

SUMMARY
Diet, genetics, and the gut microbiome are determinants of metabolic status, in part through production of metabolites by the gut microbiota. To understand the mechanisms linking these factors, we performed LC-MS-based metabolomic analysis of cecal contents and plasma from C57BL/6J, 129S1/SvImJ, and 129S6/SvEvTac mice on chow or a high-fat diet (HFD) and HFD-treated with vancomycin or metronidazole. Prediction of the functional metagenome of gut bacteria by PICRUSt analysis of 16S sequences revealed dramatic differences in microbial metabolism. Cecal and plasma metabolites showed multifold differences reflecting the combined and integrated effects of diet, antibiotics, host background, and the gut microbiome. Eighteen plasma metabolites correlated positively or negatively with host insulin resistance across strains and diets. Over 1,000 still-unidentified metabolite peaks were also highly regulated by diet, antibiotics, and genetic background. Thus, diet, host genetics, and the gut microbiota interact to create distinct responses in plasma metabolites, which can contribute to regulation of metabolism and insulin resistance.
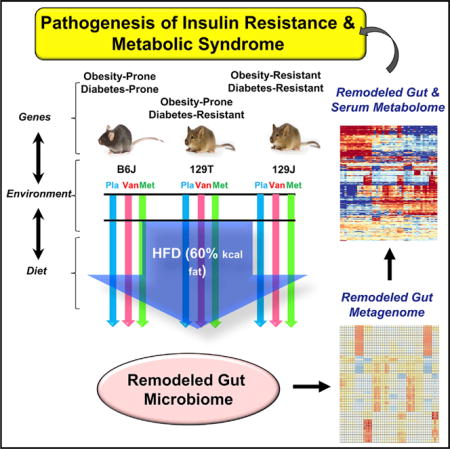
Graphical abstract
In Brief: Fujisaka et al. show that mice with differing propensities to obesity and diabetes have differing metabolomic responses to diet and antibiotic treatment. Several serum metabolites correlate with changes in the gut microbiota or with insulin resistance across strains. Thus, diet, genetics, and the gut microbiota interact to create distinct plasma metabolomic responses.
INTRODUCTION
Over the past decade, it has become clear that one factor affecting systemic metabolism is the composition of the gut microbiota (Le Chatelier et al., 2013; Lynch and Pedersen, 2016; Schroeder and Bäckhed, 2016). The nature of the microbial community and its changes in response to environmental factors such as dietary nutrients, fiber, and antibiotics is dependent on the genetic background of the host (Cho et al., 2012; Fujisaka et al., 2016; Parks et al., 2015; Tamburini et al., 2016; Ussar et al., 2016). These changes in the microbiota have been linked to development of obesity, diabetes, and metabolic syndrome, and many of these phenotypes can be transferred, at least in part, via the microbiome to germ-free mice (Ridaura et al., 2013; Turnbaugh et al., 2006). The gut microbiota can also affect intestinal function and the immune system. Intestinal microbes utilize dietary components to produce energy and metabolites, many of which are taken up into the blood stream where they can be further metabolized or affect host metabolism (Pedersen et al., 2016; Wikoff et al., 2009).
The effects of bacterial metabolites on host metabolism can be both beneficial and harmful. For example, short-chain fatty acids (SCFAs), derived from otherwise undigestible fiber, have generally beneficial effects on the host, including anti-obesity and anti-diabetic actions (Chang et al., 2014; Kimura et al., 2013; Tolhurst et al., 2012). On the other hand, N-nitroso compounds, ammonia, and hydrogen sulfide derived by bacteria from dietary protein can induce reactive oxygen species (ROS) and DNA damage and activate inflammatory pathways (Kim et al., 2013). Trimethylamine-N-oxide (TMAO), an end metabolite of dietary choline, has been shown to promote arteriosclerosis and correlate with cardiovascular disease (CVD), stroke, and death (Tang et al., 2013; Wang et al., 2011b). Deoxycholic acid, a secondary bile acid produced by the gut microbiota, promotes development of hepatocellular carcinoma (Yoshimoto et al., 2013).
C57BL/6 and 129 mice are excellent models to study the different metabolic phenotypes that occur in response to a high-fat diet (HFD). C57BL/6J (B6J) mice from Jackson Laboratories (Jax) are obesity- and diabetes-prone, whereas 129S1/SvImJ (129J) mice from the same vendor are obesity- and diabetes-resistant (Almind and Kahn, 2004). 129S6/SvEvTac (129T) mice, which are genetically similar to 129J mice but bred at Taconic Farms, are obesity-prone but remain metabolically healthy (Fujisaka et al., 2016; Ussar et al., 2015). The better insulin sensitivity in 129T and 129J mice is explained in part by their genetically based anti-inflammatory potential (Fujisaka et al., 2016). However, some of the difference between strains is due to differences in their gut microbiotas (Ussar et al., 2015). When the gut microbiotas of three mouse strains are decreased by antibiotics, bacterially derived secondary bile acids, especially deoxycholic acid, are decreased both in the cecum and plasma, and this leads to attenuation of high fat diet-induced inflammation in B6J mice, improving insulin sensitivity. But, among these three strains, this only occurs in B6J mice.
There are many classes of bacterial metabolites in addition to bile acids that can be modified by factors such as diet and antibiotic treatment (Brown and Hazen, 2017; Nieuwdorp et al., 2014). Many of these metabolites are absorbed into the circulation, where they can act directly or be further metabolized by the host, leading to bioactive compounds that can act on tissues and affect the host metabolism (Wikoff et al., 2009). In the present study, we have dissected the complex relationship between host microbiota, genetic background, and environmental factors by performing untargeted metabolomics analysis of the plasma and cecal contents of B6J, 129J, and 129T mice on chow, a HFD, and a HFD supplemented with either vancomycin or metronidazole and correlated these data with physiological responses. We have used phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) analysis and untargeted liquid chromatography-mass spectrometry (LC-MS) metabolomics to assess gut microbiota and systemic metabolic changes in response to these manipulations. We found that different classes of metabolites exhibit unique host- and microbiome-dependent changes in both the cecum and plasma and that a number of metabolites are positively or negatively associated with insulin resistance across strains, indicating an important role for the metabolome as an integrator of the effects of diet, genetics, and the microbiome.
RESULTS
Diet, Genetics, and Antibiotics Affect Gut Microbial Structure
To assess the effects of diet and antibiotics on the gut microbiota and metabolism, 6-week-old normal chow-fed (22% fat by calories) B6J, 129J, and 129T mice were given drinking water (placebo) or drinking water containing vancomycin (1 g/L) or metronidazole (1 g/L). One week later, both antibiotic-treated groups and half of the control mice were challenged with an HFD (60% fat by calories) for 4 weeks. Consistent with previous reports (Fujisaka et al., 2016; Ussar et al., 2015), body weight gain on the chow diet was greater in B6J mice than that in 129J mice, and 129T mice fell in between (Figure 1A). HFD feeding increased body weight gain, with its greatest effects in B6J > 129T > 129J, and this was not significantly affected by either vancomycin or metronidazole (Figure 1A). Blood glucose was higher in B6J mice than in either 129 strain and modestly increased by 4 weeks of HFD feeding; this did not quite reach statistical significance (Figure 1B) but does so after longer periods of HFD challenge (Fujisaka et al., 2016). We have previously shown, in 8- to 12-week studies, that antibiotics improve insulin resistance in HFD-fed B6J mice (Fujisaka et al., 2016), and, in these short-term cohorts, insulin levels and insulin resistance (homeostatic model assessment for insulin resistance [HOMA-IR]) were higher in B6J than 129 mice on a HFD and tended to be reduced by vancomycin treatment in B6J mice Figures 1C and S1).
Figure 1. Effect of Diet, Genetics, and Antibiotics on Phenotype and the Gut Microbial Community.
