncogenic transformation, such as those of MAPK and RAS signaling pathways. Other pathways, including those of oxytocin, chemokine, calcium, axon guidance, apelin amoebiasis, and lipolysis in adipocytes, were also enriched for the target mRNAs of the differentially expressed circRNAs. Since both circRNA and its target mRNAs usually show a co-regulated pattern, it is likely that the same signaling pathways are predominantly up-regulated in the bladder tumors as well (see Discussion).
Figure 5.
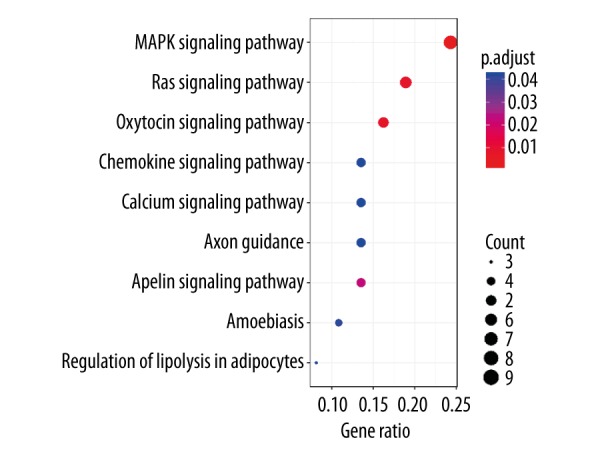
KEGG pathway analysis of the top predicted differentially expressed mRNAs. The putative biological functions of the differentially expressed circRNAs for bladder cancer are demonstrated by KEGG signaling pathway analysis of the predicted mRNA targets. KEGG – Kyoto encyclopedia of genes and genomes.
Discussion
In the present study, we profiled circRNAs that were differentially expressed in the bladder tumors in comparison with the normal tissues. By applying multiple bioinformatics filters, we identified 7 significantly differentially expressed circRNAs and predicted their mRNA targets. The KEGG pathway analysis on these predicted targets suggests that the differentially expressed circRNAs are likely to up-regulate signaling pathways of cell proliferation, oncogenic transformation, chemotaxis, and metabolism. These results reveal a complex regulatory network for the development and progression of bladder cancer, and will assist with further explorations to identify novel therapeutic targets and prognostic/diagnostic markers for bladder cancer.
To date, a large number of circRNAs have been cataloged owing to the rapidly evolving high-throughput sequencing technologies. It has been established that circRNAs usually show tissue- or developmental stage-specific expression patterns [12,25]. CircRNAs are predominantly cytoplasmic, and function as microRNA sponges, playing important regulatory roles during the development and progression of cancer [26]. A circRNA expression study revealed that abundantly expressed circRNAs may be a class of good prognostic biomarkers because they have distinct features from the low-expressing circRNAs [27]. Although various circRNAs have been documented for many cancers [23,28,29], studies of circRNAs in bladder cancer remain important since they have significant clinical implications. Furthermore, the functional implications of the differentially expressed circRNAs await systematic investigations. In the present study, implementing multiple stringent statistical filters, we identified 7 significantly differentially expressed circRNAs, 5 of which were novel and have not yet been reported. Interestingly, 6 circRNAs were up-regulated in tumors, while only 1 was down-regulated. This result is in line with the clustering results on the differentially expressed mRNAs, where a majority of the genes were up-regulated in the tumors (Figure 3D). This observation supports the previous notion that circRNAs and their mRNA targets are co-regulated. As a result, the interacting circRNA-mRNA pairs will display a unidirectional pattern. The fact that most genes were up-regulated in the tumors suggests that many pathways might have been turned on to promote the development and progression of bladder cancer. Indeed, KEGG signaling pathway analysis on the proposed targets of the differentially expressed circRNAs verified that the pathways of cell proliferation and oncogenic transformation have been enriched in the bladder tumors.
In the current KEGG pathway analysis, the top significant signaling pathways (p<0.01) – MAPK, RAS, and oxytocin signaling pathways – are enriched for the putative mRNA targets of circRNAs in the bladder tumors. The MAPK pathway is involved in many oncogenic processes, including cell proliferation, and its high activity is ubiquitously observed in the cancer cells. This result is consistent with a previous finding that p38 MAPK can promote bladder tumor cell migration and invasion by upregulating the metalloproteinases MMP-2 and MMP-9 [30]. For the RAS pathway, up-regulation of the RAS signaling is always associated with the oncogenic transformation of the malignant cells in bladder cancer. This result is also in line with previous studies in which both HRAS and KRAS were identified as frequently mutated genes in bladder cancer [4,6,15]. Lastly, the discovery of the oxytocin pathway being enriched for the mRNA genes is somewhat surprising. However, it has been reported that oxytocin can promote migration of prostate tumor cells, thus indicating a role in metastasis [31]. Therefore, the role of oxytocin in the progression of bladder cancer should be investigated in future studies.
