

Abstract
Cryopreservation is a common method used to preserve the sperm of various animal species, and it is widely used with zebrafish (Danio rerio). As with other animals, there is a possibility of paternal pathogen transmission through sperm. We evaluated the ability of five common and important pathogens of zebrafish to survive cryopreservation as used with zebrafish sperm and freezing without cryopreservant. We evaluated Mycobacterium chelonae, Mycobacterium marinum, and Edwardsiella ictaluri, each originally isolated from zebrafish, eggs of Pseuodocapillaria tomentosa, and spores of Pseudoloma neurophilia. Each mycobacterial isolate showed relatively minimal reduction in survival after freezing and thawing, particularly when subjected to cryopreservation. E. ictaluri also showed survival after cryopreservation, but exhibited a several log reduction after freezing at −80°C without cryopreservant. With P. neurophilia, two separate experiments conducted 3 years apart yielded very similar results, showing some, but reduced, survival of spores by using three different viability assays: SYTOX stain, Fungi-Fluor stain, and presence of a spore vacuole. Eggs of P. tomentosa showed no survival based on larvation of eggs when subjected to either freezing method. Given that four of the five pathogens exhibited survival after cryopreservation, we recommend that sperm samples or donor male zebrafish fish be tested for pathogens when sperm are to be stored by using cryopreservation.
Keywords: : zebrafish, cryopreservation, mycobacteria, Pseudoloma neurophilia, Pseudocapillaria tomentosa
Introduction
Zebrafish (Danio rerio) are becoming one of the most commonly used animal models in research. Their transparency and internal physiological similarities to humans1,2 make them ideal models for a variety of research areas, including immunological and cancer development studies.2,3 There are numerous wild-type, mutant, and transgenic zebrafish lines in existence, and the NIH-supported Zebrafish International Resource Center (ZIRC, Eugene, OR) is a principal supplier of such lines.
ZIRC provides the research community with a place to store and obtain various zebrafish strains.4 In 2015 alone, ZIRC received 10,950 zebrafish lines and shipped a total of 79,561 zebrafish embryo and adult strains (Z. Varga, ZIRC, personal communication). ZIRC is the main distributor of live zebrafish fish, embryos, and adults to research facilities all around the world. They provide diagnostic pathology services and knowledge about zebrafish health, husbandry practices, sperm cryopreservation, and in vitro fertilization (IVF).5 Cryopreservation of zebrafish sperm enables ZIRC to store the vast array of zebrafish strains they receive. By using this technique, they have been able to preserve more than 10,500 zebrafish strains, making up about 36,000 genetic modifications and alleles.5
Implementation of sperm cryopreservation and IVF has proved to be as beneficial in the zebrafish research community and at ZIRC6 as it has been with food fish aquaculture. Numerous microorganisms detected in semen of domestic animals pose risks of paternal transmission.7,8 These microbes associated with sperm and semen include both obligate pathogens such as viruses, parasites, and certain bacteria, and opportunistic bacterial contaminants. Hence, the risk of transmission of certain pathogens with cryopreserved fish sperm should be considered.7 There are several maternally transmitted pathogens in salmonid fishes, particularly via eggs and ovarian fluid.8 Therefore, knowledge of the pathogen history of brood fish providing gametes is a key element in the avoidance of transmission in the aquaculture industry.8,9
Pseudoloma neurophilia and Mycobacterium chelonae are two of the most common pathogens found in zebrafish facilities, including ZIRC.4 Infected fish may experience emaciation (P. neurophilia) or chronic inflammation (M. chelonae) with low rates of mortality.4 P. neurophilia is maternally transmitted.10 This parasite has been detected by using PCR on sperm stripped from intact fish and dissected testes,11 and M. chelonae is often observed in ovaries and testes of zebrafish.12,13
ZIRC categorizes Mycobacterium marinum, Mycobacterium haemophilum, Edwardsiella ictaluri, and Pseudocapillaria tomentosa as high-risk pathogens due to their ability to cause severe infections and high rates of mortality.4 M. haemophilum has been observed in the ovaries and testes of infected fish.4 Knowledge of the disease and pathogen history for adult zebrafish providing sperm for cryopreservation is often lacking, and, hence, there is concern that the sperm from these fish may contain pathogens. For example, thousands of new lines are shipped to ZIRC as cryopreserved sperm and the health status of the contributing males is often unknown.
The objective of this study was to evaluate the survival potential of zebrafish pathogens that were subjected to the freezing and thawing solutions and procedures utilized by ZIRC to cryopreserve sperm samples. We evaluated the survival potential of the following pathogens in the cryopreservant14 and without cryopreservant: M. chelonae, M. marinum, E. ictaluri, eggs of P. tomentosa, and spores of P. neurophilia. These are five of the six most common pathogens associated with disease in zebrafish research facilities.4 M. haemophilum is also recognized as a serious pathogen of zebrafish15 but this was not included in our study due to its extremely slow in vitro growth and other difficulties with culture.
Materials and Methods
Bacteria cultures
Two strains of M. chelonae (H1E1 ZF55 and H1E2 2F60)12 and one strain of M. marinum (OSU 214),16 all of which were originally isolated from zebrafish, were used in this study. These bacteria were cultured on Middlebrook 7H10 plates supplemented with 0.5% glycerol and 10% OADC enrichment. Colonies selected from plates were used to inoculate Middlebrook 7H9 broths supplemented with 0.2% glycerol, 0.05% Tween, and 10% ADC enrichment (Remel, Lenexa, Kansas) and incubated at 28°C with gentle shaking. The broth cultures were incubated for 3 days (M. chelonae) and 7 days (M. marinum), after which they were used to inoculate new broths that were then allowed to incubate for about 2 days (M. chelonae) and 5 days (M. marinum), to obtain exponentially growing cells to subject to the cryopreservation protocol.
We used an isolate of E. ictaluri (LADL11-100) obtained from Dr. John Hawke, Louisiana State University, which was from an outbreak in zebrafish.17 The bacterium was cultured on blood agar plates (TSA with 5% sheep blood; Remel). Brain Heart Infusion (BHI) porcine broth (BD Bacto, Sparks, MD) was then inoculated with a colony from the cultured E. ictaluri plates, and it was incubated at 28°C with gentle shaking for 2 days. Again, the same procedure that was used for the Mycobacterium samples to ensure the bacteria were in an exponential phase of growth when frozen was also used with E. ictaluri.
ZIRC cryopreservation (cryoprotectant): bacteria samples
We used the ZIRC protocol for cryopreserving zebrafish sperm with only a few modifications.14 Before freezing the bacterial samples, two McFarland standards No. 1 and No. 3 (3 × 108 and 9 × 108 bacterial colony-forming units [CFU]/mL, respectively) and a spectrophotometer were used to estimate the density of the cultured broths. We aimed for an absorbance between 0.2 and 0.4 nm, which is estimated to be about 3 × 108–6 × 108 CFU/mL. The bacterial broths were then diluted in 1 × phosphate-buffered saline (PBS) by using a 1:10 serial dilution.
ZIRC cryopreserves 20 μL samples, taken from solutions consisting of 5 μL of sperm, 1 μL E400, and 15 μL Raffinose freezing medium (RMMB). E400 is a high-potassium, buffered salt solution that has an osmolality of 400 mmol/kg that is used after the sperm from the zebrafish have been collected to keep urine from activating the sperm cells.14 E400 consists of 130 mM KCl, 50 mM NaCl, 2 mM CaCl2, 1 mM MgSO4, 10 mM d-(+)-Glucose, and 30 mM HEPES-KOH (7.9).14 RMMB is the cryopreservant and consists of 20% (w/v) d-(+)-Raffinose pentahydrate (R7630; Sigma), 2.5% (w/v) Difco Skim Milk (No. 232100; Difco), 6.67% (v/v) methanol (Acetone-free, Absolute, Certified ACS Reagent Grade, A412; Fisher Scientific), and 30 mM Bicine-NaOH (pH 8.0).14 The same ratio of solutions and sample volume were used in this experiment except that the bacteria took the place of the sperm.
A total of 90 μL of RMMB solution was added to a sterile 2 mL cryogenic vial (No. 430659; Corning). To that same vial, 6 μL of E400 and 30 μL of the 104 CFU/mL bacterial concentration were then added and mixed by pipetting. Then, 20 μL of this mixture was transferred into cryo-vials in triplicate. These cryo-vials were then capped and placed onto another empty cryo-vial without a cap, and both were then placed into 15 mL Polypropylene conical tubes (No. 352096; Falcon, Corning). These 15 mL tubes were then capped and placed into a container filled with powdered-dry ice until the caps were flush with the surface of the ice. The samples remained in the powdered-dry ice for at least 20 min. This configuration yields a freeze rate of about 20°C/min. After 20 min, each sample was removed from the conical tubes, placed on a cryogenic cane, and quickly placed in a liquid nitrogen (LN2) dewar. This same procedure was also done using the starting broth, which had the high bacterial concentration (108 CFU/mL).