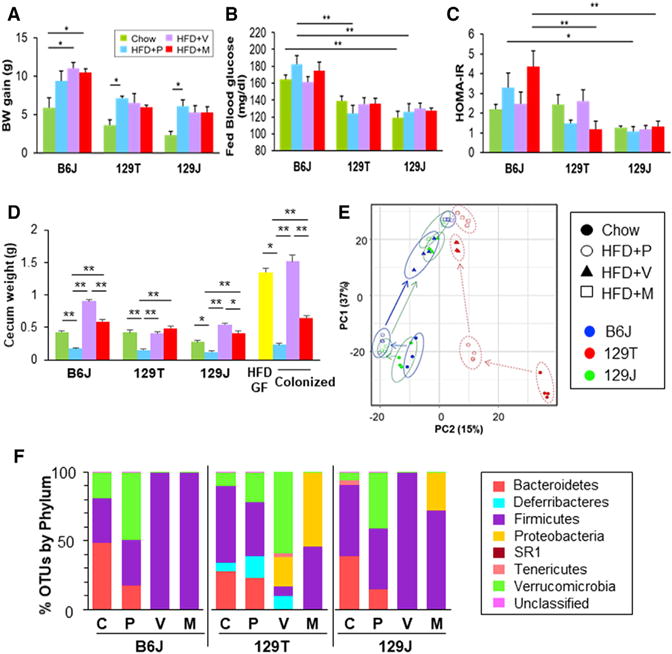
(A) Weight gain of mice on chow (green), HFD (blue), HFD+vancomycin (purple), and HFD+ metronidazole (red) from age 7 to 11 weeks (n = 3–4/group). *p < 0.05 by ANOVA, followed by Tukey-Kramer post hoc test.
(B) Blood glucose levels in the fed state at 11 weeks of age.
(C) Calculated HOMA-IR (n = 3–4/group).
(D) Cecum weight of B6J, 129T, and 129J mice on chow, HFD, HFD+vancomycin, or HFD+ metronidazole (n = 3–4/group). Right: cecal weight of HFD-fed GF B6 mice (yellow) and GF B6 mice colonized with bacteria from B6J mice on either an HFD, HFD+vancomycin, or HFD+ metronidazole, weighed 2 weeks after transfer (n = 3/GF, n = 8–9/colonized groups). *p < 0.05, **p < 0.01 by ANOVA, followed by Tukey-Kramer post hoc test. In (A)–(C), results are shown as the mean ± SEM.
(E) Principal component analysis (PCA) of cecal 16S rRNA sequencing data for B6J, 129T, and 129J mice on chow, HFD, HFD+vancomycin, or HFD+metronidazole at 11 weeks of age.
(F) Representation of bacterial phyla in the cecal bacteria of mice from each group (n = 3–4) at 11 weeks of age (4 weeks on the chow diet or HFD, 5 weeks on antibiotics).
It has been previously shown that cecum size is increased in germ-free mice (Jakobsdottir et al., 2013). Interestingly, HFD feeding markedly reduced the weight of the cecum in all strains of mice, and this was restored to chow diet levels or above by treatment with either antibiotic (Figure 1D). This effect on cecum size could be reproduced by microbiome transfer to germ-free mice. Thus, the cecum size in HFD-fed germ-free B6J mice was reduced by ~80% following colonization with fecal material from HFD conventional mice but remained large in germ-free B6 mice colonized with gut bacteria of HFD-fed mice treated with vancomycin or metronidazole (Figure 1D).
Principal-component analysis of 16S rRNA sequence data of cecal contents showed clear differences in community structure between the different experimental groups (Figure 1E), with both HFD and antibiotic treatment having a strong effect on the bacterial structure, with lesser but clear differences among strains. At the phylum level, the relative abundance of Firmicutes to Bacteroidetes was higher in both chow-fed 129 strains compared to B6J mice (Figure 1F). HFD feeding reduced Bacteroidetes in all strains of mice. Both antibiotics resulted in elimination of most of the bacteria, except for Firmicutes in B6J and 129J mice, whereas, in 129T mice, considerable Proteobacteria and Verrucomicrobia remained.
Microbial Modifications Affect Predicted Functional Pathways in the Microbiome
To understand the potential implications of the different bacterial communities, we performed PICRUSt analysis, a computational approach that predicts the functional composition of the bacterial metagenome using 16S rRNA data (Allegretti et al., 2016). As shown in Figure 2A, dynamic and consistent changes in Kyoto Encyclopedia pf Genes and Genomes (KEGG) metabolic pathways were observed in the different strains of mice depending on diet, genetic background, and antibiotic treatment. For example, carbohydrate metabolism, pyrimidine metabolism, starch and sucrose metabolism, cyanoamino acid metabolism, phenylpropanoid biosynthesis, tyrosine metabolism, pyruvate metabolism, and gluconeogenesis pathways were markedly upregulated by vancomycin treatment of HFD-fed mice but not by metronidazole treatment (Figure 2A). Interestingly, this effect occurred only in B6J and 129J mice and was not observed in 129T mice. Likewise, phenylalanine, tyrosine, and tryptophan biosynthesis pathways were decreased by vancomycin in HFD-fed B6J and 129J mice but not in HFD-fed 129T mice. By comparison, pathways of galactose metabolism and arginine and proline metabolism were downregulated by both antibiotics in all strains of mice. Metronidazole treatment increased aldehyde dehydrogenase, branched-chain amino acid degradation, butanoate metabolism, and fatty acid metabolism pathways in all strains, but the effect was greatest in B6J > 129J > 129T. Even with the HFD alone, a variety of metabolic pathways were regulated in a strain-dependent manner (Figure S2). Thus, changes in bacterial communities produced by HFD and antibiotic treatment have the potential to change on multiple microbial metabolic pathways, and these effects are different in these three strains of mice.
Figure 2. Modifications of the Gut Microbiota Affect Predicted Functional Metabolic Pathways.
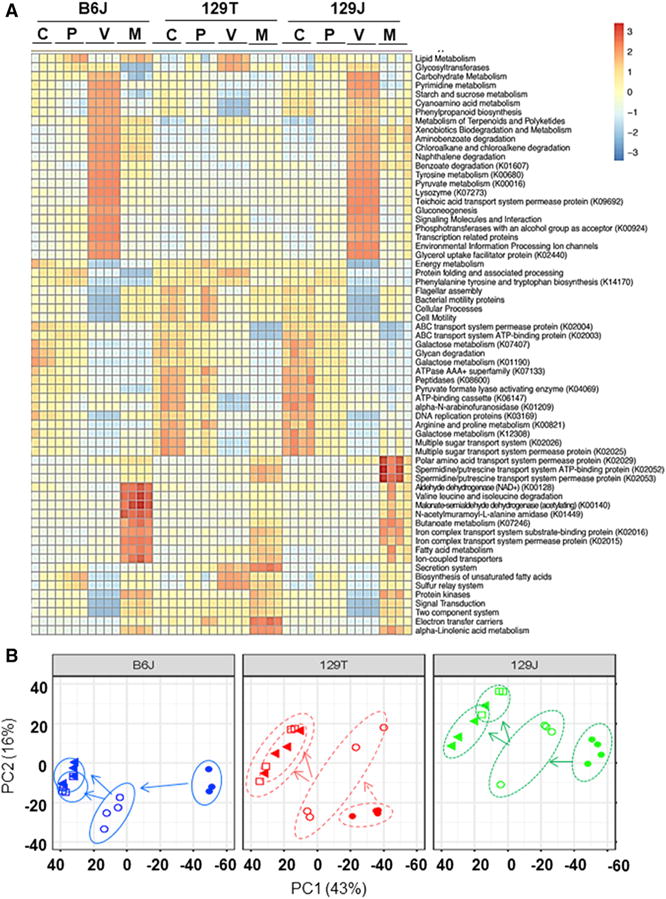
(A) Heatmap of metabolic pathways of each group obtained from PICRUSt analysis of 16S rRNA sequencing data. C, chow placebo; P, HFD+placebo; V, HFD+vancomycin; M, HFD+ metronidazole.
(B) PCA of identified metabolites in the cecum for B6J, 129T, and 129J mice on either chow, HFD, HFD+vancomycin, or HFD+metronidazole at 11 weeks of age.
Cecum and Plasma Metabolites Are Altered by Gut Microbial Modification
To assess the effects of these changes in predicted metabolic pathways of the gut microbiota, we performed an untargeted metabolomic analysis of cecal contents and plasma using a panel of LC-MS protocols. In the cecum, a total of 49,712 reproducible peaks or features were detected. Because some metabolites and contaminants may produce several peaks, this likely represents ~20,000–25,000 different low-molecular-weight (most < 1 kDa) molecules. Of these, 482 corresponded to previously identified metabolites, and the remaining represent an estimated 20,000+ unknown molecules. In the plasma, 19,627 peaks were detected, of which 374 were previously identified metabolites, and ~11,500 were unknown molecules (Table S1). Despite being on identical diets and in the same vivarium, principal-component analysis (PCA) of cecal metabolites showed a clear separation among the three strains and between the diet-and antibiotic-treated groups (Figure 2B).
