

Abstract
GV1001 is a 16-amino acid peptide derived from the human telomerase reverse transcriptase (hTERT) protein (616–626; EARPALLTSRLRFIPK), which lies within the reverse transcriptase domain. Originally developed as an anticancer vaccine, GV1001 demonstrates diverse cellular effects, including anti-inflammatory, tumor suppressive and antiviral effects. In the present study, the radioprotective and antifibrotic effects of GV1001 were demonstrated through suppressing transforming growth factor-β (TGF-β) signaling. Proliferating human keratinocytes underwent premature senescence upon exposure to ionizing radiation (IR), however, treatment of cells with GV1001 allowed the cells to proliferate and showed a reduction in senescent phenotype. GV1001 treatment notably increased the levels of Grainyhead-like 2 and phosphorylated (p-)Akt (Ser473), and reduced the activation of p53 and the level of p21/WAF1 in irradiated keratinocytes. It also markedly suppressed the level of TGF-β signaling molecules, including p-small mothers against decapentaplegic (Smad)2/3 and Smad4, and TGF-β target genes, including zinc finger E-box binding homeobox 1, fibronectin, N-cadharin and Snail, in irradiated keratinocytes. Furthermore, GV1001 suppressed TGF-β signaling in primary human fibroblasts and inhibited myofibroblast differentiation. Chromatin immunoprecipitation revealed that GV1001 suppressed the binding of Smad2 on the promoter regions of collagen type III α1 chain (Col3a1) and Col1a1. In a dermal fibrosis model in vivo, GV1001 treatment notably reduced the thickness of fibrotic lesions and the synthesis of Col3a1. These data indicated that GV1001 ameliorated the IR-induced senescence phenotype and tissue fibrosis by inhibiting TGF-β signaling and may have therapeutic effects on radiation-induced tissue damage.
Keywords: GV1001, human telomerase reverse transcriptase, ionizing radiation, fibrosis, transforming growth factor-β signaling
Introduction
Human telomerase reverse transcriptase (hTERT) is highly expressed in almost all types of cancer and maintains the integrity of telomere sequences required for an immortal phenotype (1,2). Due to this association between the level of hTERT and cancer, hTERT has been suggested as an effective target for anticancer vaccines (3). Phase I/II clinical trials using the hTERT peptide fragment, GV1001, led to immune activation when cluster of differentiation 4+ (CD4+) T cells were surveyed (4). A dose-escalating phase I/II study in patients with pancreatic cancer revealed prolonged survival rates in patients receiving the GV1001 peptide with an immunologic response, compared with those without an immune response (5). Another study showed the activation of cytotoxic T cells and an anti-leukemic response in patients with B-cell chronic lymphocytic leukemia when administered with GV1001 (6). Therefore, earlier clinical studies suggested potential anticancer effects of GV1001, primarily through immunologic activation.
GV1001 is a 16-amino acid peptide corresponding to hTERT 616–626 sequences (EARPALLTSRLRFIPK), which lies within the reverse transcriptase functional domain (5,7). Previous studies have demonstrated direct cellular effects of GV1001, beyond its immunologic functions, as an anticancer vaccine, including anti-inflammatory effects (8–10), tumor suppressive effects (11,12) and antiviral activities (13,14). GV1001 has also been found to protect against β-amyloid-induced neurotoxicity in neural stem cells (NSCs), suggesting its potential therapeutic effects for neurodegenerative diseases (15); NSCs showed enhanced cell proliferation and survival when treated with β-amyloid in the presence of GV1001. Furthermore, GV1001 administration in tumor xenografts was shown to suppress fibrotic tissue surrounding the tumor nodules, allowing for increased tumoral susceptibility to chemotherapeutic agents and cell death (12). These studies demonstrated the versatility of GV1001 with multiple biological effects, independent of the immune activation originally intended as an anticancer vaccine.
Radiation induces normal tissue injuries, which are characterized at the histologic level by loss of parenchymal cells, excessive fibrosis and tissue atrophy. Fibrotic changes occur as result of the loss of epithelial identity and function. There is a long list of systemic diseases eventually resulting in organ failure due to fibrotic changes with significant mortality rates; it has been estimated that organ fibrosis is responsible for ~45% of all chronic systemic disease-associated mortality (16). The primary driver of tissue fibrosis is transforming growth factor-β (TGF-β) signaling, as evidenced in radiation-induced pulmonary scarring, through the induction of myofibroblast differentiation (17,18). Following exposure to ionizing radiation (IR), the expression of TGF-β1 has been shown to increase in a dose-dependent manner in the rat liver (19). Similarly, in a mouse model of radiation-induced lung injury, fibrosis development was accompanied by an increase in the expression of TGF-β1 and activation of the TGF-β1 signal transduction pathway (20). As there is substantial evidence supporting the importance of TGF-β1 in the development of radiation-induced tissue damage, TGF-β1 and its signaling molecules have become logical targets for molecular therapies designed to prevent or reduce normal tissue injury following irradiation exposure. In the present study, the effects of GV1001 on radioprotection and antifibrosis were examined in primary normal human oral keratinocytes (NHOKs) and fibroblasts (NHOFs). The data indicated that GV1001 protected the NHOKs from IR-induced damage and suppressed epithelial-mesenchymal transition (EMT) in cells exposed to IR, allowing the cells continued proliferation with maintenance of the epithelial phenotype. In addition, GV1001 directly suppressed TGF-β-induced EMT in epithelial cells and myofibroblast differentiation. When GV1001 was administered by subcutaneous injection in mice exposed to bleomycin (BLM), the level of dermal fibrosis and the synthesis of collagen type III α1 chain (Col3a1) were significantly reduced in the dermal layer of the skin. Collectively, the data indicated that GV1001 may be of therapeutic benefit against tissue injury and fibrosis resulting from IR exposure through direct suppression of the TGF-β signaling pathway.