In the present study, we identified multiple circRNAs that are differentially expressed in bladder tumors. The clustering and pathway analyses on their putative mRNA targets have shown distinct enrichment patterns. This suggests the clinical potential for these circRNAs to serve as biomarkers for diagnostic and prognostic predictions for bladder cancer patients. As cancer is one of the leading causes of death worldwide, it is imperative to identify new diagnostic biomarkers and therapeutic targets to improve overall survivals [16]. Deciphering the roles of circRNAs in bladder cancer would likely facilitate the identification of new diagnostic markers and therapeutic interventions in early stages. In future studies, we will further evaluate and verify the expression patterns of these circRNAs in larger clinical sample cohorts. Consequently, follow-up studies will need to address the feasibility of using these circRNAs as biomarkers for clinical applications such as assisted diagnosis, prognostic prediction, and patient stratification for bladder cancer management.
Conclusions
In summary, by investigating the differentially expressed circRNAs for clinical tumor samples, we have revealed a complex regulatory network for bladder cancer. The signaling pathways involved in the proliferation, metastasis, and transformation of the cancer are predicted to be subject to regulations of these differential circRNAs. We expect that the present results will assist future studies on the pathogenesis, theranostics, and treatment of bladder cancer.
Footnotes
Source of support: Departmental sources
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Pasin E, Josephson DY, Mitra AP, et al. Superficial bladder cancer: An update on etiology, molecular development, classification, and natural history. Rev Urol. 2008;10(1):31–43. [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung C, Dinh T, Lee J. The health economics of bladder cancer: An updated review of the published literature. Pharmacoeconomics. 2014;32(11):1093–104. doi: 10.1007/s40273-014-0194-2. [DOI] [PubMed] [Google Scholar]
- 4.Guo G, Sun X, Chen C, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45(12):1459–63. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cordon-Cardo C, Dalbagni G, Saez GT, et al. p53 mutations in human bladder cancer: Genotypic versus phenotypic patterns. Int J Cancer. 1994;56(3):347–53. doi: 10.1002/ijc.2910560309. [DOI] [PubMed] [Google Scholar]
- 6.Jebar AH, Hurst CD, Tomlinson DC, et al. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24(33):5218–25. doi: 10.1038/sj.onc.1208705. [DOI] [PubMed] [Google Scholar]
- 7.Cappellen D, De Oliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23(1):18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 8.Platt FM, Hurst CD, Taylor CF, et al. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15(19):6008–17. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 9.Cairns P, Proctor AJ, Knowles MA. Loss of heterozygosity at the RB locus is frequent and correlates with muscle invasion in bladder carcinoma. Oncogene. 1991;6(12):2305–9. [PubMed] [Google Scholar]
- 10.Hornigold N, Devlin J, Davies AM, et al. Mutation of the 9q34 gene TSC1 in sporadic bladder cancer. Oncogene. 1999;18(16):2657–61. doi: 10.1038/sj.onc.1202854. [DOI] [PubMed] [Google Scholar]
- 11.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205–11. doi: 10.1038/nrm.2015.32. [DOI] [PubMed] [Google Scholar]
- 13.Babjuk M, Bohle A, Burger M, et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: Update 2016. Eur Urol. 2017;71(3):447–61. doi: 10.1016/j.eururo.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 14.Sylvester RJ, van der Meijden AP, Oosterlinck W, et al. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur Urol. 2006;49(3):466–65. doi: 10.1016/j.eururo.2005.12.031. discussion 675–77. [DOI] [PubMed] [Google Scholar]
- 15.Hedegaard J, Lamy P, Nordentoft I, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 2016;30(1):27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Wang F, Nazarali AJ, Ji S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am J Cancer Res. 2016;6(6):1167–76. [PMC free article] [PubMed] [Google Scholar]
- 17.Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15(7):409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly S, Greenman C, Cook PR, Papantonis A. Exon skipping is correlated with exon circularization. J Mol Biol. 2015;427(15):2414–17. doi: 10.1016/j.jmb.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Rybak-Wolf A, Stottmeister C, Glazar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–85. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 21.Bachmayr-Heyda A, Reiner AT, Auer K, et al. Correlation of circular RNA abundance with proliferation – exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057. doi: 10.1038/srep08057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–88. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Zheng F, Xiao X, et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017;18(9):1646–59. doi: 10.15252/embr.201643581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P, Chen S, Chen H, et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta. 2015;444:132–36. doi: 10.1016/j.cca.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 25.Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–38. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 26.Dong Y, He D, Peng Z, et al. Circular RNAs in cancer: An emerging key player. J Hematol Oncol. 2017;10(1):2. doi: 10.1186/s13045-016-0370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okholm TLH, Nielsen MM, Hamilton MP, et al. Circular RNA expression is abundant and correlated to aggressiveness in early-stage bladder cancer. NPJ Genom Med. 2017;2:36. doi: 10.1038/s41525-017-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kristensen LS, Hansen TB, Veno MT, Kjems J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene. 2018;37(5):555–65. doi: 10.1038/onc.2017.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong Z, Lv M, Chen J. Screening differential circular RNA expression profiles reveals the regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in bladder carcinoma. Sci Rep. 2016;6:30919. doi: 10.1038/srep30919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar B, Koul S, Petersen J, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010;70(2):832–41. doi: 10.1158/0008-5472.CAN-09-2918. [DOI] [PubMed] [Google Scholar]
- 31.Zhong M, Boseman ML, Millena AC, Khan SA. Oxytocin induces the migration of prostate cancer cells: Involvement of the Gi-coupled signaling pathway. Mol Cancer Res. 2010;8(8):1164–72. doi: 10.1158/1541-7786.MCR-09-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]