Heatmaps showing 75 of the most changed known metabolites in the cecum with their parallel changes in the plasma are shown in Figures 3A and 3B. Changes in response to diet were generally larger in the cecum than in the plasma (note the scales). As expected, a high-fat diet markedly increased the levels of multiple bile acids in the cecum in all strains of mice, and this was largely reversed by treatment with either vancomycin or metronidazole (Figure 3A). In the plasma, chow and HFD mice had similar levels of most bile acids, but antibiotic treatment did lower the levels of the secondary bile acid taurodeoxycholic acid, reflecting the change in the cecum (Figure 3B).
Figure 3. Modifications of the Gut Microbiota Affect Cecal and Plasma Metabolites.
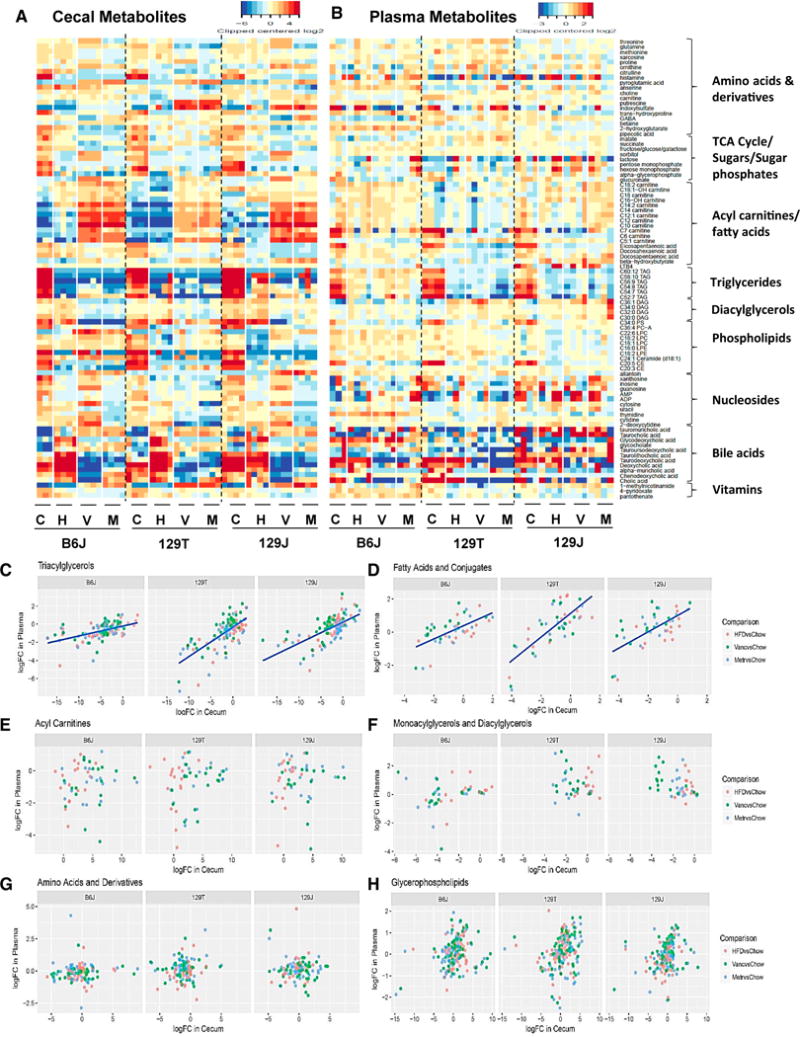
(A and B) PCA of cecal (A) and plasma (B) metabolites for B6J, 129T, and 129J mice on chow, HFD, HFD+vancomycin, or HFD+metronidazole. Shown is the correlation of the log-fold change in response to HFD between cecum contents and plasma.
(C–H) Metabolic classes of (C) triacylglycerols, (D) fatty acids and conjugates, (E) acyl carnitines, (F) monoacylglycerols and diacylglycerols, (G) amino acids and derivatives, and (H) glycerophospholipids are shown.
For some metabolites, the changes in the plasma in response to diet or antibiotics correlated with those in the cecum, but this varied by both metabolite and by strain. For example, changes in triacylglycerols (Figure 3C) and fatty acids (Figure 3D) in the plasma correlated well with changes in the cecum, suggesting that absorption of these metabolites by the intestine is a significant determinant of plasma level, although there were differences in the slope of this relationship by strain and metabolite. By contrast, acyl carnitines, mono- and diacyl-glycerols, glycerophospholipids, and amino acids showed no significant correlations between changes in the cecum and changes in the plasma when considered as a class (Figures 3E–3H).
Not unexpectedly, a high-fat diet affected lipid metabolites in the cecum, but, surprisingly, the most dramatic changes were decreases in many high-molecular-weight polyunsaturated triacylglycerols (C52:7 to C60:12), C36:4 phosphatidylcholine (PC), and C20:5 and C20:3 cholesterol ester (CE), and these were not further affected by antibiotic treatment. Both diet and antibiotics also had a considerable effect on these metabolites in the plasma. One of the most dramatically changed classes of lipids was the acylcarnitines. In the cecum, virtually all acylcarnitines, especially short-chain (C5-C9) acylcarnitines, showed modest increases on HFD and even more dramatic increases following antibiotic therapy, indicating effects of the changing microbiome on lipid metabolism in the gut (Figures 3A, 3E, and S3F–S3I). By contrast, in the plasma, the short-chain acylcarnitines decreased on an HFD, and there was little effect of antibiotics (Figures 3B and S3F–S3I).
Some changes depended on the strain or breeding site of the mouse and the resulting differences in bacterial composition. For example, both antibiotics elevated allantoin in the cecum but had a reverse effect in the plasma uniquely in Jax-bred mice (Figures 3A, 3B, and S3A). Likewise, both B6J and 129J mice showed a marked decrease in cecal γ-aminobutyric acid (GABA) levels in response to vancomycin and an increase by metronidazole, which was not seen in 129T mice (Figures 3A, 3B, and S3B). Both B6J and 129J mice also showed an increase in cecal threonine by vancomycin and a decrease by metronidazole, which was not observed in 129T mice (Figures S3C and S3D). On the other hand, antibiotic treatment of HFD-fed mice produced a decrease in asparagine in only 129T Tac-derived mice. None of the latter changes were observed in the plasma, suggesting that gut bacterial metabolism is not the primary driver controlling the plasma levels of these metabolites (Figure S3E). Hexose (fructose/glucose/galactose) levels in the cecum were decreased by an HFD and partially rescued by vancomycin in B6J and 129J mice but not in 129T mice, which matched the PICRUSt analysis; however, smaller changes were observed in plasma levels (Figures 3B and S3J).
Robust Metabolite Responses in HFD Antibiotic-Treated Mice
The complete dataset of known metabolites in the cecum and plasma and their response to diet/antibiotics is available at http://www.metabolomicsworkbench.org and highlighted in Figure 4. Compared with chow-fed mice, the levels of pyrimidine metabolites (uridine, thymidine, cytosine, and 2-deoxcytidine) in the cecum were decreased by an HFD in both Jax-derived strains, and this decrease was reversed by vancomycin treatment but potentiated by metronidazole treatment (Figure 4A). These changes matched the PICRUSt predictions (compare Figures 3A and 2A). However, the plasma levels did not reflect the cecum levels. Thus, metronidazole treatment decreased the levels of uridine in the cecum but increased the levels in the plasma. The HFD also induced large decreases in cecal levels of AMP, ADP, and cytidine monophosphate (CMP) and nucleosides such as inosine (Figures 4A, S3K, and S3L). These changes were most dramatic in B6J and 129J mice and reversed by vancomycin but not metronidazole. Interestingly, an HFD also significantly increased cyclic AMP (cAMP) levels in the cecum in all strains of mice, and these were further increased by vancomycin treatment (Figure 4A). Although some of these changes were observed in the plasma, overall, the differences were small.
Figure 4. Robust Metabolite Responses to HFD and Antibiotic Treatment across Three Strains of Mice.
