

Abstract
By taking inspiration from the catalytic properties of single‐site catalysts and the enhancement of performance through ionic liquids on metal catalysts, we exploited a scalable way to place single cobalt ions on a carbon‐nanotube surface bridged by polymerized ionic liquid. Single dispersed cobalt ions coordinated by ionic liquid are used as heterogeneous catalysts for the oxygen evolution reaction (OER). Performance data reveals high activity and stable operation without chemical instability.
Keywords: carbon nanotubes, ionic-liquid polymer, nanocomposites, oxygen evolution reaction, single atom catalysts
The development of efficient, stable, and low‐cost water oxidation catalysts continues to be centrally important for the conversion of renewable energy into chemical energy.1 Owing to its abundance and low cost, cobalt has become a popular non‐noble metal for the design of robust oxygen evolution reaction (OER) catalysts requiring moderate overpotentials.2 Redox reactions of the water molecule entail that the metal centers of the catalyst occupy multiple oxidation states. Because the most stable state of a metal capable of varying its oxidation states often exhibits different coordination environments, the same ligand leads to dynamical instability of a cobalt catalyst across the multielectron transformation of the OER.3 A recent report about Co2+ salts in phosphate electrolyte (Co‐Pi) shows an inexpensive self‐repair system.3 The dissolution process can be countered by continual catalyst formation establishing a dynamic equilibrium between Co2+‐HPO4 2− in solution and Co3+‐ HPO4 2− on the anodically poisoned electrode. This family of catalysts reduces the problem of ligand oxidative instability; however, the electrodeposited material consists of a film of micrometer‐sized particles, which is not very effective for a heterogeneous catalyst. Compared to nano‐ and subnano‐catalysts, single‐atom catalysts may achieve an atom utilization efficiency of 100 % and bridge the gap between homogeneous and heterogeneous catalysts.4 In the past, single atom catalysts have become a new frontier in heterogeneous catalysis.5 A nitrogen‐coordinated cobalt structure was reported and demonstrated to have excellent catalytic performance as a catalyst in oxygen reduction and C−C bond‐forming reactions.6 Although single‐atom catalysts have shown promising performance in catalyzing oxidations, hydrogenations, and other reactions of organic compounds,7 progress is still slow because of the synthetic challenge and of the lack detailed knowledge about the active sites. Therefore, developing scalable and diverse synthesis methods to further fundamental understanding of single metal atom or ion versions of catalysts are needed.
Ionic liquids (ILs), have undergone great development as solvents or catalysts in liquid‐phase catalysis.8 Polymerized ionic liquids or poly (ionic liquid)s (PILs) also have a role in some fields in polymer chemistry and materials science.9 It is well established that ILs can be used as the solvent and stabilizer to produce metal nanoparticles which have higher catalytic activities and selectivities than samples without the use of ILs.10 PILs, as another kind of stabilizer, act in a similar role, allowing highly dispersed small particles to be obtained.11 By forming PIL supported on electron‐conducting multi‐walled carbon nanotube (CNT) surfaces, which introduces a large number of surface functional groups on the CNTs, a uniform distribution of metal nanoparticles can be dispersed on CNTs.11b Recently, solid IL/nanocarbon composites have been developed with a controllable thin or even single IL layer on nanocarbon surface.12 By adding different metal catalysts to the IL layer, mono‐dispersed catalysts can be obtained.13
Herein, a functional IL (SSIL) with an imidazolium cation bearing both sulfates and vinyl groups was chosen as a ligand to load cobalt single sites onto the surface of CNTs (details in Supporting Information). As shown in Scheme 1, cobalt was introduced via a simple chemical reaction between sulfate groups in an ionic‐liquid precursor and CoCO3 forming a new kind of IL salt with Co species. The physisorption energy of the imidazolium cation of the IL on a single graphene layer can reach more than 230 kJ mol−1.12b With this adsorption force, ILs can assemble on the nanocarbon surfaces in liquid phase. Cobalt can then also be well distributed on the CNT surfaces. The subsequent thermal‐initiated free‐radical polymerization process occurred at 190 °C. The PIL is more strongly bound to the CNT than the IL monomer.
