

SUMMARY
Stressful events rapidly trigger activity-dependent synaptic plasticity, driving the formation of aversive memories. However, it remains unclear how stressful experience affects plasticity mechanisms to regulate appetitive learning, such as intake of addictive drugs. Using rats, we show that corticotropin-releasing factor (CRF) and α1 adrenergic receptor (α1AR) signaling enhance the plasticity of NMDA-receptor-mediated glutamatergic transmission in ventral tegmental area (VTA) dopamine (DA) neurons through distinct effects on inositol 1,4,5-triphosphate (IP3)-dependent Ca2+ signaling. We find that CRF amplifies IP3-Ca2+ signaling induced by stimulation of α1ARs, revealing a cooperative mechanism that promotes glutamatergic plasticity. In line with this, acute social defeat stress engages similar cooperative CRF and α1AR signaling in the VTA to enhance learning of cocaine-paired cues. These data provide evidence that CRF and α1ARs act in concert to regulate IP3-Ca2+ signaling in the VTA and promote learning of drug-associated cues.
In Brief
Tovar-Díaz et al. demonstrate a cellular mechanism in which corticotropin-releasing factor (CRF) and α1 adrenergic receptors act in concert to regulate the induction of synaptic plasticity in VTA dopamine neurons and enhance cocaine place conditioning.
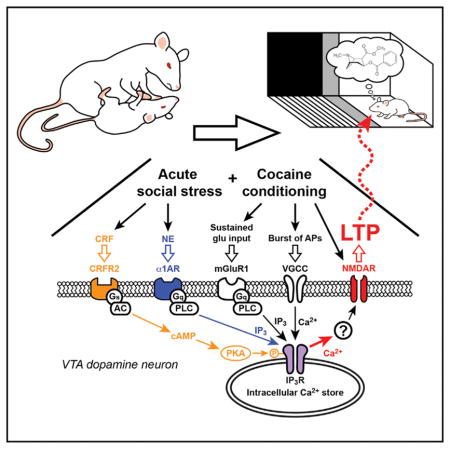
INTRODUCTION
Stress is a well-known risk factor for addiction and drug relapse. Early life or chronic stress increases addiction vulnerability (Sinha, 2001), and stress reactivity predicts relapse rates in human cocaine addicts (Back et al., 2010). Similarly, acute stress reliably reinstates drug seeking in extinguished animals (Mantsch et al., 2016; Polter and Kauer, 2014; Shaham et al., 2000). As such, studies on the immediate impact of stress on addiction have largely focused on its effects on relapse and reinstatement of drug seeking. However, how acute stressful experience regulates the acquisition of addictive behaviors is less understood. Since addiction can be viewed as a maladaptive form of reward learning (Sinha, 2008), the impact of stress on learning of drug-associated cues may be important for the development of addiction.
Dopamine (DA) neurons in the ventral tegmental area (VTA) play a key role in reward learning (Schultz, 2015). These neurons display transient burst firing in response to primary rewards (e.g., palatable food), while addictive drugs, such as cocaine, induce repetitive DA neuron bursting via pharmacological actions (Covey et al., 2014; Keiflin and Janak, 2015). During cue-reward conditioning, DA neurons “learn” to respond to reward-predicting cues, thereby encoding the positive emotional/motivational valence of those cues (Cohen et al., 2012; Schultz, 1998; Stauffer et al., 2016). Glutamatergic inputs onto DA neurons drive burst firing via activation of NMDA receptors (Overton and Clark, 1997; Paladini and Roeper, 2014); thus, cues that excite glutamatergic inputs to the VTA may contribute to conditioned bursting. We have shown previously that repeated pairing of cue-like glutamatergic input stimulation with reward-like bursting leads to long-term potentiation (LTP) of NMDA transmission (LTP-NMDA) in DA neurons (Harnett et al., 2009). LTP induction requires amplification of burst-evoked Ca2+ signals by preceding the activation of metabotropic glutamate receptors (mGluRs) coupled to the generation of inositol 1,4,5-triphosphate (IP3). Here, IP3 receptors (IP3Rs) detect the coincidence of IP3 generated by glutamatergic input activity and burst-driven Ca2+ entry. Mechanistically, IP3 enhances Ca2+ activation of IP3Rs, thereby promoting Ca2+-induced Ca2+ release from intra-cellular stores (Taylor and Laude, 2002). LTP induction also requires NMDA receptor activation at the time of postsynaptic burst, which likely accounts for the input specificity of LTP; i.e., only those inputs paired with burst undergo LTP (Harnett et al., 2009). Thus IP3-Ca2+ signaling acts as a molecular substrate for LTP and, possibly, the learning of cue-reward associations.
Numerous studies have bridged stress to addiction through the release of corticotropin-releasing factor (CRF) and norepinephrine (NE), two well-studied mediators of responses to stress (Joëls et al., 2011; Koob, 1999; Maras and Baram, 2012). CRF and NE may represent links between stress and cue-reward learning, since they are released in response to stress and regulate IP3-Ca2+ signaling in DA neurons through CRF2 and α1 adrenergic receptors (CRFR2 and α1ARs, respectively) (Paladini et al., 2001; Riegel and Williams, 2008). Whether stress induces CRF and noradrenergic signaling in the VTA to regulate glutamatergic synaptic plasticity in DA neurons and reward learning is currently unknown. Here, we investigated how CRF and α1ARs in the VTA work in concert to regulate plasticity of NMDA transmission in DA neurons and mediate social stress enhancement of conditioning to cocaine-paired cues.
RESULTS
CRF Enhances Noradrenergic Effects on IP3-Ca2+ Signaling to Promote NMDA Plasticity in VTA DA Neurons
Potentiation of NMDA excitation of DA neurons in the VTA may contribute to the learning of cues associated with rewards, including addictive drugs (Stelly et al., 2016; Wang et al., 2011; Whitaker et al., 2013; Zweifel et al., 2008, 2009). Since CRF and NE are two major mediators of acute stress effects in the brain (Joëls et al., 2011; Maras and Baram, 2012), we examined the effect of these transmitters on NMDA plasticity using ex vivo VTA slices. First, we observed that CRF, the α1AR agonist phenylephrine, and NE, at the concentrations tested, had minimal effect on NMDA transmission itself in DA neurons (Figure S1).
Induction of LTP-NMDA requires mGluR/IP3-dependent facilitation of action potential (AP)-evoked Ca2+ signals (Harnett et al., 2009). CRF enhances IP3-Ca2+ signaling by activation of CRFR2 in DA neurons (Bernier et al., 2011; Riegel and Williams, 2008; Whitaker et al., 2013), likely via protein kinase A (PKA)-mediated phosphorylation, causing increased IP3R sensitivity (Wagner et al., 2008). To first confirm this CRF effect, we assessed AP-evoked Ca2+ signals using the size of small-conductance Ca2+-sensitive K (SK) currents (IK(Ca)) and a low concentration of IP3 (1 μM × mJ), which produced no measurable SK-mediated outward current (IIP3) by itself (Figure S2). IP3 was photolytically applied into the cytosol for 100 ms immediately before evoking unclamped APs (Experimental Procedures). Bath application of CRF (100 nM) significantly increased the magnitude of IP3-induced facilitation of IK(Ca) (Figures 1A and 1B).