ZIRC cryopreservation protocol: bacteria samples
• 1:10 serial dilution of starting broth (108 CFU/mL), using 1 × PBS.
-
• Ratio of solutions used in the ZIRC cryopreservation method:
o 5 μL sperm (in our case bacteria) +1 μL E400 (sperm extender) +15 μL RMMB (Raffinose freezing medium)
-
• Prepared the following solution:
-
o Low concentration samples (104 CFU/mL):
□ 30 μL of bacteria (104 CFU/mL) +6 μL E400 + 90 μL RMMB
□ 20 μL of the earlier described solution was pipetted into 2 mL cryogenic vials (done in triplicate).
-
o High concentration samples (108 CFU/mL):
□ 30 μL of bacteria (108 CFU/mL) +6 μL E400 + 90 μL RMMB
□ 20 μL of the earlier described solution was pipetted into 2 mL cryogenic vials (done in triplicate).
-
• Final volume frozen = 20 μL/sample
• Final freezing temperature = −196°C
Freezing at −80°C (no cryoprotectant): bacteria samples
Bacterial samples were diluted in 900 μL of 1 × PBS. One hundred microliters of the same concentrations used in the ZIRC method (104 and 108 CFU/mL) were pipetted into 2 mL cryogenic vials (No. 430659; Corning), and they were then put directly in a −80°C freezer. There was no cryoprotectant or freezing medium added to these samples. This procedure was conducted in triplicate for each dilution.
Protocol for freezing at -80°C: bacteria samples
-
• Low concentration samples (104 CFU/mL):
o 100 μL of the 104 CFU/mL dilution was placed into 2 mL cryogenic vials (done in triplicate).
-
• High concentration samples (108 CFU/mL):
o 100 μL of the 108 CFU/mL broth was placed into 2 mL cryogenic vials (done in triplicate).
• Final volume frozen = 100 μL/sample.
• Final freezing temperature = −80°C.
Calculating starting bacteria concentrations
Using the bacterial concentrations generated from the 1:10 serial dilution, 100 μL of the following concentrations, 104, 103, 102, and 101 CFU/mL, were plated to estimate the number of bacterial CFU in each sample. Each concentration was plated in triplicate on the appropriate media for the corresponding bacteria, and plates were incubated at 28°C. The number of colonies on each plate were counted, averaged, and finally used to calculate the amount of CFU in each vial before freezing.
ZIRC thawing method (thawing solution): bacteria samples
For the ZIRC method, samples were removed from the LN2 dewar after about 3 weeks, placed in a 38°C water bath for 10–15 s or until completely thawed, and immediately removed. Once thawed, 150 μL of Sperm Solution 300 (SS300) was added to each cryo-vial and mixed by pipetting. SS300 is a sperm solution with an osmolality of 300 mmol/kg14 and is used to activate thawed sperm samples. It consists of 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgSO4, 10 mM d-(+)-Glucose, and 20 mM Tris-Cl (8.0).14 The low concentration samples (104 CFU/mL) were then plated in triplicate and incubated at 28°C. The high concentration samples (108 CFU/mL) were diluted out in PBS in a 1:10 dilution series. Each of the four lowest concentrations in the series (104, 103, 102, and 101 CFU/mL) were plated in triplicate and then incubated at 28°C. Once colonies on the plates began to appear, they were counted and used to determine the amount of bacteria in each sample that survived the freezing process.
Protocol for thawing ZIRC samples: bacteria samples
• Samples were thawed in a 38°C water bath.
-
• Ratio of solutions used in the ZIRC thawing method:
o 20 μL samples +150 μL SS300 (thawing solution).
• To each thawed sample, 150 μL of SS300 was added to each vial.
• The low concentration samples (104 CFU/mL) were each plated in triplicate and incubated at 28°C.
-
• The high concentration samples (108 CFU/mL) were diluted out, in 1 × PBS, by using a 1:10 serial dilution.
o The 104, 103, 102, and 101 CFU/mL dilutions from each sample were plated in triplicate, and then incubated at 28°C.
Thawing −80°C samples (no thawing solution): bacteria samples
The bacterial samples that were stored in the −80°C freezer with only 1 × PBS were removed from the freezer and placed into a 38°C water bath for 10–15 s or until the solutions were no longer frozen, and then quickly removed. Once thawed, the low concentration samples (104 CFU/mL) were plated in triplicate and incubated at 28°C. The high concentration samples (108 CFU/mL) were diluted out in 1 × PBS using a 1:10 dilution series, and as was done with the ZIRC samples, each of the four lowest concentrations in the series were plated in triplicate and incubated at 28°C. Once colonies on the plates began to appear, they were counted and used to determine the amount of bacteria that survived the freezing process.
Protocol for thawing −80°C samples: bacteria samples
• Samples were thawed in a 38°C water bath.
• The low concentration samples (104 CFU/mL) were each plated in triplicate and incubated at 28°C.
-
• The high concentration samples (108 CFU/mL) were diluted out in 1 × PBS, by using a 1:10 serial dilution.
o The 104, 103, 102, and 101 CFU/mL dilutions from each sample were plated in triplicate, and they were finally incubated at 28°C.
P. tomentosa egg collection
A 16 L plastic mouse cage, converted to a spawning tank with a stainless steel screen, was filled with system water at 28°C (derived from dechlorinated city water at ∼125 μS conductivity) and was populated with 20 P. tomentosa infected zebrafish. To obtain freshly released unlarvated P. tomentosa eggs (i.e., undeveloped eggs without vermiform larvae), the fish were kept in the tank overnight and then the next morning, they were removed and the tank water containing any shed nematode eggs was allowed to settle for a few hours.
Approximately 90% of the tank water was removed by using a vacuum pump, and the remaining water and P. tomentosa eggs were divided into 300 mL Nalgene bottles. These were then centrifuged for 45 min at 1,500 g. After that, about 90% of the supernatant was removed from the Nalgene bottles and the remaining water and eggs were divided into 50 mL plastic conical tubes and centrifuged for 30 min at 900 g. After centrifugation, most of the supernatant was carefully removed, leaving about 1–5 mL of the concentrate. Three 25 μL drops of the egg solution were examined at × 100 with a compound microscope, and the number of eggs observed were counted. This was done to calculate the total number of eggs in the 1–5 mL solution.
P. neurophilia spore collection
P. neurophilia spores were collected from 20 known infected adult zebrafish that were euthanized by rapid cooling.18 The brain and spinal cord from these 20 fish were collected and divided into 2 small petri dishes containing a 1 × penicillin-streptomycin solution. Each dish contained 10 brains and 10 spinal cords. These solutions were then homogenized by continuously passing them through successively smaller gauges of needles (18, 23, 26 G). Once homogenized, the solutions were placed into 50 mL conical tubes and filled completely by using distilled water. The tubes were then centrifuged for 20 min at 1,400 g. The pellets were collected, re-diluted to 50 mL with distilled water, and placed on a shaker for 24 h at room temperature to enhance host cell lysis and liberation of spores from tissue. The tubes were then centrifuged for 20 min at 100 g. The supernatant was removed, and the pellets were then re-suspended in about 1 mL of distilled water. The number of spores in the final solutions was estimated by using a hemocytometer.
ZIRC cryopreservation (cryoprotectant): P. tomentosa eggs and P. neurophilia spores (2017 samples)
As with bacteria, the ZIRC protocol for cryopreserving zebrafish sperm was used but higher sample volumes were frozen down to ensure that an adequate amount of P. tomentosa eggs and P. neurophilia spores were in each sample. The same solution ratios were used; however, because we used a higher volume, the rate of freezing was slower than the usual freezing rate generated by using 20 μL samples. Each sample in the trial 1 freezing of P. tomentosa eggs contained a total volume of 200 μL consisting of 48 μL of fish water containing 144 eggs, 10 μL of E400, and 142 μL RMMB. Fish water refers to the residual water from aquaria when concentrating P. tomentosa eggs. In trial 2, each sample had a total volume of 252 μL, 60 μL of fish water containing 300 P. tomentosa eggs, 12 μL E400, and 180 μL RMMB. The 2017 P. neurophilia samples also had a total volume of 200 μL, consisting of 48 μL of 580 spores in distilled water, 10 μL of E400, and 142 μL RMMB.