Scheme 1.
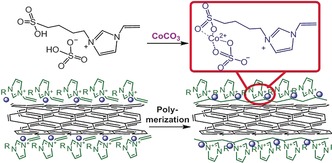
Schematic illustration of the preparation of the CoSSPIL/CNT. H2O and CO2 are byproducts of the reaction between the SSIL and CoCO3. The blue spheres represent single Co species. The image is not drawn to scale. It only shows the cation part on the CNT before and after polymerization. The structure of anion part that is covalently bonded to the CNT is given in Figure S2.
To understand whether the PIL is attached by chemical forces to the surface of the carbon nanotubes, X‐ray photoelectron spectroscopy (XPS) measurements of PIL/CNT were carried out with the sample grounded and also while applying positive and negative 5 V bias. Figure 1 a showing N1s as a representative core level of the PIL, reveals that the spectrum of the N functional group undergoes a characteristic binding energy shift, confirming the electrochemical conductivity between the PIL and the CNT surface. Compared with IL/CNT, the S1s peak of PIL/CNT shifts to higher binding energy (Figure S1 in the Supporting Information). The fitted S1s peaks of PIL/CNT are attributed to sulfate (pink peaks in Figure 1 b), sulfonate (blue peaks in Figure 1 b) and reduced sulfur (purple peak in Figure 1 b). The peak shift in Figure S1 indicates the transformation from sulfate to sulfonate, which reveals that the sulfate anion of the IL reacts with the CNT surface to form sulfonate carbon connections during the polymerization process (Figure S2). The chemical bonding between the PIL and the CNT anchors the PIL on the CNT and enhances the conductivity of the whole system. The π bonds coupling the CNTs and the PIL supply multiple binding centers and can also enhance the noncovalent interactions between polymerized IL and the CNT surfaces.12a The as‐obtained nanocomposites contain a uniform PIL coating and evenly distributed Co species on the CNT surfaces. The cation of the PIL endows charged character to the CNTs, breaking the CNT bundles by electrostatic repulsion.14 Additionally, the pure CNT and PILs cannot be dissolved in water or alcohol, but the well‐distributed PILs on the CNT surface enable the dispersion of the composites. The reason might be that the CNTs provide a high surface area to the PIL, while the PIL provides hydrophilic groups to the CNTs.
Figure 1.
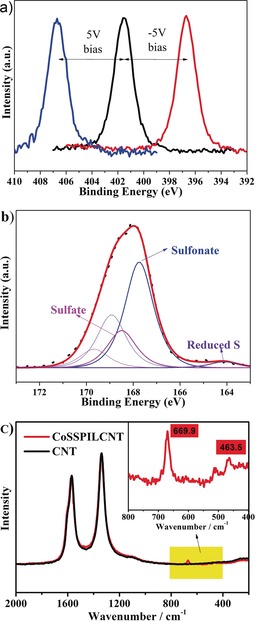
a) XPS N1s spectra of PIL/CNT undergo a characteristic 5 eV binding energy shift when applying 5 V bias (Supporting Information). b) Core level XPS S2p spectrum of the samples, and deconvolution. c) Raman spectra of pristine CNT and the composites. The inset show to the enlarged area highlighted in yellow.
The polymerization of the vinyl groups of the IL was confirmed by attenuated total reflectance infrared spectra (ATR‐IR). The main characteristic vibrations of the IL are maintained in the polymer except that the characteristic C=C peak at 855 cm−1 disappears as the C−H vibrations between 2800–3000 cm−1 increase in intensity (Figure S3). This confirms that the polymerization temperature has no obvious influence on the ionic‐liquid‐cation structure other than the vinyl group. The polymerization process is further studied by NMR spectroscopy (Figure S4). Figure 1 c shows the Raman spectra of pure CNTs and the CoSSPIL/CNT composites. No significant shift or peak shape changes for the CNTs are observed before and after the formation of polymerized IL on it, which means that no electron perturbation of the graphite π electrons is caused by the introduction of the IL polymer. Figure 1 c shows the spectrum for the Co species of the CoSSPIL/CNT sample. The main peaks of the Co species at 699.9 cm−1 and 463.5 cm−1 can be attributed to tetrahedrally and octahedrally coordinated Co2+, respectively. XPS results of different samples confirm this result. All the C1s main peaks belong to the graphitic carbon of the CNTs and no shift or obvious change occurs with the variation of the PIL content (Figure S5).