Materials and methods
Cells and cell culture
Primary NHOK and NHOF cultures were established from discarded oral mucosa without patient identification or medical information, under exemption from the Institutional Review Board of UCLA (Los Angeles, CA, USA) as described previously (21). Briefly, the discarded oral mucosal tissues of approximately 25 mm2 were collected from healthy patients who were undergoing routine dental procedures at the UCLA Dental Center between 2010 and 2016. The oral mucosal tissues were minced into 2 mm2 × 0.5 mm sections and incubated in 2.5 mg/ml dispase solution (cat. no. 17105; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in 37°C for 1 h, then the epithelial layers and connective layers were separated. The separated epithelial layers were minced and subjected to trypsin digestion (cat. no. 25200056; Gibco; Thermo Fisher Scientific, Inc.) in 37°C for 5 min to harvest the individual epithelial cells to establish NHOK. The separated connective tissue layers were minced and digested with collagenase (cat. no. 17100017; Gibco; Thermo Fisher Scientific, Inc.) in 37°C for 1 h to harvest the individual mesenchymal cells to establish NHOF. NHOKs were cultured in EpiLife supplemented with human keratinocyte growth supplement (HKGS; Cascade Biologics; Thermo Fisher Scientific, Inc.), and NHOFs were cultured in DMEM supplemented with 10% FBS (Invitrogen; Thermo Fisher Scientific, Inc.). For radiation exposure, NHOKs were exposed to varying doses of IR using a Mark I-30 Cesium-137 irradiator (JL Shepherd & Associates, San Fernando, CA, USA) with the delivery rate of 4.86 Gy min−1. IR exposure led to the induction of a senescent phenotype, described as stress-induced premature senescence (SIPS) (22). To confirm SIPS, NHOKs were stained for senescence-associated β-galactosidase (SA β-Gal) activity, which was viewed/imaged under an Olympus phase-contrast microscope (Olympus Corporation, Tokyo, Japan) (21). Squamous cell carcinomas (SCC) 4 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM/F12 (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Invitrogen; Thermo Fisher Scientific, Inc.) and 0.4 µg/ml hydrocortisol (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Reagents
The following primary antibodies were used in the present study: GAPDH (cat. no. sc-47724), zinc finger E-box binding homeobox 1 (ZEB1; cat. no. sc-25388), E-Cadherin (E-Cad; cat. no. sc-21791), P21/WAF1 (cat. no. sc-397), 53BP1 (cat. no. sc-22760), P16INK4A (cat. no. sc-468), phosphorylated (p-)Akt (Ser473; cat. no. sc-7985), collagen type I α1 chain (Col1a1; cat. no. sc-8784) and Col3a1 (cat. no. sc-28888) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); GRHL2 (cat. no. H00079977) from Abnova (Taipei City, Taiwan); N-Cadherin (N-Cad; cat. no. 610920) from BD Biosciences (San Jose, CA, USA); fibronectin (FN; cat. no. F3648), α-smooth muscle actin (α-SMA; cat. no. A5228) and Snail (cat. no. SAB1406456) from Sigma-Aldrich; Merck KGaA; p-small mothers against decapentaplegic (Smad)3 (ser423/425; cat. no. 9520), p-Smad2 (ser465/467; cat. no. 3108), Smad4 (cat. no. 38454) and p-p53 (Ser15; cat. no. 9284) from Cell Signaling Technology, Inc. (Danvers, MA, USA); and γ-H2AX (phospho S139; cat. no. ab2893) from Abcam (Cambridge, MA, USA). Alexa Fluor® anti-rabbit or anti-mouse secondary antibodies were from Thermo Fisher Scientific, Inc. TGF-β1 (cat. no. 100-21) was purchased from PeproTech, Inc. (Grand Island, NY, USA); TGF-β receptor I kinase inhibitor (TRI; cat. no. 616451) was obtained from Calbiochem; EMD Millipore (Billerica, MA, USA). BLM was purchased from Sigma-Aldrich; Merck KGaA; GV1001 was provided by Gemvax & Kael Co. Ltd. (Seoul, Korea).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, Inc.). DNA-free total RNA (5 µg) was used for the RT reaction, followed by qPCR for each 1 µl cDNA sample with 1 µl primer pair mix (5 pmol/µl each primer) and LC480 SYBR Green I master using universal cycling conditions in a LightCycler® 480 (Roche Diagnostics, South San Francisco, CA, USA). The primer sequences were obtained from the Universal Probe Library (Roche Diagnostics). The PCR cycling conditions were as follows: 45 cycles of 10 sec at 95°C, 45 sec at 55°C, and 20 sec at 72°C. The second derivative Cq value determination method was used to compare the fold-differences (23). Cq was the cycle at which the threshold was crossed. The experiments were performed in triplicate. The primer sequences used for the PCR to determine gene expression were as follows: Col1a1, forward 5′-GGGATTCCCTGGACCTAAAG-3′ and reverse 5′-GGAACACCTCGCTCTCCA-3′; Col3a1, forward 5′-CTGGACCCCAGGGTCTTC-3′ and reverse 5′-CATCTGATCCAGGGTTTCCA-3′; FN, forward 5′-GGGAGAATAAGCTGTACCATCG-3′ and reverse 5′-TCCATTACCAAGACACACACACT-3′; N-Cad, forward 5′-CTCCATGTGCCGGATAGC-3′ and reverse 5′-CGATTTCACCAGAAGCCTCTAC-3′; ZEB1, forward 5′-AACTGCTGGGAGGATGACAC-3′ and reverse 5′-TCCTGCTTCATCTGCCTGA-3′; ZEB2, forward 5′-CCAGACCGCAATTAACAATG-3′ and reverse 5′-ATGCTGACTGCATGACCATC-3′; glyceraldehyde 3-phosphate dehy drogenase (GAPDH), forward 5′-AGCCACATCGCTCAGACAC-3′ and reverse 5′-GCCCAATACGACCAAATCC-3′.
Western blot analysis
Whole cell extracts (WCEs) from the cultured cells were isolated using lysis buffer [1% Triton X-100, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 µM β-glycerophosphate, 1 mM sodium orthovanadate and 1 mg/ml PMSF]. The protein concentration of the lysates was measured using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts of protein (20 µg/lane) were separated by 8 or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto an immobilon membrane (EMD Millipore), which was incubated in blocking solution containing 5% bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) in Tris-buffered saline (TBS) for 30 min at room temperature. The membrane was then incubated with primary antibodies (e.g., GRHL2 and N-Cad) diluted in blocking solution (1:1,000) at 4°C overnight. Following washing with TBST three times, the membrane was incubated with anti-mouse or rabbit secondary antibodies (1:2,000) for 1 h at room temperature and then exposed to the chemiluminescence reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA) for signal detection.