ZIRC cryopreservation protocol: P. tomentosa eggs and P. neurophilia spores (2017) samples
-
• P. tomentosa eggs:
-
o Ratio of solutions used in the ZIRC cryopreservation method:
□ 5 μL sperm (in our case, P. tomentosa eggs in fish water) +1 μL E400 (Sperm Extender) +15 μL RMMB (Raffinose freezing medium)
-
o Kept same ratio of solutions, but increased sample volumes to ensure that sufficient numbers of P. tomentosa eggs would be available for evaluation for each sample:
-
□ Trial 1 samples: 48 μL of P. tomentosa eggs (144 eggs) +10 μL E400 + 142 μL RMMB
• Total volume frozen = 200 μL/sample (five samples).
-
□ Trial 2 samples: 60 μL of P. tomentosa eggs (300 eggs) +12 μL E400 + 180 μL RMMB
• Total volume frozen = 252 μL/sample (done in triplicate).
o Final freezing temperature = −196°C
-
-
-
• P. neurophilia spores (2017 experiment):
o 48 μL of distilled water with 580 P. neurophilia spores +10 μL E400 + 142 μL RMMB
o Total volume frozen = 200 μL/sample (done in triplicate).
o Final freezing temperature = −196°C.
2014 ZIRC cryopreservation (cryoprotectant): P. neurophilia spores
One experiment with P. neurophilia was conducted in 2014, and it used a slightly different protocol based on ZIRC's protocol at that time.19,20 The freezing media used at this time were two solutions of Ginsburg Fish Ringers, one with methanol and one without methanol.19,20 A total of 5 μL of P. neurophilia spores was put into 2 mL cryo vials (No. 430659; Corning), in triplicate. Added to each vial were 1.5 μL of Ginsburg Fish Ringers without methanol and 8.5 μL of Ginsburg Fish Ringers with methanol. The cryo-vials were then capped and placed directly into 15 mL Polypropylene conical tubes (No. 352096; Falcon, Corning), one cryo-vial per tube. The conical tubes were capped and plunged into finely crushed dry ice for 20 min. The cryo-vials were then transferred to cryogenic canes and put in LN2.
In addition, some samples from the 2014 study also contained zebrafish sperm. Sperm from two male zebrafish was collected in a glass capillary, and normalized to 3.3 μL with Ginsburg Fish Ringers without methanol. Next was the addition of Ginsburg Fish Ringers with methanol, brining the mixture up to 20 μL. This sperm and freezing medium mixture was then expelled onto a watch glass, and 10 μL of P. neurophilia spores was mixed into it. Fifty microliters of this final mixture was then pipetted into a 2 mL cryo-vial (No. 430659; Corning), and the same freezing procedure that was implemented on the samples without sperm was followed. This was done in triplicate.
ZIRC cryopreservation protocol: P. neurophilia (2014) experiment
-
• P. neurophilia spores (2014 samples):
-
o P. neurophilia spores:
□ 5 μL of P. neurophilia spores in distilled water +1.5 μL Ginsburg Fish Ringers without methanol +8.5 μL Ginsburg Fish Ringers with methanol
□ Total volume frozen = 15 μL/sample (done in triplicate).
-
o P. neurophilia spores+zebrafish sperm:
□ Sperm was collected from two male zebrafish and normalized to 3.3 μL with Ginsburg Fish Ringers without methanol.
□ This mixture was then brought up to 20 μL with the addition of Ginsburg Fish Ringers with methanol.
□ 10 μL P. neurophilia spores added to the 20 μL sperm mixture.
□ Total volume frozen = 15 μL/sample (done in triplicate).
o Final freezing temperature = −196°C.
-
Freezing at −80°C, −196°C, and −20°C (no cryoprotectant): parasite samples
For the trial 1 P. tomentosa freezing, 50 μL of fish water with 150 eggs was put into 2 mL cryo-vials, in triplicate, and to each vial 100 μL 1 × PBS was added. For trial 2, 60 μL of fish water with 300 eggs was placed into cryo-vials containing 110 μL of 1 × PBS. This was done in triplicate. All vials were then stored in a −80°C freezer.
In the 2014 experiment, 10 μL of spores in distilled water was placed into 2 mL cryo-vials in triplicate, placed on canes, and stored in LN2. Ten microliters of spores in distilled water were also put into 1.5 mL Eppendorf tubes in triplicate, capped, and placed in a −20°C freezer. For the recent freezing of P. neurophilia, 125 μL of spores (1,500 spores) kept in distilled water was put into 2 mL cryo-vials, in triplicate, and placed in a −20°C freezer.
Protocol for freezing at −80°C, −196°C, and −20°C: parasite samples
-
• P. tomentosa eggs:
-
o Trial 1 samples: 50 μL of fish water with 150 P. tomentosa eggs +100 μL 1 × PBS
□ Total volume frozen = 150 μL/sample (done in triplicate).
-
o Trial 2 samples: 60 μL of fish water with 300 P. tomentosa eggs +110 μL 1 × PBS
□ Total volume frozen = 170 μL/sample (done in triplicate).
o Final freezing temperature = −80°C.
-
-
• P. neurophilia spores (2014 samples):
-
o 10 μL of P. neurophilia spores in distilled water was placed in LN2 (done in triplicate).
□ Total volume frozen = 10 μL
□ Final freezing temperature = −196°C
-
o 10 μL of P. neurophilia spores in distilled water was placed in a −20°C freezer (done in triplicate).
□ Total volume frozen = 10 μL
□ Final freezing temperature = −20°C.
-
-
• P. neurophilia spores (2017 samples):
o 125 μL of distilled water with 1,500 P. neurophilia spores was placed in a −20°C freezer (done in triplicate).
o Total volume frozen = 125 μL
o Final freezing temperature = −20°C.
ZIRC thawing method (thawing solution): P. tomentosa eggs and P. neurophilia spores (2017 samples)
Samples were thawed in a 38°C water bath for 15–20 s or until there were no longer ice crystals and then immediately removed. To each thawed vial, SS300 was added. In fact, 150 μL is the general amount of SS300 that is added to the 20 μL frozen sperm samples. In our case, because the volume frozen down in our samples was higher than 20 μL, the amount of SS300 that was added to these samples was increased to match the ratio of solutions generally used with zebrafish sperm cryopreservation. To the trial 1 P. tomentosa eggs, we added 1.5 mL of SS300. Three of these samples were incubated at 28°C for 7 days with the SS300 left in each vial (Table 1, ZIRCB). For the other two samples (Table 1, ZIRCA), after the addition of SS300, the vials were then centrifuged for 5 min at 100 g. The supernatant was removed, and 300 μL of sterilized water from our system was added to each vial.
Table 1.