Figure 1. CRF Enhances Induction of LTP- NMDA Driven by IP3-Induced Ca2+ Signal Facilitation in VTA Dopamine Neurons.

(A) Summary time graph (left) and example traces (right) showing that bath application of CRF (100 nM) augments IP3-induced facilitation of AP-evoked IK(Ca). IP3 was photolytically applied into the cytosol for 100 ms (purple bar in example traces) immediately before evoking unclamped APs (6 cells from 4 rats).
(B) Graph plotting the magnitude of IP3-induced IK(Ca) facilitation before and after CRF application; t(5) = 3.29, *p < 0.05, two-tailed paired t test.
(C) Representative experiment to induce LTP in the presence of CRF. CRF (100 nM) was perfused for ~6 min after a 10-min baseline EPSC recording, while the LTP induction protocol, which consisted of an IP3-synaptic stimulation-burst combination (illustrated at the top), was delivered at the time indicated (10 times every 20 s; during a 3-min period starting ~3 min after the onset of CRF perfusion to allow for CRF effect to take place; see [A]). Example traces of NMDA EPSCs at times indicated are shown in inset (scale bars: 50 ms/20 pA).
(D) Summary time graph of LTP experiments in which LTP was induced using an IP3-synaptic stimulation-burst combination protocol in control solution (8 cells from 8 rats) and in CRF (11 cells from 9 rats).
(E) Summary bar graphs depicting the magnitude of IK(Ca) facilitation (left) and LTP (right) for the experiments shown in (D). IP3-induced facilitation of single AP-evoked IK(Ca) was assessed by comparing the size of IK(Ca) with and without preceding IP3 application, which was done immediately before or after delivering the LTP induction protocol (IK(Ca) facilitation: t(17) = 5.01; LTP: t(17) = 5.70; ***p < 0.0001, two-tailed unpaired t test).
(F) The magnitude of LTP is plotted versus the magnitude of IK(Ca) facilitation of individual neurons. Dashed line indicates a linear fit to all data points (n = 19, r2 = 0.64).
Data are presented as mean ± SEM.
Next, the effect of CRF on LTP-NMDA was tested using an induction protocol consisting of IP3 application (1 μM × mJ; 100 ms) prior to simultaneous pairing of a burst (5 APs at 20 Hz) with a brief train of synaptic stimulation (20 stimuli at 50 Hz), the latter being necessary to induce LTP at specific inputs, likely via activating NMDA receptors at those inputs at the time of burst (Harnett et al., 2009; Stelly et al., 2016; Whitaker et al., 2013). While this induction protocol using a low concentration of IP3 (1 μM × mJ) produced relatively small LTP in control solution, robust LTP was induced in the presence of CRF (100 nM; Figures 1C–1F).
We further examined the effect of CRF on IK(Ca) and LTP induction without IP3 application. CRF (100–300 nM) had no significant effect on basal IK(Ca) (Figures 2A and 2B). Consistent with this observation, CRF failed to enable measurable LTP when simultaneous synaptic stimulation-burst pairing without prior IP3 application was used to induce LTP (Figures 2C and 2D).
Figure 2. CRF Causes No LTP without IP3-Induced Ca2+ Signal Facilitation.

(A) Summary time graph (left) and example traces (right) illustrating that CRF (100 nM) has a small effect on IK(Ca) without preceding IP3 application (8 cells from 7 rats).
(B) Summary bar graph showing the magnitude of IK(Ca) facilitation produced by two concentrations of CRF (300 nM: 5 cells from 4 rats).
(C) Representative experiment to induce LTP in the presence of CRF using an induction protocol consisting of synaptic stimulation-burst pairing with no preceding IP3 application. Example EPSC traces at the times indicated are shown in inset (scale bars: 50 ms/20 pA).
(D) Summary time graph of LTP experiments in which LTP was induced using a synaptic stimulation-burst pairing protocol in control solution (10 cells from 10 rats) and in CRF (7 cells from 7 rats).
Data are presented as mean ± SEM.
DA neurons express α1ARs that are coupled to phospholipase-C-mediated IP3 synthesis (Cui et al., 2004; Paladini et al., 2001). Accordingly, bath application of the α1AR agonist phenylephrine (0.5–1 μM) and NE (1 μM) increased IK(Ca) in a concentration-dependent manner in the absence of exogenous IP3 application (Figures 3A and 3B). Phenylephrine and NE treatment enabled robust LTP induction with simultaneous synaptic stimulation-burst pairing (Figures 3C and 3D), in contrast to the ineffectiveness of CRF described earlier.
Figure 3. α1AR Agonists Phenylephrine and Norepinephrine Enable LTP without IP3-Induced Ca2+ Signal Facilitation.

(A) Summary time graph (left) and example traces (right; 1 μM Phe) depicting the facilitatory effects of phenylephrine and NE on IK(Ca) (0.5 μM Phe: 7 cells from 3 rats; 1 μM Phe: 9 cells from 6 rats; 1 μM NE: 5 cells from 5 rats).
(B) Summary bar graph showing the magnitude of phenylephrine-induced IK(Ca) facilitation.
(C) Representative experiment to induce LTP-NMDA in the presence of phenylephrine (1 μM) using an induction protocol consisting of synaptic stimulation-burst pairing with no preceding IP3 application. Example EPSC traces at the times indicated are shown in inset (scale bars: 50 ms/20 pA).
(D) Summary time graph of LTP experiments in which LTP was induced using a synaptic stimulation-burst pairing protocol in the presence of phenylephrine or NE (0.5 μM Phe: 7 cells from 7 rats; 1 μM Phe: 10 cells from 8 rats; 1 μM NE: 9 cells from 7 rats).
Data are presented as mean ± SEM.
We next asked whether CRF, via CRFR2-mediated IP3R sensitization, could enhance the effect of α1AR activation. CRF (100 nM), which had minimal effect on IK(Ca) by itself (Figures 2A and 2B), significantly augmented the small IK(Ca) facilitation produced by a low concentration (0.5 μM) of phenylephrine (Figures 4A and 4B), while there was no significant CRF effect on IK(Ca) facilitation caused by 1 μM phenylephrine (Figure S3). As a consequence, combined application of CRF and 0.5 μM phenylephrine enabled LTP with simultaneous synaptic stimulation-burst pairing protocol, comparable to LTP induced in the presence of 1 μM phenylephrine (Figures 4C and 4D).
Figure 4. CRF Interacts with Phenylephrine to Drive LTP without IP3-Induced Ca2+ Signal Facilitation.

(A) Summary time graph (left) and example traces (right) showing that CRF augments facilitation of AP-evoked IK(Ca) produced by a low concentration (0.5 μM) of phenylephrine (7 cells from 5 rats).