Immunofluorescence staining (IFS) of cells and tissue specimens
The cells were cultured in an Nunc™ Lab-Tek™ II Chamber Slide™ system (Thermo Fisher Scientific, Inc.) to reach 70–80% confluence, and then fixed in 2% paraformaldehyde for 20 min. The cells were permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 10 min, and then blocked for 1 h in PBS containing 2% BSA (Sigma-Aldrich; Merck KGaA) at room temperature and incubated overnight at 4°C with primary antibodies (ZEB1 and α-SMA; 1:100). Following three washes with PBS, the cells were incubated with the secondary antibodies, Alexa Fluor® 594 goat anti-rabbit IgG or Alexa Fluor® 488 goat anti-mouse IgG (1:400; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Slides were mounted in Prolong Gold w/DAPI (Invitrogen; Thermo Fisher Scientific, Inc.). Images were captured with an Olympus epifluorescence inverted microscope (Olympus Corporation).
Paraffin-embedded histological sections were stained with hematoxylin and eosin (H&E) for determination of the histological changes in the dermis. The mouse skin specimens were fixed in 4% (wt/vol) paraformaldehyde at 4°C for 24 h. The samples were embedded in paraffin, sectioned at 4 µm thickness, and stained as described previously (24). Briefly, the tissue slides were dewaxed, and antigen retrieval was performed using an unmasking solution (Vector Laboratories, Inc., Burlingame, CA, USA) boiled in a microwave for 20 min. The slides were then incubated in 3% hydrogen peroxide in PBS for 10 min to quench the ZEB endogenous peroxidase. The slides were sequentially washed with distilled water and PBS, and incubated in blocking solution (2.5% BSA in PBS) for 30 min at room temperature. The slides were incubated with Col3a1 primary antibody diluted in blocking solution (1:100) at 4°C overnight. Following washing with PBS, the slides were incubated with the secondary antibody, Alexa Fluor® 594 goat anti-rabbit IgG (1:400; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. The slides were mounted in Prolong Gold w/DAPI (Invitrogen; Thermo Fisher Scientific, Inc.).
DNA foci detection
Following irradiation, the cells were fixed in 2% paraformaldehyde for 20 min at room temperature. The cells were permeabilized with 0.5% TritonX-100 and blocked with 5% FBS in PBS. The cells were then incubated with 53BP1 or γ-H2A.X primary antibody overnight at 4°C in 1% BSA. The coverslips were mounted with ProLong® Gold antifade reagent with DAPI to counterstain the nuclei. The foci were quantified using Cell Profiler to count cells with >3 foci per nucleus in ≥10 different fields from each experiment under confocal microscopy (Olympus Corporation). Experimental data were determined as the average of three independent experiments.
Chromatin immunoprecipitation (ChIP) assay
A ChIP assay was performed based on the protocols described in our previous study (22). The sequences for the PCR primers of gene promoters were as follows, Col1a1 (-1,750 bp), forward 5′-TCTCCCAGGTGTCTGTCTCC-3′ and reverse 5′-AGGAGAGGGGTCTGCTGAG-3′; Col1a1 (-1,000 bp), forward 5′-AGGGGGAAAAACTGCTTTGT-3′ and reverse 5′-TCCGACCTCTCTCCTCTGAA-3′; Col1a1 (−200 bp), forward 5′-CCACTTGGGTGTTTGAGCA-3′ and reverse 5′-CCTCCCCTCCACTCCTTC-3′; Col3a1 promoter, forward 5′-TCTCCATTGCCAGATATAGCC-3′ and reverse 5′-TCCAGATCCTCATCCACAAA-3′. The promoter regions analyzed in the present study for p-Smad2 enrichment included the Smad-binding element (SBE), AGACA sequence.
Wound healing assay
SCC4 cells (2×105 cells per well) were seeded in 6-well plate and cultured for 48 h in culture medium. A scratch (wound) was then introduced in the confluent cell layer using a P200 yellow tip. The cells were washed three times with medium to remove detached cells. The cells were then incubated with or without TGF-β (10 ng/ml) or GV1001 (1 µM) for 48 h and images of a defined wound spot were captured with a computer-aided phase contrast microscope (Olympus). Experiments were performed in triplicate.
BLM-induced dermal fibrosis model
Pathogen-free, female C57BL/6 mice (6 week-old; Jackson Laboratory, Ben Harbor, ME, USA) were used for BLM-induced dermal fibrosis by subcutaneous injection of BLM to the shaved upper back skin for 4 weeks. The animals were housed in a pathogen-free environment with a 12 h light/dark cycle in the Division of Laboratory and Animal Medicine at UCLA (Los Angeles, CA, USA). The experiments were performed based on the protocol approved by the Institutional Animal Care and Use Committee. A total of 20 mice were divided into four groups: Control group, injected subcutaneously (s.q.) with 100 µl vehicle (PBS); BLM group, injected with 100 µl BLM solution (100 µg/ml); BLM and Low GV1001 group, injected with 1 mg/kg GV1001 and 100 µg/ml BLM; BLM and high GV1001 group, injected with 5 mg/kg GV1001 and 100 µg/ml BLM. Each group included five mice. The s.q. injection continued daily for 4 weeks and the shaved upper back skin tissues were harvested for histological assessment of the lesions.
Statistical analysis
Statistical evaluations were performed using SPSS version 11.0 (SPSS, Inc., Chicago, IL, USA). All numerical data were expressed as the mean ± standard deviation. Statistical analysis was performed using Student's t-test (two-tailed) for quantitative experiments. P<0.05 was considered to indicate a statistically significant difference.
Results
GV1001 suppresses IR-induced premature senescence by reducing the genotoxic effect
Our previous study showed that exposure of primary human keratinocytes to IR triggered a senescence response (22). To examine the effect of GV1001 on the cellular response to IR, NHOKs were exposed to 6 Gy IR and the cells were cultured in the presence or absence of GV1001. The rapidly proliferating NHOKs underwent cell proliferation arrest upon irradiation and exhibited cellular morphology consistent with senescence, including a flattened cytosolic region and perinuclear vacuolization (21), whereas these senescing effects of IR were reduced in the cells treated with GV1001 (Fig. 1A). While irradiation inhibited cell proliferation, treatment of the NHOKs with GV1001 mitigated the growth suppressive effects of IR (Fig. 1B). With increased dose of IR, the percentage of cells expressing SA β-Gal was increased in the NHOKs; however, GV1001 treatment significantly reduced the percentage of cells stained positively for SA β-Gal (Fig. 1C). Similarly, the colony forming ability of the cultured NHOKs was suppressed by exposure to IR, whereas this effect was mitigated by GV1001 treatment (Fig. 1D). These results demonstrated that GV1001 treatment ameliorated IR-induced premature senescence in cultured NHOKs.