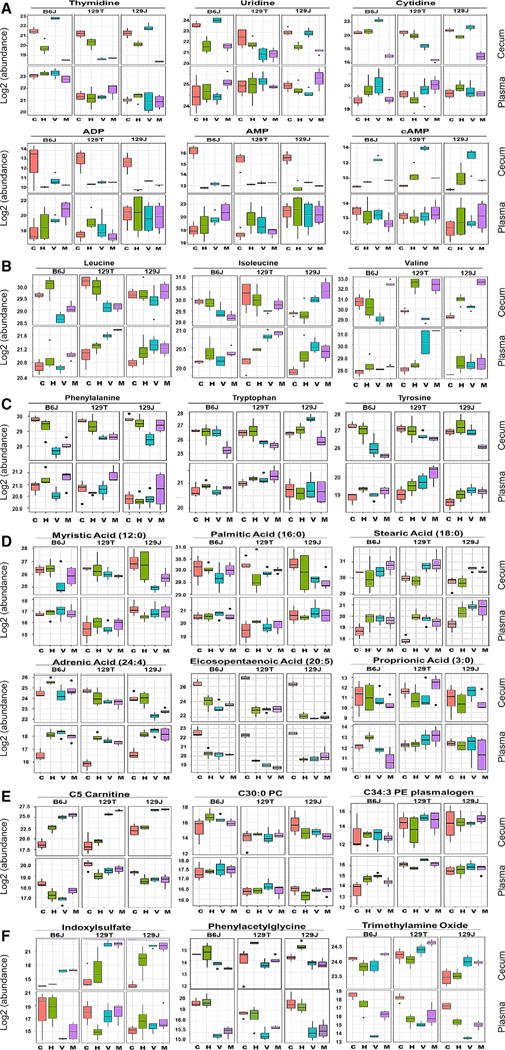
(A–F) Shown are comparisons of metabolites between the cecum (top) and plasma (bottom) in each group. Mice were 11 weeks of age (4 weeks on the chow diet or HFD, 5 weeks on antibiotics). Pink, chow+placebo, green, HFD+placebo; blue, HFD+vancomycin; purple, HFD+ metronidazole.
The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and are plotted individually.
As reported previously (Newgard et al., 2009), branched-chain amino acids (BCAAs) such as valine, leucine, and isoleucine were elevated in the plasma by an HFD. This occurred in all strains despite differences in propensity to obesity or insulin resistance and despite variable effects on cecal levels of BCAAs (Figure 4B). The effects of antibiotics to modify this response were strain- and amino acid-dependent. Thus, vancomycin decreased all BCAAs in the cecum, especially in the two obesity-prone strains (B6J and 129T), whereas metronidazole tended to decrease the levels of leucine and isoleucine and increase the levels of valine (Figure 4B). Interestingly, both antibiotics increased the levels of BCAAs in the plasma of all mice, despite the fact that antibiotics improved insulin sensitivity. With regard to aromatic amino acids, metronidazole treatment resulted in big decreases in phenylalanine, tyrosine, and tryptophan in the cecum of all mouse strains (Figure 4C), as predicted by PICRUSt analysis (Figure 2A). Despite the changes in the cecum, the levels of these metabolites in the plasma were elevated by metronidazole, suggesting that metronidazole may have additional effects on the absorption or turnover of these metabolites.
Fatty acids can have pro- or anti-inflammatory effects (Ertunc and Hotamisligil, 2016). An HFD had almost no effect on the levels of saturated fatty acids in the cecum, despite deriving 60% of calories from fat, mostly lard. An HFD accompanied by either antibiotic, but especially metronidazole, resulted in increased cecal levels of stearic acid (C18) but decreasing levels of palmitic (C16) and myristic acids (C14) (Figure 4D). In the plasma, a high-fat diet alone increased the levels of stearic acid in all strains, and this was not modified by antibiotic treatment. The effect of metronidazole on cecal fatty acids was predicted by PICRUSt analysis, but this analysis did not predict the effect of vancomycin. Ingestion of an HFD had variable effects on many of the unsaturated and short-chain fatty acids found in the cecum, but, except for adrenic acid (C24:4), these were largely unchanged by antibiotic treatment (Figure 4D). In the plasma, an HFD was associated with a major decrease in eicosopentanoic acid; this closely mirrored the changes in the cecum. By contrast, an HFD increased the plasma levels of adrenic acid independent of cecal content. The plasma levels of these unsaturated fatty acids were not influenced by antibiotic treatment. Overall, the changes in plasma fatty acid levels on an HFD reflected an increase in proinflammatory fatty acids, such as adrenic and stearic acid, and a decrease in anti-inflammatory fatty acids, such as eicosopentaenoic and docosohexanoic acids (Kuda et al., 2016; Yamada et al., 2017). However, antibiotic administration had little effect on these changes in free fatty acids in B6J mice despite improving insulin sensitivity, and the changes in free fatty acid (FFA) were similar in the insulin-sensitive 129 substrains, thus disconnecting the FFA levels from the level of insulin resistance.
The plasma levels of metabolites also showed a dynamic variation in response to strain, diet, and antibiotics, even for metabolites that were not changed or not changed in the same direction in the cecum. For example, short-chain fatty acids are well-known bacterial metabolites (Koh et al., 2016), but propionate levels were unchanged in the cecum of any strain by HFD alone, but the HFD increased propionate in the plasma of B6J mice. Both antibiotics decreased propionate in the cecum and plasma in B6J mice but had variable effects in 129 mice (Figure 4D), indicating differences dependent on genetic background, the site (vendor) where the mice were born, as well as diet and antibiotic treatment. The plasma levels of C34:3 phosphatidylethanolamine (PE) plasmalogen and C5-carnitine were 4-fold higher in both strains of 129 mice compared with B6J mice under all conditions, whereas C30:0 PC was higher in B6J compared with 129 mice (Figure 4E), indicating that genetic background/strain is an important factor affecting plasma metabolite levels.
Other plasma metabolites were mildly or not affected by diet but dramatically affected by antibiotics, indicating that they are likely direct or indirect products of intestinal bacterial metabolism. For example, indoxylsulfate, a bacterial metabolite that acts as a uremic toxin, was increased in the cecum of all three strains by both antibiotics, whereas plasma levels in B6J but not 129 mice decreased with antibiotics (Figure 4F). Both plasma and cecal levels of phenylacetylglycine were decreased about 4-to 8-fold by both antibiotics in all strains (Figure 4F). In contrast, the plasma levels of trimethylamine-N-oxide, another bacterial metabolite that has been linked to cardiovascular disease risk (Tang et al., 2013; Wang et al., 2011b), were markedly decreased by vancomycin, but not metronidazole, in B6J and 129J mice, whereas cecal levels showed no consistent pattern (Figure 4F). These results indicate that, to the extent that gut microbiota affect levels of plasma metabolites, this not only varies in a host-dependent manner but may also involve processes in regions of the intestine other than the cecum.
Metabolic Pathway Analysis of Plasma and Cecal Contents
To more completely understand the relationship between the cecal and plasma metabolite profiles and the gut microbiome, we focused on metabolic pathways for histidine and the aromatic amino acids tyrosine, tryptophan, and phenylalanine. An analysis of the histidine pathway is shown in Figure 5A, with the levels of cecal metabolites highlighted with blue boxes and the data for the same metabolites in the plasma highlighted in red.
Figure 5. Pathways for Aromatic Amino Acid Metabolism Illustrating Plasma and Cecal Contents of Each Metabolite.
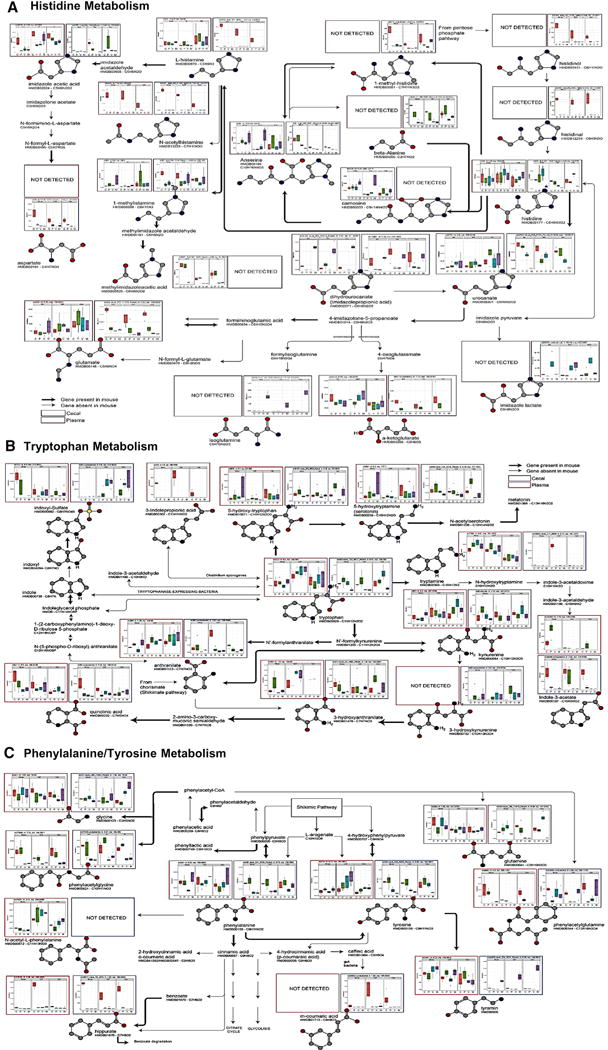
(A–C) Analysis of metabolites in (A) the histidine pathway, (B) the tryptophan pathway, and (C) the phenylalanine and tyrosine pathway. Levels of metabolites in the cecum are shown in blue boxes and those in plasma in red boxes. Thick black arrows indicate the pathway is present in the mouse, and gray arrows indicate the pathway is absent in the mouse.
The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and are plotted individually.
In the cecum, histidine levels were not affected by an HFD, but in mice from Jax (B6J and 129J), histidine was increased by vancomycin and decreased by metronidazole treatment. By contrast, in 129T mice, histidine was decreased by both antibiotics. L-histamine, N-acetyl-histamine, histadinal, histadinol, 1-methyl-histamine, and anserine in the cecum were all decreased by an HFD and not rescued by either antibiotic. Of these, only L-histamine and anserine can be made in mammalian cells and were found in the plasma, and both tended to go up, not down. However, N-acetylhistamine showed identical patterns in the plasma and cecum, suggesting that the levels of this metabolite in the plasma are purely derived from gut microbial metabolism.
Histidine can also be converted to urocanate and imidazole derivatives by both bacteria and, to some extent, mammalian cells (Figure 5A). In the cecum, urocanate showed a dramatic decrease with both antibiotics, whereas dihydrourocanate (imidazole proprionate), a product of microbial but not murine metabolism, showed a variable decrease with an HFD but rose to above chow diet levels in HFD-fed mice on metronidazole, and this was mirrored almost exactly in the plasma. Urocanate can be converted to glutamic acid, isoglutamate, and alpha-ketoglutarate, but each of these showed unique patterns. Thus, the cecal and plasma levels of alpha-ketoglutarate were decreased by the HFD and further decreased in the cecum by both antibiotics, whereas the plasma levels of alpha-ketoglutarate were rescued by antibiotics. Isoglutamate in the cecum was selectively increased by metronidazole treatment in all strains of mice, indicating a role of gut microbes resistant to metronidazole in the production of this metabolite; however, isoglutamate was not detected in the blood (Figure 5A). Thus, among histidine metabolites, only the blood levels of N-acetylhistamine and dihydrourocanate, and to some extent alpha-ketoglutarate, appear to be determined primarily by what is made or available in the gut.
Aromatic amino acids have been linked to insulin resistance (Chen et al., 2016; Wang et al., 2011a) and showed multiple pathway-specific changes (Figure 5B). Tryptophan levels in the cecum were decreased by metronidazole in all strains and by vancomycin in 129T mice, indicating the role of gut microbiota in tryptophan metabolism; however, this was not reflected by changes in the plasma. On the other hand, indole 3-acetate in the cecum decreased moderately with an HFD and markedly with both antibiotics, and this was mirrored in the plasma, indicating a strong dependence of this metabolite on the gut microbiota. 5-Hydroxytryptophan was increased in the cecum and plasma by an HFD, and anthranilate in the cecum and plasma were reduced in all vancomycin-treated groups. Likewise, although 3-indoleproprionic acid was not detected in the cecum, in the plasma, it was reduced to undetectable levels on an HFD or an HFD with antibiotics (Figure 5B), suggesting that different tryptophan metabolites are regulated by the gut microbiota, but this affects the plasma levels for only some of these.
Phenylalanine and tyrosine metabolites are illustrated in Figure 5B. In the cecum, phenylalanine was unchanged by an HFD but decreased by both antibiotics, indicating a role for the gut microbiota, whereas in the plasma, phenylalanine was decreased by vancomycin in B6J mice and increased by metronidazole in all strains. On the other hand, phenylacetylglycine in the cecum was markedly decreased by both antibiotics in all strains, consistent with its role as a known gut microbial metabolite, with virtually identical changes in the plasma. Hippurate was markedly decreased in the cecum and plasma by an HFD in all strains but not rescued by antibiotics. Tyrosine levels in the cecum were decreased by vancomycin in B6J and 129T mice and by metronidazole in all three strains but increased in the plasma. Tyramine levels in the cecum were decreased by an HFD, further decreased by vancomycin, and increased to above chow levels by metronidazole, but these occurred with no consistent changes in the plasma. Thus, although the intestinal levels of phenylalanine, phenylacetylglycine, phenylacetyl-glutamine, tyrosine, and tyramine are dependent on the gut microbiota, gut metabolism is the primary driver of blood levels of phenylacetylglycine and, to some extent, hippurate.
Many Metabolites Are Associated with Insulin Resistance
To investigate the potential role of the changes in metabolite levels with insulin resistance, we performed a Spearman correlation analysis of plasma metabolite abundances to insulin resistance scores across mouse diet and treatment groups (Supplemental Experimental Procedures). Using this approach, the plasma levels of a number of metabolites showed strong positive correlations with insulin resistance, including aminoadipate, alpha-hydroxybutyrate, acetylglycine, C16-carnitine, N-carbamoyl-beta-alanine, thymidine, carnosine, 4-pyridoxate, C34:4 PC, and C30:0 PC (Figure 6A). On the other hand, adipate, C34:2 PC plasmalogen, C36:2 PC plasmalogen, C38:6 PC plasmalogen, C58:6 triacylglycerol (TAG), C58:7 TAG, taurolithocholic acid, and guanidinoacetate all showed negative correlations with insulin resistance (Figure 6B). Importantly, 2-aminoadipoate, alpha-hydroxybutyrate, and N-acetylglycine have also been previously identified in humans as potential biomarkers for diabetes risk and insulin resistance (Gall et al., 2010; Menni et al., 2013; Wang et al., 2013), as have patterns of lipids with lower fatty acyl carbon number and double bond content (Rhee et al., 2011).
Figure 6. Metabolites Correlated with Insulin Resistance across Diets and Antibiotic Treatment.
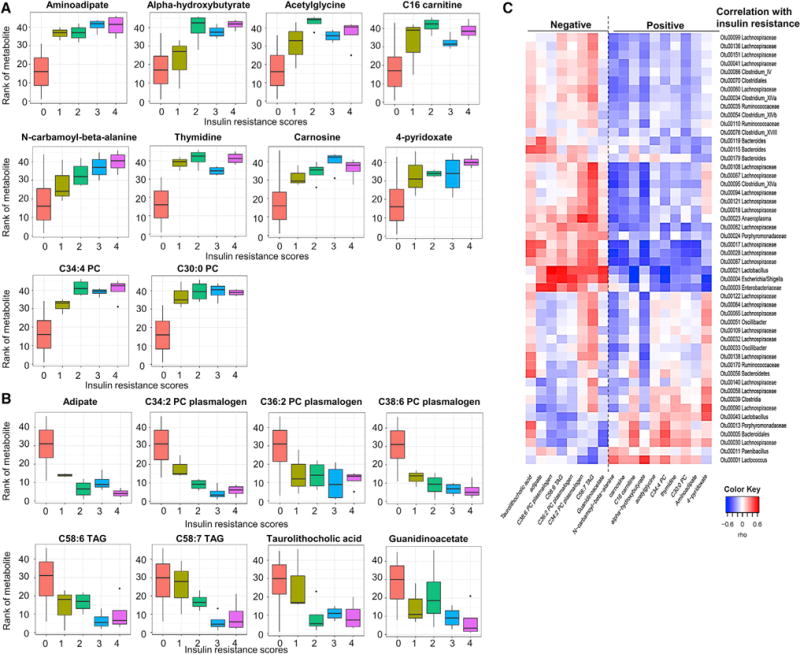
(A and B) Plasma metabolite levels that correlated with insulin resistance score positively (A) or negatively (B). Insulin resistance score were determined as described in the Supplemental Experimental Procedures.
(C) Heatmap showing OTUs that correlate with metabolites linked positively or negatively to insulin resistance. Rho in the color key represents the Spearman rank correlation coefficient.
The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and are plotted individually.
The levels of the metabolites linked to insulin resistance correlated with specific bacterial operational taxonomic units (OTUs) (Figure 6C). Most of the OTUs that were highly correlated with insulin resistance were Firmicutes of the order Clostridiales and the family Lachnospiraceae. Lachnospiraceae have been identified as over-represented in the gut microbiome of obese mice, and colonization of germ-free obese mice with Lachnospiraceae induces hyperglycemia (Kameyama and Itoh, 2014). Lachnospiraceae have also been shown to affect short- and long-chain fatty acid synthesis (Zhang and Davies, 2016). Other Clostridiales, such as Clostridium XIVa, showed a positive correlation with C58:7 TAG, a metabolite negatively correlated with insulin resistance and negative correlation with N-carbamoyl-beta-alanine and alpha-hydroxybutyrate, which correlates positively with insulin resistance. Adipate, which is negatively linked to insulin resistance, on the other hand, positively correlated with three Bacteroides species (Otu00115, Otu00119, and Otu00179). Thus, many metabolites linked with insulin resistance correlate with specific gut microbiotas, but these correlations show a complex pattern.
Dynamic Changes in Unknown Plasma Metabolites Detected by Untargeted Metabolomics
In addition to the ~400 identified metabolites profiled in the plasma using untargeted LC-MS, there were approximately 20,000 reproducible unknown peaks, and many of these showed dramatic changes in response to diet or antibiotics. Figure 7A shows a heatmap of the top 1,066 peaks that change significantly in the plasma under at least one condition (note the truncated log2 scale with many changes several log2 orders of magnitude different). Multiple interesting groups were identified, including unknowns that were markedly increased or decreased by an HFD or by one or both antibiotics. Other metabolites were markedly different in level among the three strains of mice. Using the METLIN metabolomics database (https://metlin.scripps.edu) (Smith et al., 2005) and reference standards, we have begun to identify a few of these. One unidentified peak that was uniquely increased in the three metronidazole-treated groups of mice corresponded by mass-to-charge ratio to metronidazole itself (Figure S4A), demonstrating the ability of this approach to identify unknowns. Another peak that was detected in both the cecum and plasma was identified as imidazole propionate, part of the histidine pathway (Figures 5 and S4B). Imidazole propionate in the plasma was decreased by ~80% in mice on an HFD, and this was corrected by metronidazole, but not vancomycin, in both the cecum and plasma, consistent with the notion that changes in intestinal bacteria contribute to the change in plasma level. Three other features have been identified as N-acetyllysine, cortexolone, and 5-hydroxy-4-metyluracil (Figure S4C).
Figure 7. Dynamic Changes in Unknown Plasma Metabolites.
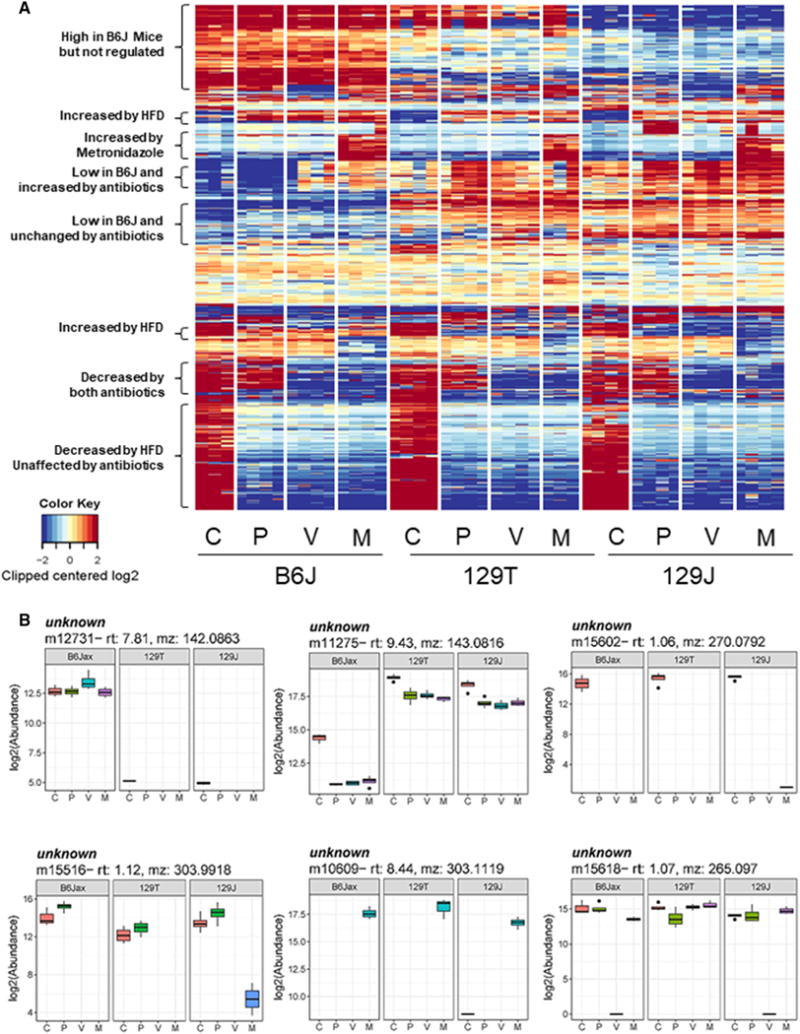
(A) Heatmap of the top 1,066 still-unidentified peaks that are significantly changed in the plasma in response to diet, antibiotics, or strain differences.
(B) Representative examples of unknown peaks that show features of diet, strain, and antibiotic dependence. The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and are plotted individually.
Although most of the unknowns remain to be identified, it is important to note that many of these changed more than 26-fold (64-fold) in response to diet and/or antibiotic treatment, and many show equally large differences among the three strains of mice (Figure 7A). Representative examples that exhibit strong biological correlates are shown in Figure 7B. For instance, metabolite m15618 was unaffected by an HFD but decreased in the plasma of B6J and 129J mice by 215-fold (32,768-fold) with vancomycin but not metronidazole treatment. Clearly, identifying these highly regulated metabolites will lead to greater insight into how the gut microbiome can affect systemic metabolism.
DISCUSSION
Microbiotas in the gastrointestinal track are seeded just after birth. Although the composition is influenced by both host genetics and environmental factors, the gut microbiome can be re-modeled throughout life, depending on many factors, including diet, antibiotics, and gastrointestinal disease (Schroeder and Bäckhed, 2016; Tamburini et al., 2016). Gut microbiotas play important roles in maintaining host homeostasis by aiding in metabolism of indigestible components of the diet, gut development and homeostasis, immune cell development, and protection from colonization by pathogenic bacteria. Gut microbiotas also produce and degrade various metabolites that can be taken up and affect the host. For example, primary bile acids are converted to secondary bile acids by gut microbiotas, and these secondary bile acids not only aid in fat absorption, but they also are reabsorbed into the circulation, where they serve as ligands for the bile acid receptors farnesoid X (FXR) and TGR5 on host cells, leading to effects on energy metabolism and the immune system (Gadaleta et al., 2011; Jiang et al., 2015; Thomas et al., 2009). Similarly, bacterially produced short-chain fatty acids such as acetate, butyrate, and propionate not only serve as important energy sources for the intestinal epithelium and the liver but can also can modify insulin secretion, immune system function, appetite, and adipose function (Bouter et al., 2017; Canfora et al., 2015; Holmes et al., 2012; Perry et al., 2016). It is not surprising, therefore, that changes in the communities of organisms in the intestine can contribute to the pathogenesis of metabolic diseases, including obesity, type 2 diabetes, and the metabolic syndrome (Mikkelsen et al., 2015; Schroeder and Bäckhed, 2016).
We and others have previously shown that different strains of mice and mice from different vendors exhibit different rates of obesity and diabetes when challenged with a high-fat diet (Parks et al., 2015; Ussar et al., 2016). On an HFD, C57BL/6J mice from Jax (B6J) and 129 mice from Taconic Farms (129T) are obesity-prone, whereas 129 mice from Jax (129J) are obesity-resistant. On the other hand, when made obese, only B6J mice develop insulin resistance with diabetes and metabolic syndrome, whereas 129T mice remain insulin-sensitive and non-diabetic. This difference between 129 mice from Jax and Taconic is, at least in part, due to differences in the gut microbiome present in these mice from these two vendors because the difference can be minimized by breeding the mice in a common facility for three generations (Ussar et al., 2015). Likewise, some of the differences between B6J and 129T mice appear to be due to influences of the gut microbiota because transfer of microbiotas from these two strains to germ-free mice can transfer some aspects of the phenotypic difference. More recently, we have shown that modifying gut microbiotas with vancomycin or metronidazole can improve HFD-induced inflammation and insulin resistance both at the physiological and signaling level in C57BL/6J mice, whereas the same antibiotic treatment has no effect on either substrain of 129 mice. At least one contributor to these effects appears to be bile acids because the metabolic phenotype closely correlates with changes in plasma levels of deoxycholic acid and its anti-inflammatory bile acid receptor TGR5 (Fujisaka et al., 2016). However, bile acids are only one class of metabolites that can be modified by the presence of different gut bacteria.