(B) Graph plotting the magnitude of IK(Ca) facilitation caused by phenylephrine (0.5 μM) alone and by phenylephrine and CRF in individual cells; t(6) = 2.22, *p < 0.05, two-tailed paired t test.
(C) Representative experiment to induce LTP in the presence of both CRF and phenylephrine (0.5 μM) using an induction protocol consisting of synaptic stimulation-burst pairing with no preceding IP3 application. Example EPSC traces at the times indicated are shown in inset (scale bars: 50 ms/50 pA).
(D) Summary time graph of LTP experiments in which LTP was induced using a synaptic stimulation-burst pairing protocol in the presence of both CRF and phenylephrine (0.5 μM) (7 cells from 4 rats).
Data are presented as mean ± SEM.
Altogether, these data in VTA slices strongly suggest that CRF and, most likely, NE acting at α1ARs promote LTP-NMDA by differentially regulating IP3-Ca2+ signaling, i.e., via CRFR2-mediated increase in IP3R sensitivity and α1AR-mediated generation of IP3. These distinct mechanisms enabled CRF and α1AR signaling to act in a cooperative fashion (Figures 4A, 4B, 5A, and 5B). Furthermore, LTP magnitude was positively correlated with the size of IK(Ca) facilitation during induction across neurons with different induction conditions (Figure 5C), supporting the notion that IP3-dependent Ca2+ signal facilitation drives LTP.
Figure 5. Summary of LTP Experiments without IP3 Application.

(A and B) Summary bar graphs demonstrating the magnitude of IK(ca) facilitation (A) and LTP (B) for all LTP experiments without IP3 application shown in Figures 2, 3, and 4. For (A): F(5, 44) = 10.22, p < 0.0001; for (B): F(5, 44) = 6.28; p = 0.0002, one-way ANOVA. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus CRF; #p < 0.05 and ##p < 0.01 versus Phe (0.5 μM), Bonferroni post hoc test).
(C) The magnitude of LTP is plotted versus the magnitude of IK(ca) facilitation in individual neurons. Dashed line indicates a linear fit to all data points (n = 50, r2 = 0.50).
Data are presented as mean ± SEM.
Acute Social Stress Enhances Cocaine-Associated Cue Learning
Since CRF and α1AR signaling promoted NMDA plasticity in DA neurons, we next investigated whether acute stress affects the learning of cocaine-associated cues using a conditioned place preference (CPP) paradigm. Rats underwent 30 min of social defeat (~5 min of direct contact/defeat followed by ~25 min of protected threat), a form of psychosocial stress that elicits strong physiological responses (Koolhaas et al., 2011). After a 10-min interval, stressed rats and handled controls were conditioned with a relatively low dose of cocaine (5 mg/kg, intraperitoneally [i.p.]; Figure 6A). This acute defeat stress-cocaine conditioning sequence was limited to a single session to eliminate the confounding effect reflecting persistent influence of stress on CPP acquisition and/or expression (Burke et al., 2011; Chuang et al., 2011; Kreibich et al., 2009; Smith et al., 2012; Stelly et al., 2016). We found that stressed rats developed a larger preference for the cocaine-paired chamber compared with control rats (Figures 6B, 6C, and 6F). Both stressed and control rats developed comparable CPP with a larger cocaine dose (10 mg/kg) during conditioning (Figures 6D–6F). Acute stress did not affect locomotor behavior during the conditioning session (Figure S4A). Defeat stress also failed to affect CPP when cocaine conditioning (5 mg/kg) was performed after a prolonged interval (1.5 hr; Figures 6G–6J). These results show that social defeat stress acutely enhances the sensitivity to cocaine conditioning.
Figure 6. Acute Exposure to Social Defeat Stress Enhances Cocaine Place Conditioning.

(A) Experimental timeline for testing the effect of acute social defeat stress on the acquisition of cocaine CPP.
(B–E) Changes in the preference for the cocaine-paired side in handled control rats and stressed rats conditioned with 5 mg/kg or 10 mg/kg cocaine. (B) Handled control rats conditioned with 5 mg/kg cocaine: t(7) = 3.14, *p < 0.05. (C) Stressed rats conditioned with 5 mg/kg cocaine: t(7) = 9.61, ***p < 0.0001. (D) Handled control rats conditioned with 10 mg/kg cocaine: t(6) = 9.97, p < 0.0001. (E) Stressed rats conditioned with 10 mg/kg cocaine: t(7) = 9.82, p < 0.0001; two-tailed paired t test; n = 7–8 rats).
(F) Summary graph demonstrating defeat-stress-induced enhancement of sensitivity to cocaine conditioning (stress: F(1, 27) = 9.81, p < 0.01; cocaine dose: F(1, 27) = 49.3, p < 0.0001; Stress × Cocaine Dose: F(1, 27) = 10.62, p < 0.01; two-way ANOVA). *p < 0.05; ***p < 0.001 (Bonferroni post hoc test).
(G) Experimental timeline for testing the effect of social defeat stress on cocaine CPP after a 1.5-hr interval.
(H and I) Changes in the preference for the cocaine-paired side in rats that underwent handling (H) or social defeat (I) 1.5 hr before cocaine conditioning (5 mg/kg). For (H): t(8) = 0.79, p = 0.45; for (I): t(8) = 1.9, p = 0.081; two-tailed paired t test; n = 9 rats. n.s., not significant.
(J) Graph illustrating the ineffectiveness of defeat stress on cocaine CPP with a prolonged interval, t(16) = 0.80, p = 0.43; two-tailed unpaired t test.
Data are presented as mean ± SEM.
CRF and α1ARs Work Together in the VTA to Drive Stress Enhancement of Cocaine Place Conditioning
Acquisition of psychostimulant CPP is inhibited by mGluR1 or NMDA blockade in the VTA, while CPP expression is attenuated by NMDA, but not mGluR1, antagonism in the VTA (Whitaker et al., 2013), supporting the potential role of LTP-NMDA in driving CPP. Since CRF and α1ARs act in concert to promote LTP-NMDA, we next explored whether CRF and α1AR actions in the VTA contribute to social-stress-induced enhancement of cocaine CPP. Low-dose cocaine (5 mg/kg) was used for conditioning in the following experiments to avoid the ceiling effect observed with a higher dose (Figure 6F). Although delivery of the CRFR2 antagonist K41498 (9 pmol/0.3 μL per side) into the VTA prior to social defeat had no significant effect, stress-enhanced cocaine conditioning was significantly suppressed by the α1AR antagonist prazosin (9 pmol/0.3 μL per side) and abolished by co-injection of K41498 and prazosin (Figures 7A–7F). Furthermore, administration of the glucocorticoid receptor antagonist mifepristone (40 mg/kg, i.p.) prior to stress had no effect on cocaine conditioning (Figure S5). Thus, acute social defeat stress recruits a cooperative CRF and NE signaling mechanism acting on CRFR2 and α1ARs in the VTA to promote cocaine conditioning, similar to our observed effects on LTP-NMDA.
Figure 7. CRF and α1ARs in the VTA Act Together to Promote Cocaine Place Conditioning.

(A) Experimental timeline for testing the effects of intra-VTA injection of CRFR2 antagonist K41498 and α1AR antagonist prazosin on defeat-stress-induced enhancement of cocaine conditioning.
(B–E) Changes in the preference for the cocaine-paired side (conditioned with 5 mg/kg cocaine) in socially defeated rats that received intra-VTA injection of PBS (B), K41498 (C), prazosin (D), or a cocktail of K41498 and prazosin (E). For (B): t(7) = 8.97, ***p < 0.0001; for (C): t(6) = 4.03, **p < 0.01; for (D): t(8) = 2.82, *p < 0.05; for (E): t(7) = 1.27, p = 0.24; two-tailed paired t test; n = 7–9 rats. n.s., not significant.
(F) Summary graph demonstrating CRFR2 and α1AR dependence of stress-induced enhancement of cocaine conditioning, F(3, 30) = 14.5, p < 0.0001, one-way ANOVA. **p < 0.01; ***p < 0.001 (Bonferroni post hoc test).
(G) Experimental timeline for testing the effects of intra-VTA injection of CRF and phenylephrine on the acquisition of cocaine CPP in non-stressed rats.
(H–L) Changes in the preference for the cocaine-paired side (conditioned with 5 mg/kg cocaine) in rats that received intra-VTA injection of PBS (H), CRF (I), low-dose phenylephrine (6 pmol/0.3 μL) (J), a cocktail of CRF and low-dose phenylephrine (K), or high-dose phenylephrine (18 pmol/0.3 μL) (L). For (H): t(5) = 0.40, p = 0.70; for (I): t(6) = 2.28, *p = 0.057; for (J): t(7) = 2.02, p = 0.083; for (K): t(8) = 8.89, ***p < 0.001; for (L): t(7) = 5.69, ***p < 0.001; two-tailed paired t test; n = 6–9 rats.
(M) Summary graph demonstrating the effects of CRF and phenylephrine on cocaine place conditioning in the absence of stress, F(4, 36) = 5.17, p < 0.01, one-way ANOVA. *p < 0.05; **p < 0.01 (Bonferroni post hoc test).
Data are presented as mean ± SEM.
We next asked whether CRF and α1AR actions in the VTA are sufficient to enhance cocaine conditioning in the absence of stress (Figure 7G). While control rats injected with vehicle (PBS) into the VTA developed inconsistent CPP, intra-VTA microinjection of CRF (1.5 pmol/0.3 μL per side) prior to cocaine conditioning enabled moderate CPP (Figures 7H, 7I, and 7M). We further found that administration of phenylephrine (18 pmol/0.3 μL per side) led to robust cocaine conditioning, although a lower dose (6 pmol/0.3 μL per side) had minimal effect (Figures 7J, 7L, and 7M). Finally, combined application of CRF with low-dose phenylephrine enabled large CPP comparable to that observed with high-dose phenylephrine (Figures 7K and 7M) and acute stress (Figures 6C and 6F). Locomotor activity was not affected by drug microinjections (Figures S4B and S4C). These data further support the idea that CRF and α1ARs cooperate in the VTA to enhance cocaine conditioning.
DISCUSSION
Our data provide strong evidence that activation of CRFR2 amplifies α1AR-driven NMDA plasticity via enhancement of IP3-Ca2+ signaling in VTA DA neurons. This cooperative action between CRFR2 and α1ARs mediated our observed stress enhancement of cocaine place conditioning and promoted conditioning in unstressed rats. Previous work has identified adaptations in glutamatergic transmission in VTA DA neurons following single episodes of stress or cocaine exposure (Saal et al., 2003; Ungless et al., 2001). In contrast to potentiation of AMPA transmission found in those experiments, we show that CRF/NE signaling promotes induction of LTP of NMDA transmission in DA neurons. It is conceivable that this form of enhanced plasticity of NMDA transmission could promote potentiation of AMPA transmission observed 24 hr later (Saal et al., 2003), serving as a potential mechanism for enhanced learning of cocaine-paired cues.
While previous studies reporting CRF/NE-induced enhancement of AMPA plasticity outside of the VTA have mostly focused on regulation of neuronal excitability (Blank et al., 2002; Liu et al., 2017) or postsynaptic AMPA receptors (Hu et al., 2007; Seol et al., 2007), our study implicates CRF/NE effects on a Ca2+-dependent induction process as the mechanism for enhancement or facilitation of NMDA plasticity. NE acting on β-adrenergic receptors (βARs) has been shown to enhance spike-timing-dependent plasticity in the hippocampus by regulating the timing of pre- and postsynaptic spikes (Lin et al., 2003; Seol et al., 2007) or the number of postsynaptic spikes (Liu et al., 2017). The present study suggests that NE acting on α1ARs to generate IP3, together with CRFR2 amplifying this α1AR effect, promotes LTP-NMDA in VTA DA neurons, potentially enabling conditioning to subthreshold doses of cocaine. Thus, these two stress mediators appear to lower the “gate” for synaptic plasticity at multiple levels in different brain areas.
Although our study has identified a critical role of CRFR2 in the VTA in promoting NMDA plasticity and cocaine conditioning, it is known that DA neurons also express CRFR1 (Sauvage and Steckler, 2001), which can control DA neuron physiology and reward/drug-driven behaviors (Henckens et al., 2016). For example, while we observed no significant effect of CRF (100–300 nM) on NMDA transmission, previous studies have reported CRF effects on NMDA and AMPA transmission in VTA DA neurons, involving multiple mechanisms via both CRFR1 and CRFR2, depending on the CRF concentration used (Hahn et al., 2009; Ungless et al., 2003; Williams et al., 2014). Moreover, as our enhanced LTP-NMDA reflected post-synaptic CRFR2 and α1AR effects, it is possible that presynaptic CRFRs are engaged to promote plasticity through glutamate or NE release. The latter is supported by data showing that NE neurons of the locus coeruleus (LC) express CRFR1 (Reyes et al., 2006), and Crhr2 mRNA is present in the nucleus of the solitary tract (NTS) (Van Pett et al., 2000), two noradrenergic regions that project to the VTA (Mejías-Aponte et al., 2009). Additionally, since CRFR2 has a greater affinity for the CRF-related peptide urocortin-3 (Lewis et al., 2001), it is possible that stress also recruits this peptide to mediate our observed effects on conditioning. In line with the known acute stress effect on DA neuron activity in vivo (Ungless et al., 2010), these CRFR1/CRFR2-dependent effects on glutamatergic excitation, together with CRF/NE effects on DA neuron firing (Grenhoff et al., 1995; Paladini et al., 2001; Wanat et al., 2008), may contribute to the acute-stress-induced enhancement of drug-seeking behavior (Holly et al., 2016; Mantsch et al., 2016; Wang et al., 2007).