Aberration‐corrected scanning transmission electron microscopy was utilized to examine the fine structure of CoSSPIL/CNT. The annular dark‐field STEM (ADF‐STEM) image shown in Figure 2 a clearly reveals a good dispersion of the Co atoms over the surface of the CNT. As a result of their higher atomic number, Co atoms appear as bright dots. For better visualization, the Co atoms were colored pink in Figure 2 b. In addition to single atoms, a small number of Co clusters were observed on the CNT (circled with a red dotted line in Figure 2 a). They result from the aggregation of the IL polymer during polymerization. Elemental analysis was also performed. The EDX spectrum in Figure 2 c shows the presence of C, N, O, S, and Co, confirming the existence of PIL and Co in the sample. The XRD pattern of the CoSSPIL/CNT shows only the characteristic peaks of the CNT (Figure S6). This also demonstrates that both PIL and Co species are well distributed on the CNT surfaces and no obvious agglomeration happens during the synthesis.
Figure 2.
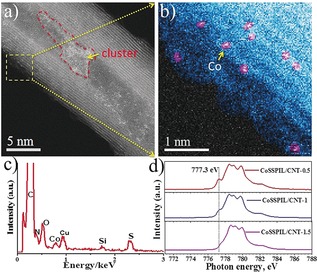
a),b) ADF‐STEM images of the CoSSPIL/CNT‐1. c) EDX spectrum of the sample. d) NEXAFS spectra of different samples.
To see whether the IL improves the catalytic performance of the metal centers in the OER, as it does in liquid‐phase catalysis, different samples were synthesized by varying the IL precursor and CoCO3 molar ratio to 0.5, 1, 1.5, 2, and 2.5, respectively. In the following, samples are denoted with these numbers. Near edge X‐ray absorption fine structure (NEXAFS) spectra of the different samples are given in Figure 2 d. The L3 edge between 775 eV and 785 eV of the spectra is due to the excitation of Co 2p core electrons to unoccupied 3d orbitals of Co2+ [2p63d7→2p53d8 for Co2+].15 The first peak of CoSSPIL/CNT‐0.5 (777.3 eV, black vertical line in Figure 2 d) is attributed to t2g‐holes3a which exist only in an octahedral environment of Co2+. With the increase of PIL content, the t2g‐hole features of CoSSPIL/CNT‐1 decrease. This indicates the configuration of the Co2+ transforms from six‐coordinated octahedral structure (splitting energy, 10 Dq) to four‐coordinated tetrahedral structure (splitting energy, 4.45 Dq). The CoSSPIL/CNT‐1.5 shows a tetrahedral configuration which has three half‐full t2g orbitals. More sulfate and sulfonate are involved in the coordination environment (as shown in Figure S7). Thus, the increase of the IL reduces the eg and t2g splitting; the Co2+ forms weak Co–ligand bonds which introduce high‐spin states. This might cause an easy oxidation of Co2+ to higher valence during the reaction.