Figure 1.

GV1001 attenuates the IR-induced premature senescence phenotype of NHOKs. (A) Rapidly proliferating NHOKs were exposed to 6 Gy IR and maintained in culture for 10 days in the presence or absence of GV1001 (1 µM) (original magnification, ×100; scale bar, 100 µm; n=3). (B) Cell proliferation kinetics were determined in NHOKs irradiated at 6 Gy IR with GV1001 (1 µM) or vehicle control. (C) NHOKs were exposed to IR at 2–10 Gy in the presence or absence of GV1001 (1 µM) and stained for SA β-Gal. Positively stained cells were counted and plotted. Error bars represent the mean ± standard deviation. (D) NHOKs exposed to 2–10 Gy IR were plated at low density for colony formation in the presence or absence of GV1001 (1 µM). After 10–14 days, the number of colonies were counted and plotted. Error bars represent the mean ± standard deviation. *P<0.05. NHOKs, normal human oral keratinocytes; IR, ionizing radiation; Cont, control; SA β-Gal, senescence-associated β-galactosidase.
Keratinocyte proliferation is regulated in part through Grainyhead-like 2 (GRHL2), which is a novel transregulator of the hTERT gene and determines epithelial phenotype through the suppression of EMT regulators (25–27). The western blot analysis showed that the endogenous level of GRHL2 was induced by GV1001, even following exposure to 6 Gy IR (Fig. 2A). GV1001 treatment led to reduction in the level of N-Cad, encoded by the CDH2 gene, which is elevated during EMT (28), consistent with the increased expression of GRHL2. As EMT is induced by TGF-β signaling, these data indicated the possible activation of the TGF-β pathway in NHOKs following IR exposure. IR also led to p53 phosphorylation (Ser15) and an increased level of p21/WAF1 cyclin-dependent kinase inhibitor, however, these protein levels were reduced in the cells treated with GV1001. p-Akt (Ser473), which is necessary for radioresistance in cells by enhancing DNA double strand break (DSB) repair (29), was elevated in the NHOKs treated with GV1001. The level of DNA DSB in the irradiated cells was measured directly by staining for 53BP1 and γ-H2AX, which form fluorescent foci at the sites of DSBs (30,31), as shown in Fig. 2B. The NHOKs were irradiated with 6 Gy IR in the presence or absence of GV1001, and the percentages of cells with intranuclear 53BP1 and γ-H2AX foci were determined at 10 days post-IR by IFS using confocal microscopy. There was elevation of DSBs in the irradiated cells, compared with the control cells without IR exposure, and the level of DSBs was decreased in cells cultured with GV1001, indicating that GV1001 enhanced the removal of DSBs in the cells (Fig. 2C). Taken together, these data indicated that GV1001 suppressed IR-induced premature senescence, in part via mitigating the genotoxic effects.
Figure 2.

GV1001 treatment reduces the level of DNA double strand breaks in NHOKs exposed to IR. (A) Western blot analysis was performed with NHOKs 10 days following exposure to 6 Gy IR with or without GV1001 (1 µM) for GRHL2, N-Cad, p-Akt (Ser473), p-p53 (Ser15) and p21/WAF1. GAPDH was used as a loading control. (B) NHOKs were exposed to 6 Gy IR in the presence or absence of GV1001 (1 µM), were stained for 53BP1 and were viewed under confocal microscopy to detect 53BP1 intranuclear foci. DAPI staining revealed the nuclei (original magnification of left 2 panels, ×100; scale bar, 100 µm; n=3; original magnification of right panels, ×500; scale bar, 20 µm; n=3). (C) Percentages of cells with 53BP1 or γ-H2AX intranuclear foci (>3 foci per nucleus) were counted in ≥10 various fields from each experiment and plotted. Error bars represent the mean ± standard deviation. *P<0.05. NHOKs, normal human oral keratinocytes; IR, ionizing radiation; GRHL2, Grainyhead-like 2; N-Cad, N-Cadherin; p-, phosphorylated.
GV1001 inhibits TGF-β signaling
The senescent phenotype in cells exposed to IR is regulated through the auto/paracrine activity of TGF-β (17,32). The irradiation of NHOKs also led to the activation of TGF-β signaling molecules, inducing the phosphorylation of Smad2/3 complex and the level of Smad4 (Fig. 3), suggesting their role in premature senescence. In addition, the irradiated cells exhibited a mesenchymal phenotype and expressed higher levels of TGF-β target genes, including N-Cad, ZEB1, FN and Snail. Treatment of the cells with GV1001 decreased the expression of these TGF-β target mesenchymal markers and TGF-β signaling molecules, including p-Smad2/3 and Smad4, compared with the control cells. The expression of target proteins and signaling induced by TGF-β were also eliminated by exposure to 1 µM TRI, which was used as the positive control for GV1001 treatment. These data indicated that the mesenchymal phenotype induced by IR was prevented by GV1001 in the NHOKs, possibly through suppressing TGF-β signaling.
Figure 3.
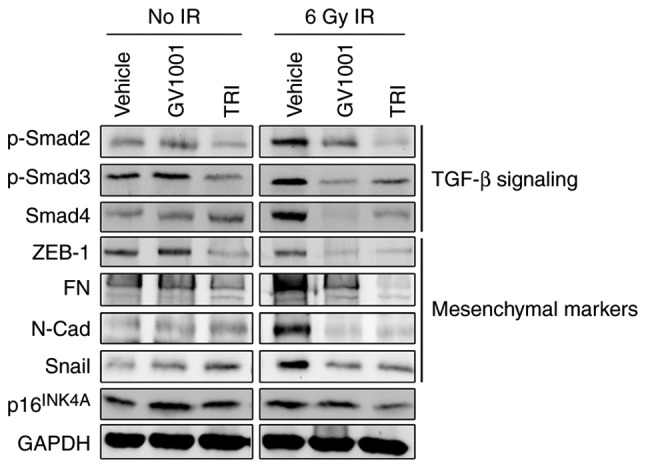
GV1001 suppresses TGF-β signaling and EMT in NHOKs exposed to IR. NHOKs were exposed to 6 Gy IR and maintained in culture for 10 days in the presence of GV1001 (1 µM) or TRI (1 µM). Western blot analysis was performed for TGF-β signaling molecules, p-Smad2/3 and Smad4, and TGF-β target mesenchymal markers, ZEB1, FN, N-Cad and Snail. GAPDH was used as a loading control. NHOKs, normal human oral keratinocytes; IR, ionizing radiation; TGF-β, transforming growth factor-β; TRI, TGF-β receptor inhibitor; Smad, small mothers against decapentaplegic; p-, phosphorylated; ZEB1, zinc finger E-box binding homeobox 1; FN, fibronectin; N-Cad, N-Cadherin.