In the present study, we have begun to dissect the complex relationship between metabolite changes, diet, host microbiota, and genetic background by performing an untargeted metabolomics analysis of the plasma and cecal contents as well as 16S rRNA sequence analysis of cecal contents from B6J, 129J, and 129T mice on chow or an HFD or HFD-treated with one of two antibiotics widely used in humans: vancomycin, a non-absorbable antibiotic that targets Gram-positive bacteria, and metronidazole, an absorbed antibiotic that targets anaerobic bacteria. In agreement with our previous study (Fujisaka et al., 2016), we find that, although an HFD and antibiotics produce shifts in the composition of the gut microbiota, each strain of mice shows a distinctly different pattern of microbes in response to these treatments. These differences are large enough to be detected with statistical significance even with a relatively small number, although one does need to be cautious about potential cage-related differences that can affect the microbiome and, thus, the metabolome.
One interesting and unexpected effect of the changing microbiome at the level of the gut is the effect on the cecum itself. It is known that mice receiving short-term antibiotics or germ-free mice show enlargement of the cecum (Furusawa et al., 2013; Savage and Dubos, 1968). This enlargement of the cecum is thought to be due to a defect in fermentation of dietary fiber, and, in the latter, can be reversed by bacterial recolonization (Jakobsdottir et al., 2013). In our study, the HFD decreased cecum size in all mouse strains, which was returned to or above normal by antibiotic treatment. This change in cecum size was reproduced by transfer of gut microbiotas, indicating that these changes in cecum size are mediated by the changing microbial composition. Although the mechanism of changing cecum size remains to be determined, the potential function effects of the different microbial communities in the three strains on different diets and antibiotics is apparent in a PICRUSt analysis of the 16S rRNA data that predicts effects of diet and antibiotics to upregulate and downregulate genes for many different metabolic pathways, often in a strain-dependent manner.
The complex interactions between diet, antibiotics, and host genetics on gut bacterial metabolism were even more apparent in the analysis of metabolites in the cecum of the mice. Indeed, of the over 20,000 known and unknown compound peaks detected in the LC-MS analyses, over 70% showed a highly significant (false discovery rate [FDR] < 0.01) change under one or more conditions in at least one strain of mice. Although it was impossible to assess these large number of metabolites in a truly quantitative manner (i.e., with standards for each metabolite), changes in many metabolites were more than 64-fold based on relative peak heights. A number of the metabolite changes in cecal contents matched well with the PICRUSt predictions. For example, the galactose metabolism pathway is mildly decreased by the HFD and further decreased by antibiotics. Another good prediction was observed for pyrimidine metabolites (uridine and cytosine), which are decreased by the HFD and restored by vancomycin treatment in Jax-derived mice but not Taconic Farms mice. Histidine levels in the cecum, on the other hand, are increased by vancomycin and decreased by metronidazole in Jax-derived mice and decreased by both antibiotics in 129T mice, reflecting intrinsic differences in the gut flora of these vendor-bred strains. The PICRUSt prediction also indicates decreased synthesis of aromatic amino acids in Jax mice on antibiotics with a lesser effect in 129T mice, and this agrees well with decreased levels of phenylalanine, tyrosine, and tryptophan in the cecum of antibiotic-treated mice, especially in B6J and 129J mice.
However, the relationship between plasma and cecal levels of metabolites is complex and depends on both the class of metabolite and the specific metabolites within a given class. For some metabolite classes, such as triacylglycerides and fatty acids, the response in the cecum is highly correlated with that in the plasma, indicating that these circulating metabolites are likely products of intestinal bacteria. For these metabolites, there is a clear potential role in crosstalk between the gut environment and systemic metabolism.
For most other classes of metabolites, the plasma levels do not correlate with cecal levels for the group as a whole, but many metabolites within a given class plasma level are still tightly linked to gut microbial metabolism. Among metabolites showing little correlation, for example, are C10–C14 carnitines, whose levels in the cecum are increased dramatically by antibiotics, with no changes being observed in the plasma. Likewise, histamine, fumarate, malate, monophosphate, AMP, ADP, and CMP levels are decreased in the cecum by an HFD, with no change or an increase in plasma levels. This suggests that intestinal metabolism is not the only or major source of these metabolites in plasma, although there are several other reasons why this may occur. First, we have assessed the levels of these metabolites only in the systemic circulation, and it is possible that some gut metabolites that enter the portal circulation are taken up or metabolized by the liver, reducing the magnitude of change in the systemic circulation. Another possible reason for a mismatch may be the fact that levels of intestinal metabolites were only analyzed in the cecum, and some of the relevant changes may be occurring elsewhere in the gut. It is also possible that some metabolites, such as lipids, are preferentially taken up into the lymphatic circulation before entering the blood. It is also important to keep in mind that changes in metabolite concentrations in the cecum may have physiological effects even when levels do not change in the plasma because some of these metabolites may act as neurotransmitters or regulatory molecules on the gut epithelium itself. In this regard, it is interesting to note that that cAMP and acetylcholine levels in the gut were significantly increased by vancomycin treatment, and cecal histamine levels were downregulated by an HFD, without parallel changes in the plasma. The potential biological consequences of these changes in cecal metabolites will need further study.
Among products of microbial metabolism, several bile acids, especially deoxycholic acid and taurodeoxycholic acid, are decreased in both the cecum and plasma following antibiotic treatment (Fujisaka et al., 2016, and this study). This is due to depletion of Clostridium clusters XI and XIVa, which have the bile acid 7α-dehydratase BaiE that converts primary bile acids to secondary bile acids. We have previously shown that antibiotic treatment also increases the level of the anti-inflammatory bile acid receptor TGR5 in the liver (Fujisaka et al., 2016), which, together with the change in bile acids, could contribute to decreasing the systemic inflammation induced by an HFD.
Dietary tryptophan is metabolized by the gut microbiota to indole and then to indoxylsulfate, which is absorbed into the circulation and eventually excreted in the urine. Interestingly, indoxylsulfate in the cecum is increased by an HFD and by both antibiotics in all three strains of mice, suggesting a role of vancomycin- and metronidazole-resistant bacteria in its production. On the other hand, plasma indoxylsulfate shows an increase with antibiotics only in 129J and 129T mice, whereas, in B6J mice, plasma indoxylsulfate decreased with antibiotic treatment. This implies another metabolic pathway of indoxyl sulfate that is unique to this strain of mice. Another bacterial metabolite reported to have a negative effect on glucose metabolism (Dambrova et al., 2016) that is decreased by antibiotics in the plasma is TMAO, and this occurs with little effect on the cecal levels of the metabolite. On the other hand, propionate, which has been reported to have a favorable effect on insulin resistance (De Vadder et al., 2014), is also decreased in antibiotic-treated mice despite their improved insulin sensitivity. These findings point to the complex changes in metabolites, which can affect metabolism.
Despite this complexity, Spearman correlation analysis of plasma metabolites reveals a number of metabolites that strongly correlate positively or negatively with insulin resistance across all strains of mice, diets, and antibiotics. Three of the metabolites identified to be positively correlated with insulin resistance have been previously associated with insulin resistance in humans. Thus, individuals with 2-aminoadipate levels in the top quartile have a more than 4-fold increased risk of future diabetes (Wang et al., 2013). On the other hand, administration of 2-aminoadipate lowers blood glucose levels in mice and enhances insulin secretion by β cells in vitro, suggesting that 2-aminoadipate is a both biomarker and a potential regulator of diabetes pathogenesis. Likewise, α-hydroxybutyrate and N-acetylglycine have also been reported as possible risk markers for type 2 diabetes mellitus (T2DM) in humans (Gall et al., 2010; Menni et al., 2013). The present study shows there is a larger family of metabolites that could be biomarkers or mediators of insulin resistance and/or type 2 diabetes mellitus (T2DM). There are other metabolites that negatively correlate with insulin resistance and could offer protective effects on metabolism. Many of these metabolites correlate well with the relative abundance of different bacterial taxa (OTUs), but many of the taxa that appear to be driving these metabolic changes are still poorly characterized. Thus, further work will be required to clarify the mechanisms of regulation of metabolites by these gut microbiota.
In summary, our data demonstrate that gut microbiota have dramatic effects on the nature and quantity of many metabolites in multiple chemical classes in the gut and that these changes are reflected to varying degrees in changes in plasma levels of the same metabolites. The changes at the level of the gut and blood are dramatically influenced by diet, exposure to antibiotics, genetic background, and site of original bacterial colonization. The differences between strains and sites of breeding are, in many cases, as great as the effects of diet and antibiotics. Although a number of known metabolites positively or negatively correlate with insulin resistance, there is even greater numbers of unknown metabolites that show equally strong differences. Understanding the full set of metabolites and their responses to perturbation will open new insights into how changes in the gut microbiome affect systemic metabolism and its alterations in diabetes and obesity.
EXPERIMENTAL PROCEDURES
See the Supplemental Experimental Procedures for detailed procedures. All mouse experiments were performed on males between 5 and 12 weeks of age, complied with regulations and ethics guidelines, and were approved by the International Animal Care and Use Committee (IACUC) of the Joslin Diabetes Center (97-05) and Harvard Medical School (05131).
Statistical Analysis
Statistical significance was evaluated using ANOVA and a post Tukey-Kramer test. A p value of less than 0.05 was considered significant.
Supplementary Material
Highlights.
Risk of diabetes and obesity in mice is associated with differences in gut bacteria
Effects of diet, host genetics, and the microbiome are reflected in the metabolome
18 plasma metabolites correlate with insulin resistance across strains and diets
More than 1,000 unidentified MS peaks are also regulated by diet, genes, and the gut microbiota
Acknowledgments
We thank Christie Penniman, Vladimir Yeliseyev, Qing Liu, and Kristina Kriauciunas for their technical assistance. This work was supported by grants from the NIH (R01DK031036 and R01DK033201 to C.R.K.) and a Mary K. Iacocca professorship (to C.R.K.). S.F. was supported by a Sunstar Foundation post-doctoral fellowship. The work was also supported by a Joslin DRC grant (P30DK036836), the Bioinformatics and Animal Physiology Cores, and the Harvard Digestive Diseases Center for Clinical and Translational Metagenomics at Brigham and Women’s Hospital.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the metabolomics data reported in this study are Metabolomics Workbench: ST000879 and ST000880. The accession number for the 16S rRNA datasets containing the analysis of gut microbiota is SRA: SRP132006.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.060.
AUTHOR CONTRIBUTIONS
S.F. designed and performed the experiments and wrote the manuscript. J.A.-P., J.M.D., H.P., and C.B.C. analyzed the metabolomics and 16S rRNA sequence data. A.K. performed the PICRUSt analysis. M.S., S.U., and E.A. contributed to discussions. N.L. and L.B. conducted the microbiome analysis. C.R.K. designed the experiments and wrote the manuscript. C.R.K. is the guarantor of this work and, as such, had full access to all data and takes responsibility for the integrity of the data and the accuracy of the data analysis.
DECLARATION OF INTERESTS
C.R.K. is a member of the Board of Directors and SAB of Kaleido Biosciences. A.K. is a co-founder and consultant for DeepBiome Therapeutics Inc. In both cases, the work presented here was a product of their academic laboratories. The other authors declare no competing interests.
References
- Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, Gerber GK, Bry L, Clish CB, Alm E, Korzenik JR. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment Pharmacol Ther. 2016;43:1142–1153. doi: 10.1111/apt.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almind K, Kahn CR. Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes. 2004;53:3274–3285. doi: 10.2337/diabetes.53.12.3274. [DOI] [PubMed] [Google Scholar]
- Bouter KE, van Raalte DH, Groen AK, Nieuwdorp M. Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology. 2017;152:1671–1678. doi: 10.1053/j.gastro.2016.12.048. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hazen SL. Targeting of microbe-derived metabolites to improve human health: The next frontier for drug discovery. J Biol Chem. 2017;292:8560–8568. doi: 10.1074/jbc.R116.765388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. 2015;11:577–591. doi: 10.1038/nrendo.2015.128. [DOI] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Zheng X, Ma X, Bao Y, Ni Y, Hu C, Rajani C, Huang F, Zhao A, Jia W, Jia W. Tryptophan Predicts the Risk for Future Type 2 Diabetes. PLoS ONE. 2016;11:e0162192. doi: 10.1371/journal.pone.0162192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E. Diabetes is Associated with Higher Trimethylamine N-oxide Plasma Levels. Exp Clin Endocrinol Diabetes. 2016;124:251–256. doi: 10.1055/s-0035-1569330. [DOI] [PubMed] [Google Scholar]
- De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Bäckhed F, Mithieux G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014;156:84–96. doi: 10.1016/j.cell.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57:2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaka S, Ussar S, Clish C, Devkota S, Dreyfuss JM, Sakaguchi M, Soto M, Konishi M, Softic S, Altindis E, et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J Clin Invest. 2016;126:4430–4443. doi: 10.1172/JCI86674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, Klomp LWJ, Siersema PD, Schipper MEI, Danese S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, Ryals JA, Milburn MV, Nannipieri M, Camastra S, et al. RISC Study Group α-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE. 2010;5:e10883. doi: 10.1371/journal.pone.0010883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Li JV, Marchesi JR, Nicholson JK. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012;16:559–564. doi: 10.1016/j.cmet.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Jakobsdottir G, Xu J, Molin G, Ahrné S, Nyman M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE. 2013;8:e80476. doi: 10.1371/journal.pone.0080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125:386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Itoh K. Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice. Microbes Environ. 2014;29:427–430. doi: 10.1264/jsme2.ME14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Coelho D, Blachier F. Review of the association between meat consumption and risk of colorectal cancer. Nutr Res. 2013;33:983–994. doi: 10.1016/j.nutres.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- Kuda O, Brezinova M, Rombaldova M, Slavikova B, Posta M, Beier P, Janovska P, Veleba J, Kopecky J, Jr, Kudova E, et al. Docosahexaenoic Acid-Derived Fatty Acid Esters of Hydroxy Fatty Acids (FAHFAs) With Anti-inflammatory Properties. Diabetes. 2016;65:2580–2590. doi: 10.2337/db16-0385. [DOI] [PubMed] [Google Scholar]
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. MetaHIT consortium Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- Lynch SV, Pedersen O. The Human Intestinal Microbiome in Health and Disease. N Engl J Med. 2016;375:2369–2379. doi: 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- Menni C, Fauman E, Erte I, Perry JRB, Kastenmüller G, Shin SY, Petersen AK, Hyde C, Psatha M, Ward KJ, et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes. 2013;62:4270–4276. doi: 10.2337/db13-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen KH, Frost M, Bahl MI, Licht TR, Jensen US, Rosenberg J, Pedersen O, Hansen T, Rehfeld JF, Holst JJ, et al. Effect of Antibiotics on Gut Microbiota, Gut Hormones and Glucose Metabolism. PLoS ONE. 2015;10:e0142352. doi: 10.1371/journal.pone.0142352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwdorp M, Gilijamse PW, Pai N, Kaplan LM. Role of the microbiome in energy regulation and metabolism. Gastroenterology. 2014;146:1525–1533. doi: 10.1053/j.gastro.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab. 2015;21:334–346. doi: 10.1016/j.cmet.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, Forslund K, Hildebrand F, Prifti E, Falony G, et al. MetaHIT Consortium Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee EP, Cheng S, Larson MG, Walford GA, Lewis GD, McCabe E, Yang E, Farrell L, Fox CS, O’Donnell CJ, et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J Clin Invest. 2011;121:1402–1411. doi: 10.1172/JCI44442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DC, Dubos R. Alterations in the mouse cecum and its flora produced by antibacterial drugs. J Exp Med. 1968;128:97–110. doi: 10.1084/jem.128.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079–1089. doi: 10.1038/nm.4185. [DOI] [PubMed] [Google Scholar]
- Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005;27:747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. 2016;22:713–722. doi: 10.1038/nm.4142. [DOI] [PubMed] [Google Scholar]
- Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61:364–371. doi: 10.2337/db11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng L, Bry L, Gordon JI, Kahn CR. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015;22:516–530. doi: 10.1016/j.cmet.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Fujisaka S, Kahn CR. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol Metab. 2016;5:795–803. doi: 10.1016/j.molmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011a;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011b;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Ngo D, Psychogios N, Dejam A, Larson MG, Vasan RS, Ghorbani A, O’Sullivan J, Cheng S, Rhee EP, et al. 2-Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest. 2013;123:4309–4317. doi: 10.1172/JCI64801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Umemoto T, Kakei M, Momomura SI, Kawakami M, Ishikawa SE, Hara K. Eicosapentaenoic acid shows anti-inflammatory effect via GPR120 in 3T3-L1 adipocytes and attenuates adipose tissue inflammation in diet-induced obese mice. Nutr Metab (Lond ) 2017;14:33. doi: 10.1186/s12986-017-0188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- Zhang LS, Davies SS. Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med. 2016;8:46. doi: 10.1186/s13073-016-0296-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.