The VTA receives several CRF inputs, some of which originate in the bed nucleus of the stria terminalis, central amygdala, and hypothalamus (Marcinkiewcz et al., 2016; Rinker et al., 2017; Rodaros et al., 2007). These brain regions, along with the LC and NTS, are activated by social defeat stress (Martinez et al., 1998). Thus, stress may recruit specific CRF and NE sources to the VTA to generate the effects we observed ex vivo, thereby enhancing cocaine conditioning. It should be noted that CRF and NE actions in other limbic structures also contribute to different aspects of reward-driven behavior (Henckens et al., 2016; Otis et al., 2015; Smith and Aston-Jones, 2008). Despite the engagement of multiple brain circuits in response to acute-stress-induced CRF/NE actions, our data implicate VTA DA neuron plasticity as the critical substrate for enhancement of cocaine conditioning.
Multiple stress mediators interact, sometimes in an antagonistic fashion, to acutely regulate synaptic plasticity and learning and memory processes (Joëls et al., 2011; Maras and Baram, 2012; McEwen, 2007). For example, corticosterone can promote or suppress the facilitatory effect of NE, acting via βARs, on synaptic plasticity, depending on the timing of application in the hippocampus and amygdala (Akirav and Richter-Levin, 2002; Pu et al., 2007, 2009), while a recent study reported a cooperative action of corticosterone and CRF that impairs hippocampal glutamatergic synapses and spatial memory (Chen et al., 2016). The present study demonstrates that CRF and NE acting on α1ARs converge on IP3-Ca2+ signaling via a CRFR2-dependent increase in IP3 sensitivity and an α1AR-dependent IP3 synthesis, respectively, driving enhanced plasticity of NMDA transmission in VTA DA neurons. Our data further implicate a collaborative action of CRFR2 and α1AR signaling in acute social-defeat-stress-induced enhancement of cocaine place conditioning. Although we specifically manipulated α1AR activity in vivo, the critical noradrenergic effects are most likely mediated by NE, the primary endogenous ligand for α1ARs. Thus, this study identifies a previously unreported mechanism in which CRF and NE act in concert to regulate a form of appetitive learning.
A number of studies have shown persistent changes in VTA synapses (both excitatory and inhibitory) lasting >1 day following single or repeated stress exposure, which are frequently linked to intensification and/or reinstatement of drug-seeking behavior (Polter and Kauer, 2014; Saal et al., 2003). While most studies have focused on the impact of stress on expression of drug-seeking behavior, there are some data on how stress affects drug conditioning. For example, repeated forced swim or social defeat stress enhances cocaine conditioning (McLaughlin et al., 2006a, 2006b; Stelly et al., 2016), and a single episode of defeat stress can enhance conditioning in mice (Montagud-Romero et al., 2015). It should also be noted that a single exposure to inescapable footshock or restraint stress has been reported to promote CPP acquisition for days (Pacchioni et al., 2002; Will et al., 1998), and acute stress can promote learning of cue-reward associations in humans (Lewis et al., 2014). We expand on these studies by demonstrating that acute stress exposure recruits cooperative CRF/α1AR signaling in the VTA, which is critical to enhanced cocaine conditioning.
Interestingly, acute stress (inescapable electric shock or swim stress) has been shown to enhance Pavlovian eyeblink conditioning (Shors, 2001; Shors et al., 1992), which may be driven by synaptic plasticity in the cerebellum that is dependent on an IP3-Ca2+ signaling mechanism similar to NMDA plasticity in DA neurons (Wang et al., 2000). This facilitatory effect on eyeblink conditioning can be observed 30 min to 24 hr after stress exposure. In the present study, the effect on cocaine CPP was observed 10 min, but not 1.5 hr, following a single episode of defeat stress, illustrating the transient nature of stress effect mediated by acute CRF/NE action in the VTA. In line with this, our previous study observed no persistent change in mGluR/IP3-Ca2+ signaling measured 1–2 days following single-defeat stress exposure (Stelly et al., 2016). Furthermore, we have shown that repeated social defeat stress engages glucocorticoid receptor signaling to produce lasting enhancement of cocaine conditioning and mGluR/IP3-Ca2+ signaling, with no changes in intrinsic firing properties or global NMDA-induced currents (Stelly et al., 2016), while glucocorticoid receptor blockade failed to suppress the acute stress effect on cocaine CPP in the present study. The role of stress mediators underlying the persistence of single-stress exposure on eyeblink conditioning has not been explored, although effects of CRF and NE on cerebellar synaptic plasticity have been reported (Carey and Regehr, 2009; Schmolesky et al., 2007).
In summary, we demonstrate a converging action of two stress mediators on synaptic plasticity in VTA DA neurons that may account for acute stress enhancement of cocaine conditioning. Our data suggest that this plasticity can promote learning of the appetitive valence of drug reward (cocaine)-associated cues during acute stress exposure, as observed in humans using monetary rewards for conditioning (Lewis et al., 2014). Thus, this study identifies a molecular target on which CRF and NE act in concert to regulate appetitive learning and suggest that this process could contribute to addiction vulnerability in humans exposed to stress.
EXPERIMENTAL PROCEDURES
Animals
Male Sprague-Dawley rats (aged 4–12 weeks; Harlan Laboratories, Houston, TX, USA) were housed in pairs on a 12-hr/12-hr light/dark cycle with food and water available ad libitum. All procedures were approved by the University of Texas at Austin Institutional Animal Care and Use Committee.
Brain Slice Electrophysiology
Midbrain slices were prepared, and recordings were made in the lateral VTA located 50–150 mm from the medial border of the medial terminal nucleus of the accessory optic tract, as in our previous studies (Stelly et al., 2016; Whitaker et al., 2013). Tyrosine-hydroxylase-positive neurons in this area (i.e., lateral part of the parabrachial pigmented nucleus) largely project to the ventrolateral striatum (Ikemoto, 2007) and show little VGluT2 co-expression (Trudeau et al., 2014). Briefly, rats were anesthetized with isoflurane, horizontal midbrain slices (200 μm) were prepared, and recordings were performed at 34°C in ACSF containing (in millimolar): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2 (pH 7.4, 295 mOsm/kg) and perfused at 2 mL/min. Patch pipettes (1.5–2.0 MΩ) were pulled from borosilicate glass and filled with internal solution containing (in millimolar): 115 K-methylsulfate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2-phosphocreatine (pH ~7.25, ~285 mOsm/kg). Putative dopamine neurons in the lateral VTA were identified by spontaneous firing of broad APs (>1.2 ms) at 1–5 Hz in cell-attached configuration and large Ih currents (>200 pA; evoked by a 1.5-s hyperpolarizing step of 50 mV) in whole-cell configuration (Ford et al., 2006; Lammel et al., 2008; Margolis et al., 2008). Cells were voltage clamped at −62 mV (corrected for −7-mV liquid junction potential). A 2-ms depolarizing pulse of 55 mV was used to elicit an unclamped AP. For bursts, 5 APs were evoked at 20Hz. The time integral of the outward tail current, termed IK(Ca) (calculated after removing the 20-ms window following each depolarizing pulse; expressed in pC), was used as a readout of AP-evoked Ca2+ transients, as it is eliminated by tetrodotoxin (TTX) and also by apamin, a blocker of Ca2+-activated SK channels (Cui et al., 2007). Series and input resistances were monitored throughout experiments, and recordings were discarded if the series resistance increased beyond 20 MΩ or if the input resistance dropped below 200 MΩ. A Multiclamp 700A amplifier (Molecular Devices) and AxoGraph X (AxoGraph Scientific) were used to record and collect data, which were filtered at 1–5 kHz and digitized at 2–10 kHz.
UV Photolysis
Cells were loaded with caged IP3 (1–10 μM) through the recording pipette. UV light (100 ms) was applied using the excitation light from the xenon arc lamp of the Olympus Disk Spinning Unit imaging system. The light was focused through a 60× objective onto a ~350-μm area surrounding the recorded neuron. Photolysis of caged compounds is proportional to the UV light intensity, which was adjusted with neutral density filters and measured at the focal plane of the objective (in milliwatts). The applied IP3 concentration is expressed in micromolar · millijoules (joules = watts × seconds). IP3, thus, applied produces concentration-dependent activation of SK-mediated outward currents (IIP3) (Bernier et al., 2011; Harnett et al., 2009; Stelly et al., 2016; Whitaker et al., 2013), which display a roughly linear relationship with bulk cytosolic Ca2+ levels in DA neurons (Morikawa et al., 2003).
LTP Experiments
Synaptic stimuli were delivered with a bipolar tungsten electrode placed ~200 μm rostral to the recorded neuron. To isolate NMDA excitatory postsynaptic currents (EPSCs), recordings were performed in DNQX (10 μM), picrotoxin (100 μM), CGP54626 (50 nM), and sulpiride (100 nM) to block AMPA/kainate, GABAA, GABAB, and D2 dopamine receptors and in glycine (20 μM) and low Mg2+ (0.1 mM) to enhance NMDA receptor activation. NMDA EPSCs were monitored every 20 s. The LTP induction protocol consisted of photolytic application of a low concentration of IP3 (1 μM × mJ; Figure S2) for 100 ms prior to the simultaneous delivery of afferent stimulation (20 stimuli at 50 Hz) and postsynaptic burst (5 APs at 20 Hz), repeated 10 times every 20 s. LTP magnitude was determined by comparing the average EPSC amplitude 30–40 min post-induction with the average EPSC amplitude pre-induction (each from a 10-min window).
Resident-Intruder Social Defeat Paradigm
Twelve-week old male resident rats were vasectomized and pair-housed with 6-week-old females. Residents (used for ~8–10 months) were screened for aggression (biting or pinning within 1 min) by introducing a male intruder to the home cage. Intruders and controls were young males (4–5 weeks old at the beginning) housed in pairs. For defeat sessions, intruders were introduced to residents’ home cages after removing females. Following ~5 min of direct contact, a perforated Plexiglass barrier was inserted for ~25 min to allow sensory contact, as in our previous study (Stelly et al., 2016). The barrier was removed for a brief period (<1 min) in certain cases to encourage residents’ threatening behavior. Handled controls were placed in novel cages for 30 min. Intruders and controls were housed separately.
Cocaine Place Conditioning
CPP boxes (Med Associates) consisting of two distinct compartments separated by a small middle chamber were used for conditioning. One compartment had a mesh floor with white walls, while the other had a grid floor with black walls. A discrete cue (painted ceramic weight) was placed in the rear corner of each compartment (black one in the white wall side, white one in the black wall side) for further differentiation. Rats were first subjected to a pretest in which they explored the entire CPP box for 15 min. The percentage of time spent in each compartment was determined after excluding the time spent in the middle chamber. Rats with initial side preference >60% were excluded. The following day, rats were given a saline injection in the morning and confined to one compartment, then in the afternoon, they were given cocaine (5 or 10 mg/kg, i.p.) and confined to the other compartment (10 min each). Compartment assignment was counterbalanced so that animals had, on average, ~50% initial preference for the cocaine-paired side. A 15-min posttest was performed 1 day after conditioning. The CPP score was determined by subtracting the preference for the cocaine-paired side during pretest from that during posttest. For experiments in Figure S5, mifepristone was dissolved in 30% propylene glycol plus 1% Tween-20 in 0.9% saline. The experimenter performing CPP experiments was blind to animal treatments.
Intra-VTA Microinjections
Rats (7–10 weeks old) were anesthetized with a mixture of ketamine and xylazine (90 mg/kg and 10 mg/kg, respectively, i.p.) and implanted with bilateral chronic guide cannulas (22G; Plastics One), with dummy cannulas (32G) inside, aimed at 1 mm above the VTA (anteroposterior, −5.3; mediolat-eral, +2.2; dorsoventral, −7.5; 10° angle). The guide cannulas were affixed to the skull with stainless steel screws and dental cement. After surgery, rats remained singly housed for a 7-day recovery before being subjected to conditioning experiments.
Intra-VTA microinjections were made via injection cannulas (28G; Plastics One) that extended 1 mm beyond the tip of the guide cannulas. Injection cannulas were connected to 1-μL Hamilton syringes mounted on a microdrive pump (Harvard Apparatus). Rats received bilateral infusions (0.3 μL per side, 0.15 μL per min) of different pharmacological agents in certain conditioning experiments. The injection cannulas were left in place for 60 s after infusion. Rats administered antagonists were subjected to social defeat stress 10 min later, and rats administered agonists underwent cocaine conditioning 10 min later (no stress).
At the end of conditioning experiments, rats were anesthetized with a mixture of ketamine and xylazine (90 mg/kg and 10 mg/kg, i.p.) and transcardially perfused with 4% paraformaldehyde. Brains were then carefully removed and stored in 4% paraformaldehyde. Coronal sections (100 μm) were cut using a vibratome and stained with cresyl violet for histological verification of injection sites (Figure S6). Data from rats with injection sites outside the VTA were excluded from the analysis.
Drugs
DNQX, picrotoxin, CGP55845, sulpiride, CRF, K41498, and mifepristone were obtained from Tocris Biosciences. Caged IP3 was a generous gift from Dr. Kamran Khodakhah (Albert Einstein College of Medicine). All other chemicals were from Sigma-RBI.
Statistical Analysis
Data are presented as mean ± SEM. Statistical significance was determined by Student’s t test or ANOVA followed by Bonferroni post hoc test. The difference was considered significant at p < 0.05.
Supplementary Material
Highlights.
CRF and α1ARs converge on IP3-Ca2+ signaling to enhance NMDA plasticity
Acute social defeat stress promotes cocaine place conditioning
CRF and α1ARs work in concert in the VTA to enhance cocaine conditioning
Acknowledgments
We thank Dr. Kamran Khodakhah for the generous gift of caged IP3 made in his lab at the Albert Einstein College of Medicine. We thank Dr. Claire Stelly and Simone Giovanetti for comments on figures and the manuscript. We also thank Jerome John Garcia for continuing inspiration that moves us brightly. This work was supported by NIH grants DA015687 and AA015521 to H.M. and NSF fellowship DGE-1110007 to M.B.P.
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.039.
AUTHOR CONTRIBUTIONS
J.T.-D. conducted the electrophysiology experiments and data analysis, as well as stereotaxic surgery, and assisted in behavioral experiments. M.B.P., R.K., and B.P. performed CPP experiments. H.M., M.B.P., and J.T.-D. designed the experiments and wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Akirav I, Richter-Levin G. Mechanisms of amygdala modulation of hippocampal plasticity. J Neurosci. 2002;22:9912–9921. doi: 10.1523/JNEUROSCI.22-22-09912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SE, Hartwell K, DeSantis SM, Saladin M, McRae-Clark AL, Price KL, Moran-Santa Maria MM, Baker NL, Spratt E, Kreek MJ, Brady KT. Reactivity to laboratory stress provocation predicts relapse to cocaine. Drug Alcohol Depend. 2010;106:21–27. doi: 10.1016/j.drugalcdep.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier BE, Whitaker LR, Morikawa H. Previous ethanol experience enhances synaptic plasticity of NMDA receptors in the ventral tegmental area. J Neurosci. 2011;31:5205–5212. doi: 10.1523/JNEUROSCI.5282-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Eckart K, Spiess J. Priming of long-term potentiation in mouse hippocampus by corticotropin-releasing factor and acute stress: implications for hippocampus-dependent learning. J Neurosci. 2002;22:3788–3794. doi: 10.1523/JNEUROSCI.22-09-03788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke AR, Watt MJ, Forster GL. Adolescent social defeat increases adult amphetamine conditioned place preference and alters D2 dopamine receptor expression. Neuroscience. 2011;197:269–279. doi: 10.1016/j.neuroscience.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MR, Regehr WG. Noradrenergic control of associative synaptic plasticity by selective modulation of instructive signals. Neuron. 2009;62:112–122. doi: 10.1016/j.neuron.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Lauterborn JC, Trieu BH, Bolton JL, Patterson KP, Gall CM, Lynch G, Baram TZ. Converging, synergistic actions of multiple stress hormones mediate enduring memory impairments after acute simultaneous stresses. J Neurosci. 2016;36:11295–11307. doi: 10.1523/JNEUROSCI.2542-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Perello M, Sakata I, Osborne-Lawrence S, Savitt JM, Lutter M, Zigman JM. Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest. 2011;121:2684–2692. doi: 10.1172/JCI57660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature. 2012;482:85–88. doi: 10.1038/nature10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DP, Roitman MF, Garris PA. Illicit dopamine transients: reconciling actions of abused drugs. Trends Neurosci. 2014;37:200–210. doi: 10.1016/j.tins.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Okamoto T, Morikawa H. Spontaneous opening of T-type Ca2+ channels contributes to the irregular firing of dopamine neurons in neonatal rats. J Neurosci. 2004;24:11079–11087. doi: 10.1523/JNEUROSCI.2713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Bernier BE, Harnett MT, Morikawa H. Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci. 2007;27:4776–4785. doi: 10.1523/JNEUROSCI.0139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenhoff J, North RA, Johnson SW. Alpha 1-adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci. 1995;7:1707–1713. doi: 10.1111/j.1460-9568.1995.tb00692.x. [DOI] [PubMed] [Google Scholar]
- Hahn J, Hopf FW, Bonci A. Chronic cocaine enhances cortico-tropin-releasing factor-dependent potentiation of excitatory transmission in ventral tegmental area dopamine neurons. J Neurosci. 2009;29:6535–6544. doi: 10.1523/JNEUROSCI.4773-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnett MT, Bernier BE, Ahn KC, Morikawa H. Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron. 2009;62:826–838. doi: 10.1016/j.neuron.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckens MJ, Deussing JM, Chen A. Region-specific roles of the corticotropin-releasing factor-urocortin system in stress. Nat Rev Neurosci. 2016;17:636–651. doi: 10.1038/nrn.2016.94. [DOI] [PubMed] [Google Scholar]
- Holly EN, Boyson CO, Montagud-Romero S, Stein DJ, Gobrogge KL, DeBold JF, Miczek KA. Episodic social stress-escalated cocaine self-administration: role of phasic and tonic corticotropin releasing factor in the anterior and posterior ventral tegmental area. J Neurosci. 2016;36:4093–4105. doi: 10.1523/JNEUROSCI.2232-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Fernandez G, Roozendaal B. Stress and emotional memory: a matter of timing. Trends Cogn Sci. 2011;15:280–288. doi: 10.1016/j.tics.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Keiflin R, Janak PH. Dopamine prediction errors in reward learning and addiction: from theory to neural circuitry. Neuron. 2015;88:247–263. doi: 10.1016/j.neuron.2015.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46:1167–1180. doi: 10.1016/s0006-3223(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flügge G, Korte SM, Meerlo P, Murison R, Olivier B, Palanza P, et al. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev. 2011;35:1291–1301. doi: 10.1016/j.neubiorev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Kreibich AS, Briand L, Cleck JN, Ecke L, Rice KC, Blendy JA. Stress-induced potentiation of cocaine reward: a role for CRF R1 and CREB. Neuropsychopharmacology. 2009;34:2609–2617. doi: 10.1038/npp.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci USA. 2001;98:7570–7575. doi: 10.1073/pnas.121165198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Porcelli AJ, Delgado MR. The effects of acute stress exposure on striatal activity during Pavlovian conditioning with monetary gains and losses. Front Behav Neurosci. 2014;8:179. doi: 10.3389/fnbeh.2014.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–4181. doi: 10.1523/JNEUROSCI.23-10-04173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cui L, Schwarz MK, Dong Y, Schlüter OM. Adrenergic gate release for spike timing-dependent synaptic potentiation. Neuron. 2017;93:394–408. doi: 10.1016/j.neuron.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Funk D, Lê AD, Shaham Y. Stress-induced reinstatement of drug seeking: 20 years of progress. Neuropsycho-pharmacology. 2016;41:335–356. doi: 10.1038/npp.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012;35:315–324. doi: 10.1016/j.tins.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewcz CA, Mazzone CM, D’Agostino G, Halladay LR, Hardaway JA, DiBerto JF, Navarro M, Burnham N, Cristiano C, Dorrier CE, et al. Serotonin engages an anxiety and fear-promoting circuit in the extended amygdala. Nature. 2016;537:97–101. doi: 10.1038/nature19318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J Neurosci. 2008;28:8908–8913. doi: 10.1523/JNEUROSCI.1526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M, Phillips PJ, Herbert J. Adaptation in patterns of c-fos expression in the brain associated with exposure to either single or repeated social stress in male rats. Eur J Neurosci. 1998;10:20–33. doi: 10.1046/j.1460-9568.1998.00011.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsycho-pharmacology. 2006a;31:787–794. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C. Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology. 2006b;31:1241–1248. doi: 10.1038/sj.npp.1300872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejías-Aponte CA, Drouin C, Aston-Jones G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: prominent inputs from medullary homeostatic centers. J Neurosci. 2009;29:3613–3626. doi: 10.1523/JNEUROSCI.4632-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagud-Romero S, Aguilar MA, Maldonado C, Manzanedo C, Miñarro J, Rodríguez-Arias M. Acute social defeat stress increases the conditioned rewarding effects of cocaine in adult but not in adolescent mice. Pharmacol Biochem Behav. 2015;135:1–12. doi: 10.1016/j.pbb.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Werner CT, Mueller D. Noradrenergic regulation of fear and drug-associated memory reconsolidation. Neuropsychopharmacology. 2015;40:793–803. doi: 10.1038/npp.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Pacchioni AM, Gioino G, Assis A, Cancela LM. A single exposure to restraint stress induces behavioral and neurochemical sensitization to stimulating effects of amphetamine: involvement of NMDA receptors. Ann N Y Acad Sci. 2002;965:233–246. doi: 10.1111/j.1749-6632.2002.tb04165.x. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Roeper J. Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience. 2014;282:109–121. doi: 10.1016/j.neuroscience.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Fiorillo CD, Morikawa H, Williams JT. Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci. 2001;4:275–281. doi: 10.1038/85124. [DOI] [PubMed] [Google Scholar]
- Polter AM, Kauer JA. Stress and VTA synapses: implications for addiction and depression. Eur J Neurosci. 2014;39:1179–1188. doi: 10.1111/ejn.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Z, Krugers HJ, Joëls M. Corticosterone time-dependently modulates beta-adrenergic effects on long-term potentiation in the hippocampal dentate gyrus. Learn Mem. 2007;14:359–367. doi: 10.1101/lm.527207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Z, Krugers HJ, Joëls M. Beta-adrenergic facilitation of synaptic plasticity in the rat basolateral amygdala in vitro is gradually reversed by corticosterone. Learn Mem. 2009;16:155–160. doi: 10.1101/lm.1272409. [DOI] [PubMed] [Google Scholar]
- Reyes BA, Fox K, Valentino RJ, Van Bockstaele EJ. Agonist-induced internalization of corticotropin-releasing factor receptors in noradrenergic neurons of the rat locus coeruleus. Eur J Neurosci. 2006;23:2991–2998. doi: 10.1111/j.1460-9568.2006.04820.x. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Williams JT. CRF facilitates calcium release from intracellular stores in midbrain dopamine neurons. Neuron. 2008;57:559–570. doi: 10.1016/j.neuron.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinker JA, Marshall SA, Mazzone CM, Lowery-Gionta EG, Gulati V, Pleil KE, Kash TL, Navarro M, Thiele TE. Extended amygdala to ventral tegmental area corticotropin-releasing factor circuit controls binge ethanol intake. Biol Psychiatry. 2017;81:930–940. doi: 10.1016/j.biopsych.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience. 2007;150:8–13. doi: 10.1016/j.neuroscience.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sauvage M, Steckler T. Detection of corticotropin-releasing hormone receptor 1 immunoreactivity in cholinergic, dopaminergic and noradrenergic neurons of the murine basal forebrain and brainstem nuclei–potential implication for arousal and attention. Neuroscience. 2001;104:643–652. doi: 10.1016/s0306-4522(01)00137-3. [DOI] [PubMed] [Google Scholar]
- Schmolesky MT, De Ruiter MM, De Zeeuw CI, Hansel C. The neuropeptide corticotropin-releasing factor regulates excitatory transmission and plasticity at the climbing fibre-Purkinje cell synapse. Eur J Neurosci. 2007;25:1460–1466. doi: 10.1111/j.1460-9568.2007.05409.x. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz W. Neuronal reward and decision signals: from theories to data. Physiol Rev. 2015;95:853–951. doi: 10.1152/physrev.00023.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J. Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev. 2000;33:13–33. doi: 10.1016/s0165-0173(00)00024-2. [DOI] [PubMed] [Google Scholar]
- Shors TJ. Acute stress rapidly and persistently enhances memory formation in the male rat. Neurobiol Learn Mem. 2001;75:10–29. doi: 10.1006/nlme.1999.3956. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Weiss C, Thompson RF. Stress-induced facilitation of classical conditioning. Science. 1992;257:537–539. doi: 10.1126/science.1636089. [DOI] [PubMed] [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Aston-Jones G. Noradrenergic transmission in the extended amygdala: role in increased drug-seeking and relapse during protracted drug abstinence. Brain Struct Funct. 2008;213:43–61. doi: 10.1007/s00429-008-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C. Stress-induced activation of the dynorphin/κ-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci. 2012;32:1488–1495. doi: 10.1523/JNEUROSCI.2980-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer WR, Lak A, Yang A, Borel M, Paulsen O, Boyden ES, Schultz W. Dopamine neuron-specific optogenetic stimulation in rhesus macaques. Cell. 2016;166:1564–1571e6. doi: 10.1016/j.cell.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelly CE, Pomrenze MB, Cook JB, Morikawa H. Repeated social defeat stress enhances glutamatergic synaptic plasticity in the VTA and cocaine place conditioning. eLife. 2016;5:e15448. doi: 10.7554/eLife.15448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW, Laude AJ. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+ Cell Calcium. 2002;32:321–334. doi: 10.1016/s0143416002001859. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Hnasko TS, Wallén-Mackenzie A, Morales M, Rayport S, Sulzer D. The multilingual nature of dopamine neurons. Prog Brain Res. 2014;211:141–164. doi: 10.1016/B978-0-444-63425-2.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Argilli E, Bonci A. Effects of stress and aversion on dopamine neurons: implications for addiction. Neurosci Biobehav Rev. 2010;35:151–156. doi: 10.1016/j.neubiorev.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–3596. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Denk W, Häusser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- Wang B, You ZB, Rice KC, Wise RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacology (Berl) 2007;193:283–294. doi: 10.1007/s00213-007-0782-3. [DOI] [PubMed] [Google Scholar]
- Wang LP, Li F, Wang D, Xie K, Wang D, Shen X, Tsien JZ. NMDA receptors in dopaminergic neurons are crucial for habit learning. Neuron. 2011;72:1055–1066. doi: 10.1016/j.neuron.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker LR, Degoulet M, Morikawa H. Social deprivation enhances VTA synaptic plasticity and drug-induced contextual learning. Neuron. 2013;77:335–345. doi: 10.1016/j.neuron.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will MJ, Watkins LR, Maier SF. Uncontrollable stress potentiates morphine’s rewarding properties. Pharmacol Biochem Behav. 1998;60:655–664. doi: 10.1016/s0091-3057(98)00027-6. [DOI] [PubMed] [Google Scholar]
- Williams CL, Buchta WC, Riegel AC. CRF-R2 and the hetero-synaptic regulation of VTA glutamate during reinstatement of cocaine seeking. J Neurosci. 2014;34:10402–10414. doi: 10.1523/JNEUROSCI.0911-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci USA. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.