To examine the effects of GV1001 on TGF-β signaling, rapidly proliferating NHOKs were exposed to TGF-β in the presence or absence of GV1001. Exposure to TGF-β for up to 10 days led to induction of a mesenchymal phenotype through EMT (Fig. 4A). It was also found that GV1001 treatment reduced the expression of Snail and α-SMA in the cells exposed to TGF-β, in addition to TGF-β signaling molecules, including p-Smad2/3 and Smad 4 (Fig. 4B). TGF-β treatment also led to enhanced cell migration of the SCC4 cell line, reminiscent of the mesenchymal phenotype, and GV1001 almost completely prevented the enhanced migratory effects of TGF-β (Fig. 4C). In the NHOFs, the expression of TGF-β target molecules and mesenchymal markers, including Col1a1, Col3a1, FN, N-Cad, ZEB1 and ZEB2, was suppressed in the cells treated with GV1001 (Fig. 5A). The effect of GV1001 on binding of p-Smad to the SBE was also confirmed by a ChIP assay of NHOFs exposed to TGF-β (Fig. 5B and C). Treatment of the cells with GV1001 markedly suppressed binding of Smad2 on the Col1a1 and Col3a1 gene promoter regions, indicating that GV1001 interfered with TGF-β-mediated target gene transcription in NHOFs.
Figure 4.

GV1001 suppresses TGF-β-induced EMT in NHOKs. (A) Rapidly proliferating NHOKs were exposed to TGF-β (10 ng/ml) for 10 days to induce EMT (shown in islet, arrows). The same cultures were also maintained in the presence or absence of GV1001 (1 µM) (original magnification, ×100; scale bar, 100 µm; n=3). (B) Western blot analysis was performed with NHOKs exposed to TGF-β (10 ng/ml) with GV1001 (1 µM) or the control for Snail, α-SMA, p-Smad2/3 and Smad4. GAPDH was used as a loading control. (C) Cell migration of SCC4 cells exposed to TGF-β (10 ng/ml) with or without GV1001 (1 µM) for 24 h (original magnification, ×40; scale bar, 500 µm; n=3). NHOKs, normal human oral keratinocytes; EMT, epithelial-mesencyhmal transition; TGF-β, transforming growth factor-β; α-SMA, α-smooth muscle actin; Smad, small mothers against decapentaplegic; p-, phosphorylated; GV, GV1001.
Figure 5.

GV1001 inhibits Smad2 binding to target gene promoters. (A) Reverse transcription-quantitative polymerase chain reaction analysis was performed on NHOFs exposed to TGF-β (10 ng/ml) with GV1001 (1 µM) or control for 10 days, for the gene expression of Col1a1, Col3a1, FN, N-Cad, Zeb1 and Zeb2. (B and C) NHOFs were cultured with 10 ng/ml TGF-β and GV1001 (1 µM) for 10 days. Chromatin immunoprecipitation was performed for Smad2 enrichment on Col1a1 and Col3a1 promoter regions. Error bars indicate the mean ± standard deviation. *P<0.05. NHOKs, normal human oral keratinocytes; TGF-β, transforming growth factor-β; Col1a1, collagen type I α1 chain; Col3a1, collagen type III α1 chain; ZEB1, zinc finger E-box binding homeobox 1; FN, fibronectin; N-Cad, N-Cadherin; Smad, small mothers against decapentaplegic; Cont, control; GV, GV1001.
GV1001 inhibits BLM-induced dermal fibrosis
In the subsequent experiments, the effects of GV1001 in fibrotic tissue formation were determined. Whether GV1001 treatment inhibited TGF-β signaling in myofibroblast differentiation was assessed. Rapidly proliferating NHOFs were exposed to TGF-β, which triggered the induction of α-SMA, specialized actin molecules found in myofibroblasts, in addition to Col3a1, Col1a1, Snail, FN and N-Cad (Fig. 6A). These changes occurred with marked induction of p-Smad2. However, when the cells were co-treated with GV1001, there were marked reductions in the levels of α-SMA, N-Cad, FN and p-Smad in the cells. Similarly, IFS revealed induced α-SMA in the NHOFs by TGF-β treatment, in addition to ZEB1, a mesenchymal transcription factor directly regulating the expression of α-SMA (33), whereas the levels of α-SMA and ZEB1 were suppressed by GV1001 treatment (Fig. 6B). These data indicated that GV1001 inhibited TGF-β-induced myofibroblast differentiation through the suppression of ZEB.
Figure 6.

GV1001 attenuates the dermal fibrotic lesions induced by BLM. (A) NHOF cultures were treated with 10 ng/ml TGF-β for 10 days and GV1001 was added at varying concentrations between 1 and 8 µM. Western blot analysis was performed with whole cell extracts for α-SMA, Snail, FN, N-Cad, Col1a1 and Col3a1, and p-Smad2 and Smad4. GAPDH was used as a loading control. NHOFs with ectopic overexpression of GRHL2 were also included as a negative control, as GRHL2 inhibited fibrogenic differentiation and TGF-β signaling. (B) IFS was performed for α-SMA and ZEB1 in NHOFs treated with 10 ng/ml TGF-β and GV1001 (1 µM) for 10 days (original magnification, ×100; scale bar, 100 µm; n=3). (C) C57BL/6 mice were exposed to BLM (100 µg/ml) with or without GV1001, administered at low (1 mg/kg) or high (5 mg/kg) doses, by daily subcutaneous injection into the dorsal flank. Following 4 weeks of BLM injection, mice were sacrificed for histological examination by H&E staining. IFS was also performed with the skin biopsy samples for Col3a1 deposition in mice exposed to BLM with or without GV1001 (original magnification, ×100; scale bar, 250 µm; n=5). (D) Quantitation of dermal thickness was plotted in the groups with or without BLM and GV1001 administration. Error bars indicate the mean ± standard deviation. *P<0.05. NHOKs, normal human oral keratinocytes; BLM, bleomycin; IFS, immunofluorescence staining; TGF-β, transforming growth factor-β; α-SMA, α-smooth muscle actin; Col1a1, collagen type I α1 chain; Col3a1, collagen type III α1 chain; ZEB1, zinc finger E-box binding homeobox 1; FN, fibronectin; N-Cad, N-Cadherin; Smad, small mothers against decapentaplegic; p-, phosphorylated; GRHL2, Grainyhead-like 2; H&E, hematoxylin and eosin; GV, GV1001; C, control.