Electrochemical impedance spectroscopy (EIS, Figure S8) gives the uncompensated resistance of the system as 35 Ω. EIS also shows that commercial CoCO3 has a much higher resistance than the CNT‐containing samples. CNT‐containing samples undoubtedly have lower resistance owing to the excellent conductivity of the CNTs, which implies CNTs are not only the support for different samples, but also provide the conductivity for the reaction. Introduction of the PIL in SSPIL/CNT and CoSSPIL/CNT‐1 does not increase the resistance. Figure 3 a and Figure S9 show the polarization curves from different samples with and without iR‐drop correction, respectively. Owing to the high dispersion of the Co species, CoSSPIL/CNT show much better OER activity than the other two Co‐containing samples (Co3O4/CNT and CoCO3), although with a much lower Co content. The comparison between the CNTs and SSPIL/CNT confirms that the polymerized IL has almost no OER improvement on the CNT sample. The key performance indicators (KPI) of the electrochemistry data are given in Table S1. As a result of the atomic dispersion of Co in the sample, all the Co atoms can be seen as active sites. A relative turnover frequency (TOF) of 0.29 s−1 is obtained at overpotential 400 mV (details in Supporting Information). This is comparable with some homogeneous catalysts.16 Figure S10 shows a decreasing capacitance with increasing IL/Co ratio. Comparing with the pristine CNT, all the IL containing samples show steeper current/potential curves, because the introduction of IL might increase the ion conductivities of the relative composites. Ion conductivity is a fundamental property of PILs. Ohno et al. found that the polymerization of IL generally lowers the ionic conductivity, but there are still some mobile ions after covalent bonding of the polymerizable groups which can provide ionic conductivity to the IL polymer.17 The cyclic voltammogram (CV) from 1 VRHE to 1.4 VRHE (Figure S11) shows a Co activation process of the CoSSPIL/CNT series.
Figure 3.
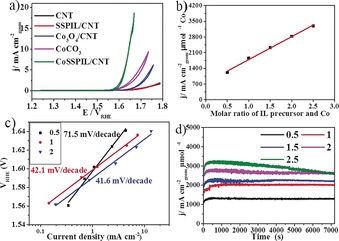
a) CV of different samples normalized to the geometric area of the active electrode area. b) Correlation of the IL content and Co activity. The content of Co and IL are values calculated from synthesis steps. c) Tafel plots of different samples. d) Stability of different samples.
The resistances of the samples have no clear relationship with the content of the polymerized IL (Figure S12), but as shown in Figure S13, the activity of per unit Co clearly improves with increasing PIL content. Figure 3 b shows a good linear relationship between Co activity (the current at 1.8 VRHE) and PIL content. This result demonstrates that the introduction of the PIL not only improves the dispersion of Co on the CNT surface, but can also enhance catalytic activity in the OER by changing the coordination of the cobalt species. NEXAFS spectra of the different samples confirm this. Tafel slopes of sample 0.5, 1, and 2 are 71.5 mV decade−1, 42.1 mV decade−1, and 41.6 mV decade−1, respectively (Figure 3 c). This also confirms the change of the electronic structure and coordination of the Co improves the Co activity. This improvement might be caused by the charge transfer between Co and IL.
The stability is another critical parameter that determines the practicability of electrocatalysts. To assess this, we performed chronoamperometric (CA) measurements by maintaining a constant potential at 1.8 VRHE for 2 h (Figure 3 d). All samples (except the sample 2.5) survived the 2 h measurement without any current changes, demonstrating the durability of the materials. Inductively coupled plasma optical emission spectrometry (ICP‐OES) detects no trace Co in the electrolyte after the CA test. The OER measurements show a good stability because the PIL ligand shows no solubility in water thus keeping the counter Co ions stable on the CNT surfaces during the reaction. Additionally, the IL is more stable compared to organic ligands because of its wide electrochemistry window. When an excess of PILs are on the CNT surfaces (sample 2.5), the excess PIL aggregate, forming polymer/CNT agglomerations. The material then disperses poorly, which leads to a poor coating on a glassy electrode. Therefore, the detachment (black particles in the electrolyte) leads to the decrease in the current of the sample (Figure 3 d). Thus, PIL improves the Co activity in the OER; the more IL used, the higher the obtainable activity. In addition, the tangled PIL on the CNT surface acts as a stable counterion and ligand of Co, stabilizing the Co ions during the reaction. However, the IL content cannot be increased indefinitely as there comes a point when dispersion problems occur. Nevertheless, a scalable way to synthesize a stable and highly active catalyst with single sites was demonstrated with a high potential of knowledge based modifications and numerous applications.
