
Figure 1. Workflow for cryo‐EM structure determination.
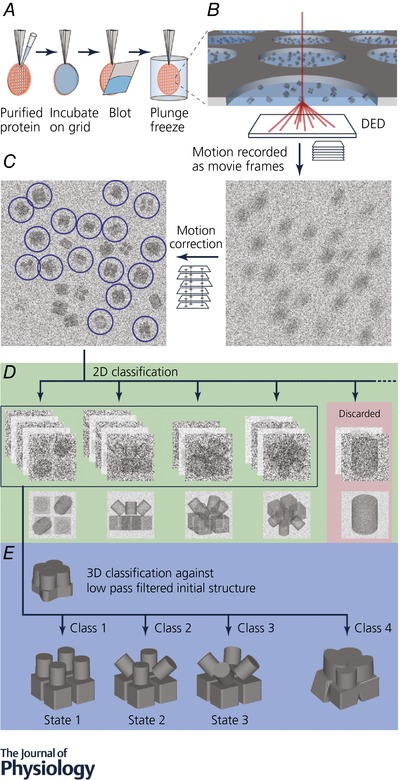
Proteins are overexpressed recombinantly in the expression system of choice, but purification from natural source is also possible as cryo‐EM requires less sample than X‐ray crystallography (Yan et al. 2015, 2017). A, purified protein is applied on a holey carbon grid. Excess protein is removed by blotting with a filter paper immediately before being plunge frozen in liquid ethane. B, protein vitrified in a thin layer of ice in the holey carbon grid is transferred to an electron microscope and imaged. Typically, thousands of micrographs are collected. Micrographs are dose fractionated into subframes. C, computational processing after image acquisition to recover information lost in the imaging process (e.g. motion correction). Particles are selected and extracted for processing. D, extracted particles are classified and averaged in two dimensions to enhance signal. Noise is significantly reduced in averaged particles and therefore ‘bad’ particles can be identified and removed. E, ‘good’ particles (green background in D) are classified in three dimensions against an initial model to generate subsets of particles with homogeneous conformation. These homogeneous subsets are used for further processing to generate the final high resolution EM map in different conformational states.