To confirm the antifibrogenic property of GV1001 in vivo, dermal fibrosis was induced in C57BL/6 mice by s.q. injection of BLM with or without co-administration of GV1001. BLM treatment for 4 weeks led to marked dermal sclerosis in the mice, which was characterized histologically by a thickened dermis with increased deposition of collagen bundles (Fig. 6C). When GV1001 was administered to these mice, there was significant reduction in the thickness of the fibrotic lesions, and a reduction in dermal levels of Col3a1, as determined by IF staining (Fig. 6C and D). Therefore, GV1001 exerted antifibrotic effects in vivo and may directly impair the fibrogenic differentiation of dermal fibroblasts.
Discussion
The present study demonstrated a novel role of GV1001, a 16-amino acid peptide sequence of hTERT protein, originally developed as an immunogenic anticancer vaccine (5), in modulating the cellular responses to IR. The irradiation of NHOKs led to suppressed cell proliferation and EMT, possibly through the activation of TGF-β signaling, and these phenotypic changes induced by IR were notably reduced following GV1001 treatment. These data suggested that GV1001 may exert its protective and anti-EMT effects through suppressing TGF-β signaling, however, the detailed mechanism requires further investigation. In addition, the radioprotective effects of GV1001 reported in the present study were based on in vitro experiments and require validation by further experiments in vivo. The radioprotective effects of GV1001 were also linked to elevated DNA repair activities. This was evidenced as the increased level of p-Akt (Ser473), which has been shown to regulate cellular radioprotection against IR (29,34), and reduced level of active p53/p21WAF1 proteins in the NHOKs treated with GV1001. This was in accordance with an earlier report showing the protective effects of GV1001 against oxidative stress in neural stem cells, allowing for continued cell proliferation and reduced cell death following exposure to H2O2 (35). Further investigations aim to elucidate the detailed mechanism by which GV1001 modulates the genotoxic effects of IR.
Of note, GV1001 treatment markedly elevated the level of GRHL2, a transcription factor with diverse biological functions and a multitude of target genes, including hTERT, proliferating cell nuclear antigen, p63, microRNA-200 family genes and epidermal cell differentiation complex genes (24–26,36). GRHL2 promotes epithelial cell proliferation in part through the transcription regulation of hTERT; the ectopic overexpression of GRHL2 in primary NHOKs led to marked extension of cell life span with continued expression of hTERT and telomerase activity (25). As GV1001 is derived from the hTERT protein sequence, the fact that GV1001 enhanced GRHL2 may indicate a feed-forward mechanism by which it further elevates the level of hTERT through the expression of GRHL2.
In the present study, GV1001 treatment suppressed TGF-β signaling, including the expression of p-Smad2/3 and Smad4, and reduced the expression of mesenchymal markers, including ZEB1, FN, N-Cad and Snail, in NHOKs exposed to IR. These findings may be linked to the effect that GV1001 enhanced the level of GRHL2, which is also known to be a master regulator of epithelial phenotype, inhibiting TGF-β-induced EMT (26,37). The present study also provided evidence that GV1001 suppressed the fibrogenic differentiation of NHOFs treated with TGF-β; GV1001 suppressed the phosphorylation of Smad2 and decreased the level of α-SMA, a marker of myofibroblasts involved in wound contraction (38). GV1001 markedly inhibited p-Smad2 binding to the Col1a1 and Col3a1 promoter regions, which may be associated with a reduced level of tissue fibrosis. As GRHL2 is an epithelial-specific protein, the suppressive effects of GV1001 against TGF-β signaling in NHOFs were not GRHL2-dependent, however, the mechanisms remain to be elucidated.
Tissue fibrosis commonly occurs with terminal stages of organ failure, which results from chronic inflammatory reactions, including chronic wound and radiation tissue damage, and fibrotic changes can be observed in several organ systems. One of the core cellular constituents of fibrosis is myofibroblasts, marked by α-SMA, which migrate into the wound area and cause wound contraction and the secretion of extracellular matrix proteins. Myofibroblasts were originally considered to arise from the resident fibroblasts in local tissues upon activation (39), however, several studies have indicated the role of adjacent epithelial and endothelial cells as the source of myofibroblasts for tissue fibrosis through EMT (40,41). EMT may also account for radiation-induced tissue fibrosis in patients exposed to therapeutic radiation. Pulmonary fibrosis in patients with lung cancer occurs with increased α-SMA-positive cells in the lung interstitium and EMT in alveolar epithelial cells, driven by Forkhead Box M1 transcription factor (17,42). In addition, head/neck irradiation in patients leads to salivary gland atrophy, termed xerostomia, resulting in salivary gland dysfunction, primarily through fibrotic changes (43). The involvement of EMT for xerostomia is not documented, however, similar glandular fibrotic tissues of the lacrimal gland have been reported to show increased markers of EMT, including Snail, α-SMA, and reduced E-cadherin, in patients with Mikulicz's syndrome, which causes abnormal enlargement of glands in the head/neck area (44). Therefore, by suppressing IR-induced EMT, GV1001 may have therapeutic benefit against the tissue fibrosis that results from IR exposure.
Acknowledgments
The authors are thankful to the Gemvax & Kael Co. Ltd. (Seoul, Korea) for providing the GV1001 peptide.
Abbreviations
- TGF-β
transforming growth factor-β
- GRHL2
Grainyhead-like 2
- FN
fibronectin
- hTERT
human telomerase reverse transcriptase
- E-Cad
E-cadherin
- N-Cad
N-cadherin
- NHOF
normal human oral fibroblast
- NHOK
normal human oral keratinocyte
- Col1a1
collagen type I α1 chain
- Col3a1
collagen type III α1 chain
- IR
ionizing radiation
- NSC
neural stem cell
- IPF
idiopathic pulmonary fibrosis
- EMT
epithelial-mesenchymal transition
- SA β-Gal
senescence-associated β-galactosidase
- WCE
whole cell extract
- BLM
bleomycin
- RT
reverse transcription
Footnotes
Funding
This study was supported in part by grants from the NIDCR/NIH (grant nos. 1R56DE024593 and R03DE024259), the University of California Cancer Research Coordinating Committee (grant no. 20152529), and Gemvax & Kael Co. Ltd. (Seoul, Korea). MKK was also supported by the Jack A. Weichman Endowed Fund.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
WC contributed to conception, design, data acquisition, analysis and interpretation, and critically revised the manuscript; KHS analyzed and interpreted the in vivo data about GV1001 inhibiting dermal fibrosis, and critically revised the manuscript; SK contributed to conception of research question and data interpretation; WJS interpreted data and critically revised the manuscript; RHK contributed to conception, data interpretation and critically revised the manuscript; NHP interpreted data and critically revised the manuscript; MKK contributed to conception, design, data analysis, interpretation, drafted and critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The animal experiments were performed based on the protocol approved by the Institutional Animal Care and Use Committee of UCLA. The discarded human tissues were obtained without patient identification or medical information, under exemption from the Institutional Review Board of UCLA.
Consent for publication
Not applicable.
Competing interests
SK is employed at Gemvax & Kael Co. Ltd. (Seoul, Korea) and TELOID Inc (Los Angeles, CA, USA). Other authors declare that there are no competing interests.
References
- 1.Kim HR, Christensen R, Park NH, Sapp P, Kang MK, Park NH. Elevated expression of hTERT is associated with dysplastic cell transformation during human oral carcinogenesis in situ. Clin Cancer Res. 2001;7:3079–3086. [PubMed] [Google Scholar]
- 2.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 3.Greener M. Telomerase: The search for a universal cancer vaccine. Mol Med Today. 2000;6:257. doi: 10.1016/S1357-4310(00)01731-7. [DOI] [PubMed] [Google Scholar]
- 4.Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Møller M, Eriksen JA, Gaudernack G. Telomerase peptide vaccination: A phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55:1553–1564. doi: 10.1007/s00262-006-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhardt SL, Gjertsen MK, Trachsel S, Møller M, Eriksen JA, Meo M, Buanes T, Gaudernack G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br J Cancer. 2006;95:1474–1482. doi: 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kokhaei P, Palma M, Hansson L, Osterborg A, Mellstedt H, Choudhury A. Telomerase (hTERT 611-626) serves as a tumor antigen in B-cell chronic lymphocytic leukemia and generates spontaneously antileukemic, cytotoxic T cells. Exp Hematol. 2007;35:297–304. doi: 10.1016/j.exphem.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Nicholls C, Li H, Wang JQ, Liu JP. Molecular regulation of telomerase activity in aging. Protein Cell. 2011;2:726–738. doi: 10.1007/s13238-011-1093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang JE, Kim HJ, Yi E, Jheon S, Kim K. Reduction of ischaemia-reperfusion injury in a rat lung transplantation model by low-concentration GV1001. Eur J Cardiothorac Surg. 2016;50:972–979. doi: 10.1093/ejcts/ezw135. [DOI] [PubMed] [Google Scholar]
- 9.Choi J, Kim H, Kim Y, Jang M, Jeon J, Hwang YI, Shon WJ, Song YW, Kang JS, Lee WJ. The anti-inflammatory effect of GV1001 mediated by the downregulation of ENO1-induced pro-inflammatory cytokine production. Immune Netw. 2015;15:291–303. doi: 10.4110/in.2015.15.6.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko YJ, Kwon KY, Kum KY, Lee WC, Baek SH, Kang MK, Shon WJ. The anti-inflammatory effect of human telomerase-derived peptide on P. gingivalis lipopolysaccharide-induced inflammatory cytokine production and its mechanism in human dental pulp cells. Mediators Inflamm. 2015;2015:385127. doi: 10.1155/2015/385127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim BK, Kim BR, Lee HJ, Lee SA, Kim BJ, Kim H, Won YS, Shon WJ, Lee NR, Inn KS, Kim BJ. Tumor-suppressive effect of a telomerase-derived peptide by inhibiting hypoxia-induced HIF-1α-VEGF signaling axis. Biomaterials. 2014;35:2924–2933. doi: 10.1016/j.biomaterials.2013.12.077. [DOI] [PubMed] [Google Scholar]
- 12.Park JK, Kim Y, Kim H, Jeon J, Kim TW, Park JH, Hwnag YI, Lee WJ, Kang JS. The anti-fibrotic effect of GV1001 combined with gemcitabine on treatment of pancreatic ductal adenocarcinoma. Oncotarget. 2016;7:75081–75093. doi: 10.18632/oncotarget.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H, Choi MS, Inn KS, Kim BJ. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Sci Rep. 2016;6:28896. doi: 10.1038/srep28896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SA, Kim J, Sim J, Kim SG, Kook YH, Park CG, Kim HR, Kim BJ. A telomerase-derived peptide regulates reactive oxygen species and hepatitis C virus RNA replication in HCV-infected cells via heat shock protein 90. Biochem Biophys Res Commun. 2016;471:156–162. doi: 10.1016/j.bbrc.2016.01.160. [DOI] [PubMed] [Google Scholar]
- 15.Park HH, Lee KY, Kim S, Lee JW, Choi NY, Lee EH, Lee YJ, Lee SH, Koh SH. Novel vaccine peptide GV1001 effectively blocks β-amyloid toxicity by mimicking the extra-telomeric functions of human telomerase reverse transcriptase. Neurobiol Aging. 2014;35:1255–1274. doi: 10.1016/j.neurobiolaging.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 16.Bitterman PB, Henke CA. Fibroproliferative disorders. Chest. 1991;99(3 Suppl):81S–84S. doi: 10.1378/chest.99.3_Supplement.81S. [DOI] [PubMed] [Google Scholar]
- 17.Judge JL, Owens KM, Pollock SJ, Woeller CF, Thatcher TH, Williams JP, Phipps RP, Sime PJ, Kottmann RM. Ionizing radiation induces myofibroblast differentiation via lactate dehydrogenase. Am J Physiol Lung Cell Mol Physiol. 2015;309:L879–L887. doi: 10.1152/ajplung.00153.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, Gauldie J, Camus P, Bonniaud P. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J Immunol. 2007;179:6043–6051. doi: 10.4049/jimmunol.179.9.6043. [DOI] [PubMed] [Google Scholar]
- 19.Anscher MS, Crocker IR, Jirtle RL. Jirtle, transforming growth factor-beta 1 expression in irradiated liver. Radiat Res. 1990;122:77–85. doi: 10.2307/3577586. [DOI] [PubMed] [Google Scholar]
- 20.Franko AJ, Sharplin J, Ghahary A, Barcellos-Hoff MH. Immunohistochemical localization of transforming growth factor beta and tumor necrosis factor alpha in the lungs of fibrosis-prone and 'non-fibrosing' mice during the latent period and early phase after irradiation. Radiat Res. 1997;147:245–256. doi: 10.2307/3579426. [DOI] [PubMed] [Google Scholar]
- 21.Kang MK, Bibb C, Baluda MA, Rey O, Park NH. In vitro replication and differentiation of normal human oral keratinocytes. Exp Cell Res. 2000;258:288–297. doi: 10.1006/excr.2000.4943. [DOI] [PubMed] [Google Scholar]
- 22.Dong Q, Oh JE, Chen W, Kim R, Kim RH, Shin KH, McBride WH, Park NH, Kang MK. Radioprotective effects of Bmi-1 involve epigenetic silencing of oxidase genes and enhanced DNA repair in normal human keratinocytes. J Invest Dermatol. 2011;131:1216–1225. doi: 10.1038/jid.2011.11. [DOI] [PubMed] [Google Scholar]
- 23.Larionov A, Krause A, Miller W. A standard curve based method for relative real time PCR data processing. BMC Bioinformatics. 2005;6:62. doi: 10.1186/1471-2105-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Xiao Liu Z, Oh JE, Shin KH, Kim RH, Jiang M, Park NH, Kang MK. Grainyhead-like 2 (GRHL2) inhibits keratinocyte differentiation through epigenetic mechanism. Cell Death Dis. 2012;3:e450. doi: 10.1038/cddis.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Dong Q, Shin KH, Kim RH, Oh JE, Park NH, Kang MK. Grainyhead-like 2 enhances the human telomerase reverse transcriptase gene expression by inhibiting DNA methylation at the 5′-CpG island in normal human keratinocytes. J Biol Chem. 2010;285:40852–40863. doi: 10.1074/jbc.M110.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, Yi JK, Shimane T, Mehrazarin S, Lin YL, Shin KH, Kim RH, Park NH, Kang MK. Grainyhead-like 2 regulates epithelial plasticity and stemness in oral cancer cells. Carcinogenesis. 2016;37:500–510. doi: 10.1093/carcin/bgw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013;73:6299–6309. doi: 10.1158/0008-5472.CAN-12-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma T, Zhao Y, Wei K, Yao G, Pan C, Liu B, Xia Y, He Z, Qi X, Li Z, et al. MicroRNA-124 functions as a tumor suppressor by regulating CDH2 and epithelial-mesenchymal transition in non-small cell lung cancer. Cell Physiol Biochem. 2016;38:1563–1574. doi: 10.1159/000443097. [DOI] [PubMed] [Google Scholar]
- 29.Holler M, Grottke A, Mueck K, Manes J, Jücker M, Rodemann HP, Toulany M. Dual Targeting of Akt and mTORC1 impairs repair of DNA double-strand breaks and increases radiation sensitivity of human tumor cells. PLoS One. 2016;11:e0154745. doi: 10.1371/journal.pone.0154745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging (Albany NY) 2014;6:481–495. doi: 10.18632/aging.100673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandevoorde C, Vral A, Vandekerckhove B, Philippé J, Thierens H. Radiation sensitivity of human CD34(+) cells versus peripheral blood T lymphocytes of newborns and adults: DNA Repair and Mutagenic Effects. Radiat Res. 2016;185:580–590. doi: 10.1667/RR14109.1. [DOI] [PubMed] [Google Scholar]
- 32.Liakou E, Mavrogonatou E, Pratsinis H, Rizou S, Evangelou K, Panagiotou PN, Karamanos NK, Gorgoulis VG, Kletsas D. Ionizing radiation-mediated premature senescence and paracrine interactions with cancer cells enhance the expression of syndecan 1 in human breast stromal fibroblasts: The role of TGF-β. Aging (Albany NY) 2016;8:1650–1669. doi: 10.18632/aging.100989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang YC, Tsai CH, Lai YL, Yu CC, Chi WY, Li JJ, Chang WW. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J Cell Mol Med. 2014;18:698–708. doi: 10.1111/jcmm.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park HS, You GE, Yang KH, Kim JY, An S, Song JY, Lee SJ, Lim YK, Nam SY. Role of AKT and ERK pathways in controlling sensitivity to ionizing radiation and adaptive response induced by low-dose radiation in human immune cells. Eur J Cell Biol. 2015;94:653–660. doi: 10.1016/j.ejcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Park HH, Yu HJ, Kim S, Kim G, Choi NY, Lee EH, Lee YJ, Yoon MY, Lee KY, Koh SH. Neural stem cells injured by oxidative stress can be rejuvenated by GV1001, a novel peptide, through scavenging free radicals and enhancing survival signals. Neurotoxicology. 2016;55:131–141. doi: 10.1016/j.neuro.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Mehrazarin S, Chen W, Oh JE, Liu ZX, Kang KL, Yi JK, Kim RH, Shin KH, Park NH, Kang MK. The p63 gene is regulated by grainyhead-like 2 (GRHL2) through reciprocal feedback and determines the epithelial phenotype in human keratinocytes. J Biol Chem. 2015;290:19999–20008. doi: 10.1074/jbc.M115.659144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cieply B, Riley P, IV, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, Denvir J, Frisch SM. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012;72:2440–2453. doi: 10.1158/0008-5472.CAN-11-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penke LR, Huang SK, White ES, Peters-Golden M. Prostaglandin E2 inhibits α-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J Biol Chem. 2014;289:17151–17162. doi: 10.1074/jbc.M114.558130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinz B. The role of myofibroblasts in wound healing. Curr Res Transl Med. 2016;64:171–177. doi: 10.1016/j.retram.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts Ab, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 42.Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32:231–244. doi: 10.1038/emboj.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saleh J, Figueiredo MA, Cherubini K, Salum FG. Salivary hypofunction: An update on aetiology, diagnosis and therapeutics. Arch Oral Biol. 2015;60:242–255. doi: 10.1016/j.archoralbio.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Fukui M, Ogawa Y, Shimmura S, Hatou S, Ichihashi Y, Yaguchi S, Hirayama M, Kawakita T, Tsubota K. Possible involvement of epithelial-mesenchymal transition in fibrosis associated with IgG4-related Mikulicz's disease. Mod Rheumatol. 2015;25:737–743. doi: 10.3109/14397595.2014.1002968. [DOI] [PubMed] [Google Scholar]