Experimental Section
Characterizations: The XPS and NEXAFS experiments were performed at the ISISS beamline at the synchrotron radiation facility BESSY II of the Helmholtz Zentrum Berlin. The measurements were carried out in a stainless steel NAP‐XPS chamber, the details of which are described elsewhere.18 The powder samples were pressed into a pellet of 8 mm diameter. Samples were placed between a stainless steel backplate and lid (with 6 mm hole) and mounted onto a sapphire sample holder. The NEXAFS spectra were obtained by recording the total electron yield (TEY). The energy resolution of the monochromator in the range of the Co l‐edge was 0.3 eV. The energies were calibrated using the C 1s first and second order peaks. The accuracy of energy calibration was estimated to be around 0.1 eV. Additional XPS experiments were carried out using a non‐monochromatized AlKα excitation in a home‐built XPS machine with SPECS (Phoibos 150) hemispherical analyzer. Raman spectra were measured by a Thermo Scientific DXR Raman Microscope with a 50× magnification and a 532 nm laser. The aberration‐corrected ABF‐STEM and ADF‐STEM image were observed with a double Cs‐corrected JEOL ARM 200 transmission electron microscope with a cold field emission gun.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
T.R and T.J acknowledge funding by the Alexander‐von‐Humboldt Foundation. The authors thank for support of the Max Planck society and Fabian Wachholz for the german translation.
Y. Ding, A. Klyushin, X. Huang, T. Jones, D. Teschner, F. Girgsdies, T. Rodenas, R. Schlögl, S. Heumann, Angew. Chem. Int. Ed. 2018, 57, 3514.
References
- 1. Lewis N. S., Nocera D. G., Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Aijaz A., Masa J., Rosler C., Xia W., Weide P., Botz A. J. R., Fischer R. A., Schuhmann W., Muhler M., Angew. Chem. Int. Ed. 2016, 55, 4087–4091; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4155–4160; [Google Scholar]
- 2b. Feng J. X., Xu H., Dong Y. T., Ye S. H., Tong Y. X., Li G. R., Angew. Chem. Int. Ed. 2016, 55, 3694–3698; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3758–3762; [Google Scholar]
- 2c. Wurster B., Grumelli D., Hötger D., Gutzler R., Kern K., J. Am. Chem. Soc. 2016, 138, 3623–3626. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hadt R. G., Hayes D., Brodsky C. N., Ullmann A. M., Casa D. M., Upton M. H., Nocera D. G., Chen L. X., J. Am. Chem. Soc. 2016, 138, 11017–11030; [DOI] [PubMed] [Google Scholar]
- 3b. Kanan M. W., Nocera D. G., Science 2008, 321, 1072–1075. [DOI] [PubMed] [Google Scholar]
- 4. Schlögl R., Angew. Chem. Int. Ed. 2015, 54, 3465–3520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3531–3589. [Google Scholar]
- 5.
- 5a. Li C., Chin. J. Catal. 2016, 37, 1443–1445; [Google Scholar]
- 5b. Thomas J. M., Nature 2015, 525, 325–326; [DOI] [PubMed] [Google Scholar]
- 5c. Buller S., Strunk J., J. Energy Chem. 2016, 25, 171–190. [Google Scholar]
- 6.