Viability of Pseudocapillaria Eggs After Cryopreservation (Zebrafish International Resource Center) and Freezing Without Cryopreservant (−80°C)
| Viability by larvation | ||||||
|---|---|---|---|---|---|---|
| Trial | Method | Replicate | N | Larvated (%) | Unlarvated (%) | Dead (%) |
| 1 | ZIRCA day 0 | 1 | 28 | 0 | 92.9 | 7.1 |
| 2 | 55 | 0 | 98.2 | 1.8 | ||
| 3 | — | — | — | — | ||
| ZIRCA day 7 | 1 | 33 | 0 | 81.8 | 18.2 | |
| 2 | 38 | 0 | 92.1 | 7.9 | ||
| 3 | — | — | — | — | ||
| ZIRCB day 0 | 1 | 55 | 0 | 100 | 0 | |
| 2 | 47 | 2.1 | 93.6 | 4.3 | ||
| 3 | 39 | 0 | 100 | 0 | ||
| ZIRCB day 7 | 1 | 28 | 0 | 85.7 | 14.3 | |
| 2 | 60 | 0 | 95 | 5 | ||
| 3 | 29 | 0 | 89.7 | 10.3 | ||
| −80°C day 0 | 1 | 58 | 0 | 96.6 | 3.4 | |
| 2 | 87 | 0 | 97.7 | 2.3 | ||
| 3 | 76 | 0 | 97.4 | 2.6 | ||
| −80°C day 7 | 1 | 18 | 0 | 100 | 0 | |
| 2 | 41 | 0 | 95.1 | 4.9 | ||
| 3 | 30 | 0 | 90 | 10 | ||
| 28°C day 0 | 1 | 208 | 0 | 89.9 | 10.1 | |
| 2 | 215 | 0 | 96.7 | 3.3 | ||
| 3 | 378 | 0 | 96.6 | 3.4 | ||
| 28°C day 7 | 1 | 56 | 82.1 | 12.5 | 5.4 | |
| 2 | 101 | 93.1 | 5 | 1.9 | ||
| 3 | 91 | 79.1 | 18.7 | 2.2 | ||
| 2 | ZIRC day 0 | 1 | 22 | 0 | 100 | 0 |
| 2 | 19 | 0 | 94.7 | 5.3 | ||
| 3 | 15 | 0 | 100 | 0 | ||
| ZIRC day 7 | 1 | 3 | 0 | 100 | 0 | |
| 2 | 7 | 0 | 100 | 0 | ||
| 3 | 17 | 0 | 100 | 0 | ||
| −80°C day 0 | 1 | 61 | 0 | 88.5 | 11.5 | |
| 2 | 86 | 0 | 94.2 | 5.8 | ||
| 3 | 46 | 0 | 100 | 0 | ||
| −80°C day 7 | 1 | 7 | 0 | 100 | 0 | |
| 2 | 15 | 0 | 100 | 0 | ||
| 3 | 19 | 0 | 94.7 | 5.3 | ||
| 28°C day 0 | 1 | 172 | 0.6 | 94.2 | 5.2 | |
| 2 | 134 | 0 | 92.5 | 7.5 | ||
| 3 | 132 | 0 | 93.2 | 6.8 | ||
| 28°C day 7 | 1 | 54 | 66.7 | 16.7 | 16.6 | |
| 2 | 92 | 73.9 | 15.2 | 10.9 | ||
| 3 | 76 | 65.8 | 27.6 | 6.6 | ||
Two separate trials were conducted. In trial 1, ZIRCA are samples where the SS300 thawing solution was added to the vials after thawing and then removed before they were incubated. ZIRCB are samples where the SS300 thawing solution was added to the vials after thawing, and not removed. In trial 2, the SS300 thawing solution was added to each thawed ZIRC sample and then removed before they were incubated.
SS300, Sperm Solution 300; ZIRC, Zebrafish International Resource Center.
For trial 2 (Table 1), 1.9 mL of SS300 was added to the P. tomentosa samples. They were then centrifuged for 5 min at 100 g. As with trial 1, the supernatant was removed and 300 μL of sterilized system water was added to each vial. For all samples, half of the eggs were counted immediately after thawing (Table 1, day 0) whereas the rest were incubated at 28°C for 7 days (Table 1, day 7) and then counted.
The same thawing procedure was employed with P. neurophilia spores, with 1.5 mL of SS300 added to each thawed cryo-vial. They were then centrifuged, the supernatant was removed, and 100 μL of sterilized system water was added. They were kept at room temperature (23°C–25°C).
ZIRC thawing method steps: P. tomentosa eggs and P. neurophilia spores (2017 samples)
-
• P. tomentosa eggs:
o Samples were thawed in a 38°C water bath.
-
o Ratio of solutions used in the ZIRC thawing method:
□ 20 μL samples +150 μL SS300 (thawing solution)
o Used the same ratio of solutions, but because the volumes of our parasite samples were increased, the amount of thawing solution added to each sample was increased.
-
o Trial 1 samples: 200 μL sample +1.5 mL SS300
□ Trial 1, ZIRCA samples: After the addition of SS300, each vial was centrifuged, the SS300 was removed, and 300 μL of sterilized water from our system was added.
□ Trial 1, ZIRCB samples: The SS300 was never removed from the vials after it was added.
-
o Trial 2 samples: 252 μL samples +1.9 mL SS300
□ After the addition of SS300, each sample was centrifuged, the SS300 was removed, and 300 μL of sterilized system water from our system was added.
o For all samples, half were examined for signs of larvation (day 0 count).
o The rest of the eggs were incubated at 28°C for 7 days, and then counted for signs of larvation (day 7 count).
-
• P. neurophilia spores (2017 samples):
o Samples were thawed in a 38°C water bath.
-
o Ratio of solutions used in the ZIRC thawing method:
□ 20 μL samples +150 μL SS300 (thawing solution)
o Used the same ratio of solutions, but because the volumes of our parasite samples were increased, the amount of thawing solution added to each sample was increased.
o 200 μL samples +1.5 mL SS300
o Samples were centrifuged, SS300 was removed, and 100 μL of sterilized system water was added.
o Samples were kept at room temperature until they were counted.
2014 ZIRC thawing method (thawing solution): P. neurophilia spores
The 2014 P. neurophilia experiment used the ZIRC thawing protocol at that time.19,20 The cryo-vials were taken out of the LN2, the caps were removed, and each vial was placed half way into a 33°C water bath for 8–10 s. To this, 10 μL of Hank's Balanced Salt Solution (HBSS) was added. The spores were then examined for viability.
ZIRC thawing method steps: P. neurophilia spores (2014 samples)
-
• P. neurophilia spores (2014) samples:
o Samples were thawed in a 33°C water bath.
o 15 μL samples +10 μL HBSS
o Samples were then examined for viability.
Thawing −80°C, −196°C, and −20°C samples (no thawing solution): parasite samples
Both P. neurophilia spores and P. tomentosa eggs were thawed in a 38°C water bath until the solutions were no longer frozen and then immediately removed. As in the ZIRC method, half of the P. tomentosa eggs were examined immediately after being thawed and the rest were incubated for 7 days at 28°C and then counted. For the 2014 experiment, spores were thawed in a 33°C water bath.
Steps for thawing −80°C, −196°C, and −20°C samples: parasite samples
-
• P. tomentosa eggs:
o Thawed in a 38°C water bath
o Half of the eggs were immediately examined for larvation (day 0 count).
o The other half were incubated at 28°C for 7 days and then examined for larvation (day 7 count).
-
• P. neurophilia spores (2017) samples:
o Thawed in a 38°C water bath and then examined for viability.
-
• P. neurophilia spores (2014) samples:
o Thawed in a 33°C water bath and then examined for viability.
Viability of P. tomentosa eggs
Eggs of P. tomentosa exhibit distinctive vermiform larvae after about 5–6 days,21 and larvation was used as an indicator of parasite survival. The number of larvated, unlarvated and obviously dead eggs in each vial were determined after the samples were thawed out (day 0) and these numbers were compared with the same endpoints at day 7. The eggs were viewed on a microscope by using × 100 and × 200 magnification.
Viability of P. neurophilia spores
The viability of P. neurophilia spores was assessed by using two fluorescent stains: SYTOX Green nucleic acid stain (No. S7020; Thermo Fisher Scientific) and Fungi-Fluor stain (No. 174421; Polysciences). We followed the same procedure described by Ferguson et al., in which positive staining with SYTOX indicates dead spores, whereas extrusion of the polar filament after incubation in Fungi-Flour indicates that the spores are viable.22 On a microscope slide, 5 μL of 100 μM SYTOX Green was added to 5 μL of spore solution. These spores were then viewed under oil ( × 1,000) by using a Lecia DMR fluorescent microscope with an FITC green filter (480–490 nm excitation, 527/30 emission). Spores that fluoresced bright green were counted as dead whereas spores that showed no signs of fluorescence were considered alive.
For our Fungi-Fluor stained spores, 5 μL of solution A of the Fungi-Fluor stain kit was added to 5 μL of spores on a microscope slide. They were then viewed under oil ( × 1,000) with a DAPI filter (340–380 nm excitation, 425 nm emission). Spores were considered alive if they extruded their polar filament when exposed to ultraviolet light within 20 s. The presence or absence of a posterior vacuole was recorded for all spores from both the SYTOX and Fungi-Fluor assays.
Statistical analysis
To compare the differences between the initial bacterial concentrations and the post-thaw bacterial concentration among each strain of bacteria, a one-sample z-test was used with the level of significance set at a p-value of <0.05.
For the P. tomentosa day 7 ZIRC method results, we wanted to determine the minimum number of larvated eggs that would be expected if the population size was set at 100,000 eggs. We used the following equation:
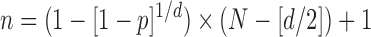 |
where n = the required samples size, N = total population size, p = probability of detecting one infected fish (in our case, one larvated egg) (p = 0.95), and d = maximum number of infected fish (in our case, larvated eggs at day 7) expected given a presumed prevalence (P) so that d = P × N. This equation is commonly used to calculate how many fish should be sampled (n) to detect at least one diseased fish presuming that 1% of the total population is infected with a disease.23,24 In our case, we already had n, which was the total number of eggs examined in all ZIRC day 7 samples, so we used this equation to first solve for d. Solving for d provides an estimate of the maximum number of larvated eggs at day 7 that would be predicted to occur out of a large population (e.g., 100,000 eggs), subjected to the ZIRC cryopreservation method, with a 95% confidence interval. Then, we used the calculated value for d to solve for P, which is the percentage of the total population of eggs at day 7 that we can expect to be larvated.
For the 2014 and 2017 P. neurophilia spore results, to assess the difference between the control and ZIRC method samples, we used a two-sample t-test with the significance level set at a p-value of <0.05.
Results
All of the pathogens tested, with the exception of P. tomentosa, showed survival after cryopreservation.
E. ictaluri
Processing samples of this bacterium through the ZIRC cryopreservation method resulted in substantial survival (73%) compared with unfrozen controls (Fig. 1). In contrast, only 2%–6% of the E. ictaluri subject to freezing at −80°C without a cryopreservant survived compared with the unfrozen control.
FIG. 1.

Bacterial survival after freezing by ZIRC cryopreservation and −80°C. Low bacterial concentrations (104 CFU/mL) represented by top graphs and high concentration (108 CFU/mL) represented by bottom graphs. White bars are the starting bacterial concentrations, and the gray bars are the average bacterial concentrations calculated after thawing. Black dots represent individual plate counts and show the range of data. One sample z-test was performed, for the high concentrations the H1E1, H1E2, and OSU 214 ZIRC samples were not statistically significant (one-sample z-test, p > 0.05). For low concentrations, and H1E2 ZIRC sample was not statistically significant (p = 1.00). All the other samples were found to be statistically significant (one-sample z-test, p < 0.05). CFU, colony-forming units; ZIRC, Zebrafish International Resource Center.
Mycobacterium spp.
The mycobacteria (two strains of M. chelonae and one strain of M. marinum) showed minimal reduction in survival after freezing with the ZIRC method (Fig. 1). Some samples showed evidence of greater growth after cryopreservation compared with the unfrozen controls. However, this increase was not statistically significant (M. chelonae H1E1: p = 0.89, H1E2: p = 1.00, M. marinum OSU 214: p = 0.81). For the −80°C samples, which contained no cryopreservant, each mycobacterium experienced a decline in bacterial concentration, but exhibited higher bacterial concentrations compared with the E. ictaluri −80°C samples (Fig. 1). Mycobacteria survival for the three strains frozen with the ZIRC method ranged between 30% and 70% compared with the −80°C controls, where survival ranged between 30% and 60% (Fig. 1).
P. tomentosa
Egg larvation was used as an indication of viability for the P. tomentosa eggs. Figure 2 shows the typical appearance of a larvated egg compared with an undeveloped (unlarvated) egg and an egg scored as dead. In trial 1, no eggs showed larvation after the samples were thawed and allowed to incubate for 7 days at 28°C (Table 1, trial 1); whereas 80%–93% of the control, non-frozen eggs larvated. However, there was one instance of a larval worm that partially hatched out of its shell at day 0 in one of the ZIRC method samples (Fig. 2e) (Table 1, trial 1). As this fully developed larva was observed at time zero, it was likely present in the egg before freezing.
FIG. 2.
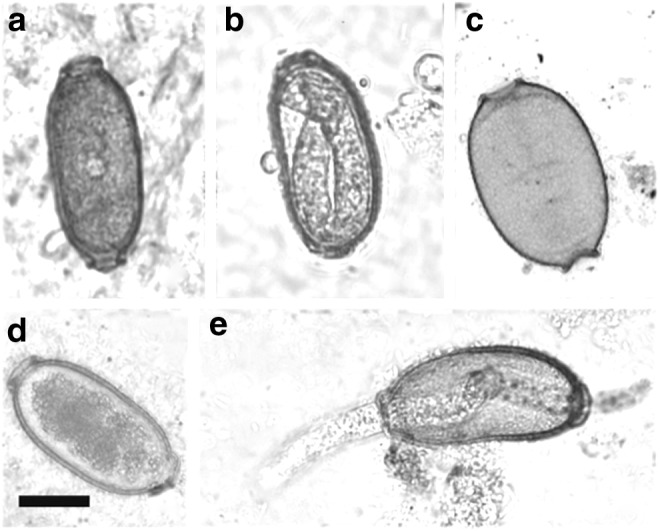
Pseudocapillaria tomentosa eggs. (a) An unlarvated egg. (b) A larvated egg. (c) Egg devoid of contents, scored as dead. (d) Egg 7 days after thawing from freezing by using the ZIRC method. These eggs typically exhibited contracted internal material with more prominent granulation. (e) Partially hatched P. tomentosa eggs observed in trial 1 on day 0. Bar 10 μm.
Many of the unlarvated eggs in either freezing method appeared intact as shown in Figure 2a. But, there were instances where the internal material of the eggs was concentrated to the middle of the egg and had more of a prominent granular appearance (Fig. 2d). Eggs that were scored as dead were devoid of internal material (Fig. 2c), and these were the second most commonly seen eggs in our frozen samples, the first most common being unlarvated eggs. The percentage of these eggs scored as “dead” increased slightly from day 0 to 7 in each sample (Table 1).
Results from trial 2 were similar to those from the first trial. No larvation was observed on day 7 by using either freezing method (Table 1, trial 2). In the ZIRC samples, 100% of the eggs observed at day 7 were unlarvated.
In the −80°C samples, at day 7, the number of unlarvated eggs was about 95%–100%, with the remaining scored as dead (Table 1). In contrast, control eggs showed between 66% and 74% larvation at day 7. There was one larvated egg that was seen at day 0 in one of our control samples, but again, this was likely an egg that had already larvated before being collected. In other words, eggs were collected from fresh feces released within 24 h, whereas this egg was likely an older egg that had been ingested by a fish and released in a larvated state.
The probability of larvation in ZIRC method
Because the results of freezing P. tomentosa showed no larvation, we used the probability of detecting a positive sample23 in a given population to assess the power of this negative result. We only examined 215 eggs. However, if we were to examine 100,000 P. tomentosa eggs subjected to the ZIRC cryopreservation method, we wanted to determine what would be the maximum number of larvated eggs that we would predict to observe on day 7, based on our negative result with 215 eggs with a 95% confidence level. Using the formula described in the Materials and Methods section, and solving for d, we obtained a value of about 1,400. We then solved for P and obtained a value of 1.4%:
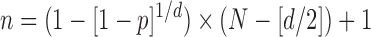 |
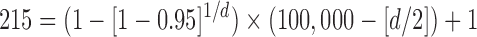 |
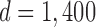 |
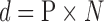 |
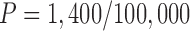 |
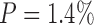 |
Therefore, if we had a large population of eggs (e.g., 105), which were subjected to the ZIRC cryopreservation method, and having examined 215 of these eggs and observing no larvation, we can only conclude with 95% confidence that no more than 1.4% of the total population of eggs would be larvated on day 7.
P. neurophilia
We conducted two separate trials, 3 years apart and using slightly different cryopreservants. In the 2014 trial, Ginsburg Fish Ringers both with and without methanol was used as the cryopreservant; whereas in the 2017 trial, Raffinose was the cryopreservant. Although the 2014 protocol utilized a membrane-permeating cryopreservant and the 2017 protocol used a non-permeating cryopreservant, these two trials yielded similar results, with some spores surviving the ZIRC protocols, regardless of the scoring method (Fig. 3).
FIG. 3.

Spores of Pseudoloma neurophilia. (a) The top spore has a vacuole, and the vacuole is absent in the lower spore. (b, c) A spore stained with SYTOX. (b) Shows the spore under brightfield, and (c) is the same spore but under fluorescence. This spore would be scored as dead. (d) Spore that expelled its polar tube (white arrow) after staining with Fungi-Fluor and exposure to ultraviolet light. This spore would be marked as alive. Bar = 5 μm. Color images available online at www.liebertpub.com/zeb
In the 2014 trial, control spores (held at 4°C in distilled water) showed more than 80% survival using the two vital stains, SYTOX and Fungi-Fluor, and about 60% had vacuoles (Fig. 4). Likewise, spores held with cryopreservant and sperm but not frozen also showed high survival. Other controls (boiling or holding spores at −20°C or −196°C) showed no survival using all three viability methods, except about 50% of the spores evaluated by SYTOX and 2% of the spores evaluated by Fungi-Fluor were scored as positive when held at −196°C. With both ZIRC-S and ZIRC cryopreservation samples, spores stained with SYTOX displayed a much higher percentage of survival than those scored by Fungi-Fluor and the presence of vacuoles (Fig. 4). However, there was considerable variability between replicate samples for each exposure method. Using a two-sample t-test to compare the 4°C samples with the ZIRC-S and ZIRC groups, the Fungi-Fluor and vacuole results were significantly different from the unfrozen controls (Fungi-Fluor: 4°C vs. ZIRC-S, p = 0.003; 4°C vs. ZIRC, p = 0.002; Vacuoles: 4°C vs. ZIRC-S and ZIRC, p = 0.03).
FIG. 4.

P. neurophilia 2014 cryopreservation results. The ZIRC sperm cryopreservation method used at that time was used in this assay. Each bar is the average spore survival based on SYTOX (white bars), Fungi-Fluor (gray bars), or presence of spore vacuoles (striped bars). Black dots represent each of the replicate samples. All of the other samples contained only spores with distilled water. The 4°C+ZIRC-S samples contained spores, zebrafish sperm, and the freezing medium (Ginsburg Fish Ringers), but they were kept at 4°C. ZIRC, without sperm; ZIRC-S, cryopreservation with sperm.
The same endpoints were used to assess spore survival in the 2017 trial, but here we used the current ZIRC sperm cryopreservation freezing and thawing methods and solutions. We observed similar spore survival results as in the 2014 assay (Fig. 5). In this trial, the controls were held at 20°C, and again viability of spores was >50% by using all three methods for scoring spore survival. There was slightly less survival observed with the spores in the ZIRC group stained with SYTOX compared with unfrozen controls (66% vs. 80%). Fungi-Fluor showed only 19% survival. There was a significant decrease in the number of spores with vacuoles in the ZIRC samples compared with the positive control (p = 0.005), with only 8% of spores in the ZIRC samples showing vacuoles (Fig. 5). Overall, both vital stains and the vacuole presence method revealed that the number of live spores in the samples that were frozen was less than the number of spores that were kept under unfrozen conditions (4°C or 20°C).
FIG. 5.

P. neurophilia 2017 cryopreservation results. The current ZIRC sperm cryopreservation method (2017) was used in this assay. Each bar is the average spore survival based on SYTOX (white bars), Fungi-Fluor (gray bars), or presence of spore vacuoles (striped bars). Each black dot equals one of three replicate samples. There was a significant decrease in the percentage of spores scored alive by the presence of vacuoles or by Fungi-Fluor in the ZIRC samples compared with the 20°C samples. (Two-sample t-test, p = 0.005.)
Discussion
The bacterial species utilized in this study survived ZIRC's sperm cryopreservation process and freezing at −80°C with no cryopreservant. Although there was variation in the percentage of survival indicated by each viability stain, P. neurophilia spores also survived the ZIRC cryopreservation method. The only pathogen that did not survive either freezing condition tested in this study was P. tomentosa eggs, which exhibited no larvation on day 7.
Both species of mycobacteria showed a relatively minimal reduction in survival when subjected to either freezing conditions. Research involving the cryopreservation and freezing of mycobacteria has been conducted since the 1960s,25 and these studies have consistently demonstrated the ability of mycobacteria to withstand subzero temperatures with minimal loss in viability.26,27 Mycobacteria are naturally found in soils, water and only a few species are restricted to their vertebrate host (e.g., Mycobacterium tuberculosis).28,29 Dwelling in what are often very harsh and variable environments, these organisms have evolved to survive various environmental stressors. One characteristic unique to Mycobacterium is their cellular wall that comprises long-chain mycolic acids ligated to arabinogalactan that surrounds a thick peptidoglycan layer.30 This thick, waxy, impermeable barrier30 likely played a significant role in the survival of each Mycobacterium species in both the ZIRC and −80°C samples (Fig. 1).
During freezing, the extracellular medium freezes first, causing the external osmolarity of the cells to increase and the internal water of the cells to cool but remain transiently unfrozen.31,32 This imbalance in which external osmolarity of the cells is higher than the internal osmolarity of the cells causes the cooled water inside the cells to travel across the cell membrane and cell wall, where it subsequently freezes with the extracellular medium.31,32 If the cells are unable to dehydrate quick enough, internal ice-crystals may form (often the case with rapid cooling), which can cause significant damage to the cells.32 Cryoprotectants are used to reduce the development of ice crystals inside of the cells.31 Cryoprotectants increase the solute concentration in the solution, which, in turn, decreases the freezing point of the cells.33 Therefore, mycobacteria survived well after freezing without a cryopreservant, whereas survival was higher with the cryopreservant.
Sample freezing rate can also impact pathogen survival. The freezing rate generated by the ZIRC method lies between what some would consider fast (100–400°C/min) and slow cooling rates (2–4°C/min).31,34 The configuration of the 20 μL ZIRC samples in the 15 mL conical tubes placed in powdered-dry ice created a cooling rate of about 20°C/min. Another study found the cooling rate of vials placed directly in a −80°C freezer to be similar.31 It is plausible that the mycobacteria were able to generate internal and external osmotic balance because their thick cell walls slow the internal cooling rate of the cells, therefore giving them ample time to dehydrate and remain internally unfrozen. In turn, the bacterial cells that did not survive freezing likely developed intracellular crystals that caused damage to the cells' structure either during freezing or when the cells were thawed.
Although not statistically significant, we surprisingly observed increased bacterial counts after cryopreservation compared with unfrozen controls for some mycobacteria samples. Iivanainen et al.28 also reported this phenomenon with environmental samples, and they attributed an apparent increase in mycobacteria counts to disruption of bacterial aggregates or a decrease of other bacterial organisms after the freeze/thaw process. It is possible that the freezing and thawing procedure used with our mycobacteria samples caused the bacteria to disaggregate and therefore, when we plated the bacteria after thawing the colony counts were higher.
E. ictaluri also showed survival under both freezing conditions, but not to the extent seen with the mycobacteria. Studies involving the freezing of E. ictaluri as well as other members of the Enterobacteriaceae have shown that these bacteria can survive freezing at −20°C and −80°C, but a substantial decrease in bacterial concentrations is expected.35–37 In many of these studies, there was no cryoprotectant utilized and instead the bacteria were frozen while in tissues. In our study, the ZIRC method samples contained a specific cryoprotectant (Raffinose Freezing Medium) and this was probably an important contributing factor for the survival of E. ictaluri compared with being frozen at −80°C with no cryoprotectant.
P. tomentosa eggs showed no survival with either freezing protocol. Nematodes and their eggs are reportedly capable of surviving these types of freezing conditions. However, these studies have been conducted with terrestrial nematodes, which have evolved to survive freezing temperatures.38–40 P. tomentosa has a very broad host and geographic range in aquatic environments,41 but it has unlikely evolved mechanisms for its eggs to survive freezing in water as they would not regularly experience extreme temperature fluctuations. In our study, observation of larvation (Fig. 2b) within eggs at 7 days (7 days after being shed or thawed) was a direct indication of survival, and most of the P. tomentosa eggs in the control samples were larvated by this time (66%–93%). Based on these results, we conclude that P. tomentosa eggs cannot survive freezing, even in the presence of a cryoprotectant such as raffinose. Nematode eggs, with their thick shell walls, are generally quite resistant to external agents. The addition of raffinose causes a solution to become hypertonic, and cells in such solutions will dehydrate to achieve osmotic balance. The shells of P. tomentosa eggs provide a strong barrier to factors that are detrimental to the developing worms, whereas perhaps the shells inhibited such an osmotic response either by not allowing the internal water of the eggs to permeate out or more likely by preventing any osmotic signals from reaching the internal regions of the eggs.
In previous studies, nematode eggs were considered non-viable if damage to the eggs was observed or if the eggs were not intact.40,42 In our study, unlarvated and dead (empty shell) eggs (Fig. 2a, c, respectively) were intact and showed no signs of damage. It is possible that some of the eggs designated unlarvated but still with intact contents were still viable and delayed in their development. However, this is unlikely because we conducted later observations of these eggs (e.g., 10–14 days after thawing) and larvation was never observed. Moreover, it is very unlikely that the empty eggs scored as dead after freezing are hatched eggs, as we saw no larvae free or within eggs at 7 days.
The survival of microsporidian spores at subzero temperatures has previously been examined, and certain species have shown the ability to survive in LN2, without a cryoprotectant, for up to 25 years.43 Some species of microsporidia maintain infectivity after freezing.44,45 These obligate intracellular parasites infect a wide range of organisms, including numerous fish species.46,47
To evaluate the survival of P. neurophilia spores after freezing, we followed the same procedure used by Ferguson et al., which involved the use of two fluorescent stains: Fungi-Fluor and SYTOX.22 The presence of a vacuole was also recorded for all of the spores examined. Some of the P. neurophilia spores in both of our assays (2014 and 2017) were able to survive the ZIRC method of freezing as well as freezing at −196°C and −20°C without cryopreservant. This supports what was observed in the Maddox and Sotlter study, in which the spores of various microsporidian survived freezing in LN2. Interestingly, Nosema spp. from terrestrial insects survived better than Nosema algerae from aquatic stages of mosquitos,43 which is further evidence that terrestrial microorganisms have evolved to survive freezing conditions better than those from aquatic environments. Although only the 2014 cryoprotectant provided for penetration of membranes (i.e., with methanol), spore survival results with the 2014 and 2017 experiments were remarkably similar. This suggests that improved survival of spores compared with freezing without cryopreservant is not related to incorporation of the cryopreservant.
One unique feature of these parasites is the presence of a large, conspicuous posterior vacuole inside their spores.48 The presence of this vacuole is an indicator that the internal structure of the spore is intact, and hence is an indicator of spore viability. Microsporidian spores also contain a long, coiled, polar filament or tube. This tube can form between 4 and 30 coils inside the spore and on excitation, this polar tube is expelled, and this is believed to be the primary mechanism that the spores use to infect the cytoplasm of host cells.49 Therefore, the ability of a spore to extrude its polar tube also indicates that a spore is viable and infectious. Exclusion of the SYTOX dye suggests that the spore wall is intact.
There was some variation in our results between the three viability methods. We observed similar results as reported by Ferguson et al.,22 in which spores from the same treatment consistently showed higher survival scores with SYTOX compared with those with Fungi-Fluor. And our study showed similar results with the latter stain and the presence of a spore vacuole. With SYTOX, spores that are scored as dead should exhibit internal green fluorescence. As with other vital dyes that rely on cell permeability, for the SYTOX stain to reach the internal area of the spore, there has to have been damage to the spore wall. In other words, some intact spores could still be dead, or otherwise non-viable, but would be scored as alive with SYTOX if the spores' wall were still not permeable. Amigó et al. observed a very similar situation with spores of the fish microsporidium Glugea stephani, where spores subjected to freezing showed 58%–97% viability based on exclusion of a propidium iodide; whereas the same samples showed only 9%–48% viability based on polar tube extrusion.45 Similar challenges regarding interpretation of results with vital dyes that rely on permeability have been reported with other eukaryotic microorganisms with thick protective outer walls, such as Cryptosporidium spp.50,51
Although there were slight differences with a few samples, overall the results for Fungi-Fluor and vacuole presence, scoring spores as alive were more similar to each other than they were to the SYTOX method. Even with the SYTOX results, which may erroneously score spores as alive, we still observed spore survival with both ZIRC cryopreservation tests. Although considerably reduced compared with non-frozen controls, there were generally higher percentages of survival in the ZIRC cryopreserved samples compared with freezing at either −196°C or −20°C. It should also be noted that we observed few differences among all three viability methods with unfrozen control samples, where most of the spores were scored as alive.
Overall, our results demonstrated the ability of certain zebrafish pathogens to survive subzero temperatures and more specifically, the sperm cryopreservation method utilized by ZIRC. Although we visually determined the survival of these pathogens, the next step would be to examine their infectivity after freezing with in vivo transmission studies. Future studies might also address the minimum infective dose of these pathogens and the likelihood that this dose would contaminate sperm samples. The type of vessel that samples are stored in should also be examined to prevent possible contamination from sample preparation, surrounding samples in the same storage containers, or from contaminated LN2. Sperm samples are commonly stored in straws, which could make them susceptible to contamination, especially when stored in LN2 dewars.52 Cryogenic vials such as the ones used at ZIRC and in this study are better storage vessels compared with straws for containing and avoiding pathogens. That being said, regarding sperm cryopreservation protocols, it is recommended that sperm samples or donor fish be tested for pathogens either before or during the time at which they are used for cryopreservation.53,54
Acknowledgments
The authors of this article would like to thank Jennifer Matthews, Katy Murray, and Zoltan M. Varga of the ZIRC for providing their sperm cryopreservation protocol and reagents, as well as general guidance throughout this project. They would also like to thank Thomas Sharpton for his advice with statistical analysis and Katy Murray for review of the article. This project was partially funded by the NIH ORIP R24 Kent and Whipps, and the NIH ORIP R24 KENT and Whipps Research Supplements to Promote Diversity in Health-Related Research (Admin Suppl.).
Disclosure Statement
No competing financial interests exist.
References
- 1.Kalueff AV, Stewart AM, Gerlai R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol Sci 2014;35:63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng Y, Martin P. Imaging innate immune responses at tumour initiation: new insights from fish and flies. Nat Rev Cancer 2015;15:556–562 [DOI] [PubMed] [Google Scholar]
- 3.Vittori M, Breznik B, Gredar T, Hrovat K, Bizjak Mali L, Lah TT. Imaging of human glioblastoma cells and their interactions with mesenchymal stem cells in the zebrafish (Danio rerio) embryonic brain. Radiol Oncol 2016;50:159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray KN, Varga ZM, Kent ML. Biosecurity and health monitoring at the Zebrafish International Resource Center. Zebrafish 2016;13:S30–S38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zebrafish International Resource Center. http://zebrafish.org/home/guide.php (accessed on July19, 2017)
- 6.Yang H, Tiersch TR. Current status of sperm cryopreservation in biomedical research fish models: zebrafish, medaka, and Xiphophorus. Comp Biochem Physiol Part C Toxicol Pharmacol 2009;149:224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiersch TR, Jenkins JA: Biosecurity and regulatory considerations for transfer of cryopreserved sperm and early life stages of aquatic species. In: Biosecurity in Aquaculture Production Systems: Exclusion of Pathogens and Other Undesirables. Lee C-S. and O'Bryen PJ. (eds), pp. 171–198, The World Aquaculture Society, Baton Rouge, LA, 2003 [Google Scholar]
- 8.Jenkins JA: Infectious disease and quality assurance considerations for the transfer of cryopreserved fish gametes. In: Cryopreservation in Aquatic Species. Tiersch TR. (ed), pp. 939–959, The World Aquaculture Society, Baton Rouge, LA, 2011 [Google Scholar]
- 9.Kent ML, Kieser D: Avoidance of introduction of exotic pathogens with Atlantic salmon reared in British Columbia. In: Biosecurity in Aquaculture Production Systems: Exclusion of Pathogens and Other Undesirables. Lee C-S. and O'Bryen PJ. (eds), pp. 43–50, The World Aquaculture Society, Baton Rouge, LA, 2003 [Google Scholar]
- 10.Sanders JL, Watral V, Clarkson K, Kent ML. Verification of intraovum transmission of a microsporidium of vertebrates: Pseudoloma neurophilia infecting the Zebrafish, Danio rerio. PloS One 2013;8:e76064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray KN, Dreska M, Nasiadka A, Rinne M, Matthews JL, Carmichael C, et al. . Transmission, diagnosis, and recommendations for controls of Pseudoloma neuophlia infections in laboratory zebrafish (Danio rerio) facilities. Comp Med 2011;61:322–329 [PMC free article] [PubMed] [Google Scholar]
- 12.Whipps CM, Matthews JL, Kent ML. Distribution and genetic characterization of Mycobacterium chelonae in laboratory zebrafish Danio rerio. Dis Aquat Organ 2008;82:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kent ML, Watral VG, Kirchoff NS, Spagnoli ST, Sharpton TJ. Effects of subclinical Mycobacterium chelonae infections on fecundity and embryo survival in zebrafish. Zebrafish 2016;13:S88–S95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews J, Carmichael C. ZIRC E400/RMMB sperm cryopreservation protocol. 2015. https://zebrafish.org/documents/protocols.php (accessed on July2, 2017)
- 15.Whipps CM, Dougan ST, Kent ML. Mycobacterium haemophilum infections of zebrafish (Danio rerio) in research facilities. FEMS Microbiol Lett 2007;270:21–26 [DOI] [PubMed] [Google Scholar]
- 16.Ostland V, Watral V, Whipps C, Austin F, St-Hilaire S, Westerman M, et al. . Biochemical, molecular, and virulence characteristics of select Mycobacterium marinum isolates in hybrid striped bass Morone chrysops × M. saxatilis and zebrafish Danio rerio. Dis Aquat Organ 2008;79:107–118 [DOI] [PubMed] [Google Scholar]
- 17.Hawke JP, Kent M, Rogge M, Baumgartner W, Wiles J, Shelley J, et al. . Edwardsiellosis caused by Edwardsiella ictaluri in laboratory populations of zebrafish Danio rerio. J Aquat Anim Health 2013;25:171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson JM, Bunte RM, Carty AJ. Evaluation of rapid cooling and tricaine methanesulfonate (MS222) as methods of euthanasia in zebrafish (Danio rerio). J Am Assoc Lab Anim Sci 2009;48:785–789 [PMC free article] [PubMed] [Google Scholar]
- 19.Draper BW, Moens CB. A high-throughput method for zebrafish sperm cryopreservation and in vitro fertilization. J Vis Exp 2009;pii: 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmichael C, Westerfield M, Varga Z: Cryopreservation and in vitro fertilization at the Zebrafish International Resource Center. In: Zebrafish: Methods and Protocols. Lieschke GJ, Oates AC, and Kawakami K. (eds), pp. 45–65, Human Press, Totowa, NJ, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martins ML, Watral V, Rodrigues-Soares JP, Kent ML. A method for collecting eggs of Pseudocapillaria tomentosa (Nematoda: Capillariidae) from zebrafish Danio rerio and efficacy of heat and chlorine for killing the nematode's eggs. J Fish Dis 2017;40:169–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferguson JA, Watral V, Schwindt AR, Kent ML. Spores of two fish microsporidian (Pseudoloma neurophilia and Glugea anomala) are highly resistant to chlorine. Dis Aquat Organ 2007;76:205–214 [DOI] [PubMed] [Google Scholar]
- 23.Simon RC, Schill WB. Tables of sample size requirements for detection of fish infected by pathogens: three confidence levels for different infection prevalence and various population sizes. J Fish Dis 1984;7:515–520 [Google Scholar]
- 24.Kent ML, Buchner C, Watral VG, Sanders JL, Ladu J, Peterson TS, et al. . Development and maintenance of a specific pathogen-free (SPF) zebrafish research facility for Pseudoloma neurophilia. Dis Aquat Organ 2011;95:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruft H, Clark ME, Osterhout M. Preservation of mycobacterial cultures. Appl Microbiol 1968;16:355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TH, Kubica GP. Preservation of mycobacteria: 100% viability of suspensions stored at −70°C. Appl Microbiol 1973;25:956–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikuta CY, Ambrosio SR, Filho AFS, Grisi-Filho JHH, Heinemann MB, Neto JSF, et al. . Cryopreservation of Mycobacterium bovis isolates. Semina Ciênc Agrár 2016;37:3701–3708 [Google Scholar]
- 28.Iivanainen E, Martikainen PJ, Katila M-L. Effect of freezing of water samples on viable counts of environmental mycobacteria. Lett Appl Microbiol 1995;21:257–260 [DOI] [PubMed] [Google Scholar]
- 29.Niederweis M. Nutrient acquisition by mycobacteria. Microbiol Read Engl 2008;154:679–692 [DOI] [PubMed] [Google Scholar]
- 30.Kieser KJ, Rubin EJ. How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol 2014;12:550–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shu Z, Weigel KM, Soelberg SD, Lakey A, Cangelosi GA, Lee K-H, et al. . Cryopreservation of Mycobacterium tuberculosis complex cells. J Clin Microbiol 2012;50:3575–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazur P. Freezing of living cells: mechanisms and implications. Am J Physiol 1984;247:C125–C142 [DOI] [PubMed] [Google Scholar]
- 33.Pegg DE. Principles of cryopreservation. Methods Mol Biol 2007;368:39–57 [DOI] [PubMed] [Google Scholar]
- 34.Djuwantono T, Wirakusumah FF, Achmad TH, Sandra F, Halim D, Faried A. A comparison of cryopreservation methods: slow-cooling vs. rapid-cooling based on cell viability, oxidative stress, apoptosis, and CD34+ enumeration of human umbilical cord blood mononucleated cells. BMC Res Notes 2011;4:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brady YJ, Vinitnantharat S. Communications: viability of bacterial pathogens in frozen fish. J Aquat Anim Health 1990;2:149–150 [Google Scholar]
- 36.Bebak J, Shoemaker C, Arias C, Klesius P. Assay performance during validation of freezing channel catfish Ictalurus punctatus (Rafinesque) infected with a Gram-negative bacterium. Aquac Res 2011;42:169–176 [Google Scholar]
- 37.Popelka P, Nagy J, Pipová M, Marcinčák S, Lenhardt Ľ. Comparison of chemical, microbiological and histological changes in fresh, frozen and double frozen rainbow trout (Oncorhynchus mykiss). Acta Vet Brno 2014;83:157–161 [Google Scholar]
- 38.Torrini G, Landi S, Tarasco E, Roversi PF. Evaluation of Steinernema carpocapsae survival and infectivity after cryopreservation. BioControl 2016;61:461–469 [Google Scholar]
- 39.Carlsson AM, Irvine RJ, Wilson K, Coulson SJ. Adaptations to the Arctic: low-temperature development and cold tolerance in the free-living stages of a parasitic nematode from Svalbard. Polar Biol 2013;36:997–1005 [Google Scholar]
- 40.Schurer J, Davenport L, Wagner B, Jenkins E. Effects of sub-zero storage temperatures on endoparasites in canine and equine feces. Vet Parasitol 2014;204:310–315 [DOI] [PubMed] [Google Scholar]
- 41.Moravec F: Revision of Capillarid Nematodes (Subfamily Capillariinae) Parasitic in Fishes. Praha: Acad Natkadatelstvai Ceskoslovenskae Akad Ved 1987:273 [Google Scholar]
- 42.Van Wyk JA, Van Wyk L. Freezing of sheep faeces invalidates Haemonchus contortus faecal egg counts by the McMaster technique. Onderstepoort J Vet Res 2002;69:299–304 [PubMed] [Google Scholar]
- 43.Maddox JV, Solter LF. Long-term storage of infective microsporidian spores in liquid nitrogen. J Eukaryot Microbiol 1996;43:221–225 [Google Scholar]
- 44.McGowan J, De la Mora A, Goodwin PH, Habash M, Hamiduzzaman MM, Kelly PG, et al. . Viability and infectivity of fresh and cryopreserved Nosema ceranae spores. J Microbiol Methods 2016;131:16–22 [DOI] [PubMed] [Google Scholar]
- 45.Amigó JM, Garcia MP, Rius M, Savlvadó H, Maillo PA, Vivarés CP. Longevity and effects of temperature on the viability and polar-tube extrusion of spores of Glugea stephani, a microsporidian parasite of commercial flatfish. Parasitol Res 1996;82:211–214 [DOI] [PubMed] [Google Scholar]
- 46.Lom J. A catalogue of described genera and species of microsporidians parasitic in fish. Syst Parasitol 2002;53:81–99 [DOI] [PubMed] [Google Scholar]
- 47.Lom J, Nilsen F. Fish microsporidia: fine structural diversity and phylogeny. Int J Parasitol 2003;33:107–127 [DOI] [PubMed] [Google Scholar]
- 48.Sanders JL, Watral V, Kent ML. Microsporidiosis in zebrafish research facilities. ILAR J 2012;53:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y, Weiss LM. The microsporidian polar tube: a highly specialised invasion organelle. Int J Parasitol 2005;35:941–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campbell AT, Robertson LJ, Smith HV. Viability of Cryptosporidium parvum oocysts: correlation of in vitro excystation with inclusion or exclusion of fluorogenic vital dyes. Appl Environ Microbiol 1992;58:3488–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robertson LJ, Campbell AT, Smith HV. Letter to the editor viability of Cryptosporidium parvum oocysts: assessment by the dye permeability assay. Appl Environ Microbiol 1998;64:3544–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bielanski A, Bergeron H, Lau PCK, Devenish J. Microbial contamination of embryos and semen during long term banking in liquid nitrogen. Cryobiology 2003;46:146–152 [DOI] [PubMed] [Google Scholar]
- 53.Torres L, Hu E, Tiersch TR. Cryopreservation in fish: current status and pathways to quality assurance and quality control in repository development. Reprod Fertil Dev 2016;28:1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cabrita E, Martínez-Páramo S, Gavaia PJ, Riesco MF, Valcarce DG, Sarasquete C, et al. . Factors enhancing fish sperm quality and emerging tools for sperm analysis. Aquaculture 2014;432:389–401 [Google Scholar]