- 6a. Yin P., Yao T., Wu Y., Zheng L., Lin Y., Liu W., Ju H., Zhu J., Hong X., Deng Z., Zhou G., Wei S., Li Y., Angew. Chem. Int. Ed. 2016, 55, 10800–10805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10958–10963; [Google Scholar]
- 6b. Zhang L., Wang A., Wang W., Huang Y., Liu X., Miao S., Liu J., Zhang T., ACS Catal. 2015, 5, 6563–6572. [Google Scholar]
- 7. Liu W., Zhang L., Yan W., Liu X., Yang X., Miao S., Wang W., Wang A., Zhang T., Chem. Sci. 2016, 7, 5758–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Dupont J., de Souza R. F., Suarez P. A. Z., Chem. Rev. 2002, 102, 3667–3691; [DOI] [PubMed] [Google Scholar]
- 8b. Wasserscheid P., Keim W., Angew. Chem. Int. Ed. 2000, 39, 3772–3789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3926–3945; [Google Scholar]
- 8c. Welton T., Chem. Rev. 1999, 99, 2071–2083. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Armand M., Endres F., MacFarlane D. R., Ohno H., Scrosati B., Nat. Mater. 2009, 8, 621–629; [DOI] [PubMed] [Google Scholar]
- 9b. Lu W., Fadeev A. G., Qi B., Smela E., Mattes B. R., Ding J., Spinks G. M., Mazurkiewicz J., Zhou D., Wallace G. G., MacFarlane D. R., Forsyth S. A., Forsyth M., Science 2002, 297, 983–987; [DOI] [PubMed] [Google Scholar]
- 9c. Yuan J., Antonietti M., Polymer 2011, 52, 1469–1482; [Google Scholar]; Mecerreyes D., Prog. Polym. Sci. 2011, 36, 1629–1648. [Google Scholar]
- 10.
- 10a. Ding Y. X., Zhu W. S., Li H. M., Jiang W., Zhang M., Duan Y. Q., Chang Y. H., Green Chem. 2011, 13, 1210–1216; [Google Scholar]
- 10b. Chen W., Zhang Y. Y., Zhu L. B., Lan J. B., Xie R. G., You J. S., J. Am. Chem. Soc. 2007, 129, 13879–13886. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Mu X.-d., Meng J.-q., Li Z.-C., Kou Y., J. Am. Chem. Soc. 2005, 127, 9694–9695; [DOI] [PubMed] [Google Scholar]
- 11b. Wu B., Hu D., Kuang Y., Liu B., Zhang X., Chen J., Angew. Chem. Int. Ed. 2009, 48, 4751–4754; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4845–4848; [Google Scholar]
- 11c. Gao M.-R., Yu S.-H., Yuan J., Zhang W., Antonietti M., Angew. Chem. Int. Ed. 2016, 55, 12812–12816; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13004–13008. [Google Scholar]
- 12.
- 12a. Ding Y., Su D. S., ChemSusChem 2014, 7, 1542–1546; [DOI] [PubMed] [Google Scholar]
- 12b. Ding Y., Sun X., Zhang L., Mao S., Xie Z., Liu Z. W., Su D. S., Angew. Chem. Int. Ed. 2015, 54, 231–235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 233–237. [Google Scholar]
- 13. Ding Y., Zhang B., Gupta N., Su D. S., Green Chem. 2015, 17, 1107–1112. [Google Scholar]
- 14. Fukushima T., Kosaka A., Ishimura Y., Yamamoto T., Takigawa T., Ishii N., Aida T., Science 2003, 300, 2072–2074. [DOI] [PubMed] [Google Scholar]
- 15. de Groot F. M. F., Abbate M., Elp J. V., Sawatzky G. A., Ma Y. J., Chen C. T., Sette F., J. Phys. Condens. Matter 1993, 5, 2277. [Google Scholar]
- 16. Yin Q. S., Tan J. M., Besson C., Geletii Y. V., Musaev D. G., Kuznetsov A. E., Luo Z., Hardcastle K. I., Hill C. L., Science 2010, 328, 342–345. [DOI] [PubMed] [Google Scholar]
- 17. Ohno H., Macromol. Symp. 2007, 249, 551–556. [Google Scholar]
- 18. Knop-Gericke A., Kleimenov E., Havecker M., Blume R., Teschner D., Zafeiratos S., Schlogl R., Bukhtiyarov V. I., Kaichev V. V., Prosvirin I. P., Nizovskii A. I., Bluhm H., Barinov A., Dudin P., Kiskinova M., Adv. Catal. 2009, 52, 213–272. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary