

Abstract
Skin sensitization associated with the development of allergic contact dermatitis occurs via a number of specific key events at the cellular level. The molecular initiating event (MIE), the first in the sequence of these events, occurs after exposure of the skin to an electrophilic chemical, causing the irreversible haptenation of proteins within skin. Characterization of this MIE is a key step in elucidating the skin sensitization adverse outcome pathway and is essential to providing parameters for mathematical models to predict the capacity of a chemical to cause sensitization. As a first step to addressing this challenge, we have exposed complex protein lysates from a keratinocyte cell line and human skin tissue with a range of well characterized sensitizers, including dinitrochlorobenzene, 5-chloro-2-methylisothiazol-3-one, cinnamaldehyde, and the non (or weak) sensitizer 6-methyl coumarin. Using a novel stable isotope labeling approach combined with ion mobility-assisted data independent mass spectrometry (HDMSE), we have characterized the haptenome for these sensitizers. Although a significant proportion of highly abundant proteins were haptenated, we also observed the haptenation of low abundant proteins by all 3 of the chemical sensitizers tested, indicating that within a complex protein background, protein abundance is not the sole determinant driving haptenation, highlighting a relationship to tertiary protein structure and the amino acid specificity of these chemical sensitizers and sensitizer potency.
Keywords: cinnamaldehyde, DNCB, keratinocytes, MCI, DIA, HDMSE, proteomics, sensitization, skin
Skin sensitization, which leads to the development of allergic contact dermatitis (ACD), is the most common manifestation of immunotoxicity found in humans. Approximately 15%–20% of people living in North America and Western Europe become sensitized to at least 1 contact allergen (Thyssen et al., 2007) in an occupational or a domestic setting. Contact allergy occurs in 2 stages: first, the sensitization phase, whereby a chemical penetrates through the stratum corneum and reaches the viable epidermis, covalently modifying (haptenating) skin proteins, inducing the generation of allergen-specific T cells; second, the elicitation phase in which re-exposure to the same (or crossreactive) chemical allergen leads to a cascade of biochemical and cellular processes, effectively recalling the allergen-specific T cells to the exposure site, resulting in a clinical manifestation of ACD (Basketter et al., 1995; Landsteiner and Jacobs, 1935; Lepoittevin, 2006). Our current understanding of the sequence of events involved in the development of sensitization and ACD is reflected in the more recent literature (Karlberg et al., 2008; Koppes et al., 2017; Martin, 2015) aspects of which are used in defining the adverse outcome pathway (AOP) for skin sensitization (OECD, 2012). This framework links our existing knowledge of the direct molecular initiating event (MIE), the haptenation of proteins within skin, to the adverse outcome, ACD, via a number of specific key events at the cellular level (Ezendam et al., 2016; Vinken, 2013). Although a simplified view, the use of AOPs provides the basis for a mechanistic understanding of the effect of a chemical at the molecular and subcellular level. Through use of mathematical modeling, AOPs underpin the development and improvement of strategies for chemical and drug safety assessment (Burden et al., 2015; Maxwell and Mackay, 2008; Strickland et al., 2016). Characterization of the MIE, ie, skin protein haptenation, is a key step in understanding the skin sensitization AOP leading to more reliable mathematical models and their use in risk assessment (Jaworska et al., 2013; MacKay et al., 2013).
Most sensitizers are electrophilic in nature, or can easily be converted to an electrophile. As such, they are likely to react with nucleophilic side chains of protein amino acid residues, mainly lysine and cysteine, and to a lesser extent tyrosine, histidine and arginine (Ahlfors et al., 2003). The modification of proteins by chemical sensitizers is generally regarded as an irreversible reaction and, given the importance of this step to skin sensitization, has been studied extensively. To this end, a number of experimental approaches have been utilized to determine the reactivity of sensitizing chemicals and the reaction rates of chemicals with model nucleophiles. Previously, researchers studied chemical reactivity using nucleophilic chemicals analogous to side chains of nucleophilic amino acids (Alvarez-Sanchez et al., 2003; Chipinda et al., 2011; Sanderson et al., 2016) whereas others used simple short peptides with single or multiple nucleophilic amino acids as biological target surrogates (Aeby et al., 2010; Aleksic et al., 2009; Gerberick et al., 2004, 2007; Natsch and Gfeller, 2008; Roberts and Natsch, 2009). Further understanding was obtained using a variety of model proteins (Aleksic et al., 2007; Alvarez-Sanchez et al., 2004b; Parkinson et al., 2014). These studies define 3 main factors that determine the binding of sensitizers to nucleophiles: electrophilicity of the sensitizer, nucleophilicity of the target and steric constraints. However, these simple experimental systems used to determine protein haptenation differ from the complex milieu of skin in a number of ways, such as: competition for binding between the proteins present, differences in protein expression levels, differences in local pH, micro-bioavailability, and steric hindrance.
The identity and location of haptenated skin proteins is currently unknown, e.g. whether intra- or extracellular space or location within the membrane of a specific cell type may provide optimum conditions for haptenation. However, the epidermis and dermis are generally regarded as the skin sites where these modifications become available to the immune system (Kimber et al., 2011; Kimber and Dearman, 2003; Pickard et al., 2009).
The limited number of detailed investigations of protein haptenation in complex protein mixtures, including cell lines and tissues such as human skin, have so far focused on the use of antibodies to specific sensitizer adduct(s) (Elahi et al., 2004), biotin-tagged electrophiles (Codreanu et al., 2009; Hong et al., 2005), click chemistry (Jacobs and Marnett, 2010), derivatization of protein bound carbonyls and aldehydes with biotin hydrazides (Conrad et al., 2001; Mello et al., 2007; Shearn et al., 2016; Spiess et al., 2011) or dependent upon intrinsic features of certain sensitizers (such as fluorescent adducts of monobromobimane) to pinpoint the amino acid site of haptenation (Simonsson et al., 2011).
Identifying sites of protein modification in complex mixtures clearly represents a considerable analytical challenge and, to date, there is no globally applicable methodology. We have previously demonstrated an increased sensitivity in detecting haptenated peptides within the model protein human serum albumin (HSA) by combining a stable isotope labeling approach with data-independent acquisition mass spectrometry (MS) (Parkinson et al., 2014). This approach revealed more about the modification of HSA by a range of sensitizing chemicals than had previously been known. To further advance our understanding of the qualitative and quantitative aspects of skin protein haptenation, an assessment of protein modification by sensitizers within the complex skin proteome is required. To address this challenge, we have exposed protein lysates from a keratinocyte cell line and human skin tissue to the well characterized sensitizers, dinitrochlorobenzene (DNCB), 5-chloro-2-methylisothiazol-3-one (MCI), cinnamaldehyde (CA), and 6-methyl coumarin (6-MC), which has been classed as a nonsensitizer in the murine local lymph node assay (Ashby et al., 1995).
The results presented here demonstrate that in a complex protein mixture, protein abundance is not the sole determinant of protein haptenation. We observe a degree of specificity of some chemicals towards binding certain amino acid side chains and a possible effect of the protein tertiary structure on the likelihood of haptenation. Additionally, we observe a relationship between the extent of haptenation and sensitizer potency; however we refrain from making firm conclusions due to a low number of chemicals tested. Based upon these experimental data, we have highlighted useful parameters for advancing the development of in silico mathematical models of skin sensitization (Maxwell et al., 2014).
MATERIALS AND METHODS
Chemical Sensitizers
DNCB (99% purity; MW 202.55 Da) was obtained from Sigma-Aldrich (Poole, UK), and DNCB-D3 (99% purity; MW 205.57 Da) was obtained from QMX Laboratories (Dunmow, UK).
Trans-CA (99% purity; MW 132.16 Da), Diphenylcyclopropenone (DPCP) (98% purity; MW 206.24 Da) and 6-MC (99% purity; MW 160.17 Da) were obtained from Sigma-Aldrich and trans-CA-D5 (98% purity; MW 137.12 Da), DPCP-D10 (97.1% purity; MW 211.27 Da) and 6-MC-3 D (99% purity; MW 163.15 Da) were custom synthesized by Quotient Amersham Radiochemicals (Irvine, California).
MCI (MW 149.60 Da) and 13 C labeled MCI (MW 150.8 Da) were synthesized and kindly donated by Prof Jean-Pierre Lepoittevin and Dr Elena Gimenez Arnau, Labarotoire de Dermatochimie, Strasbourg. Isotopically modified atoms for each chemical are shown in Table 1.
Table 1.
Structures, Position of Stable Isotope, Potency Category (Including EC3 Value, as Derived From the Local Lymph Node Assay), Δ Mass (Da) Expected Following Haptenation, Potential Reactivity Domain, and Possible Amino Acid Residue for Modification, Based on Data Shown in Parkinson et al. (2014)
| Chemical | Structure and Position of Stable Isotope Labels (*) | Potency Category (% EC3) | Δ Mass (Da) Expected for Unlabeled and (Labeled) Adduct | Potential Reactivity Domains | Residue |
|---|---|---|---|---|---|
| 5-chloro-2-methyl-4-isothiazol-3-one (MCI) | 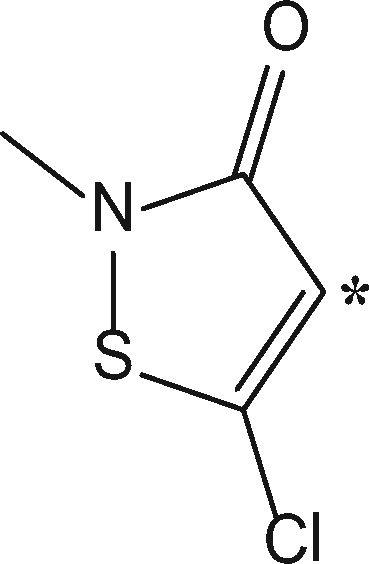 |
Extremea (0.0009) | +99.032b (+100.035) | Amide adductb | Cys, Lys, His, Tyra |
| +112.9935b (+113.9965) | Addition-Eliminationb | Cys, Lys, Tyra | |||
| +115.0092b (+116.0122) | Thioamide adductb | Cys, Lys, His, Tyra | |||
| 1-chloro-2, 4-dinitrobenzene (DNCB) | 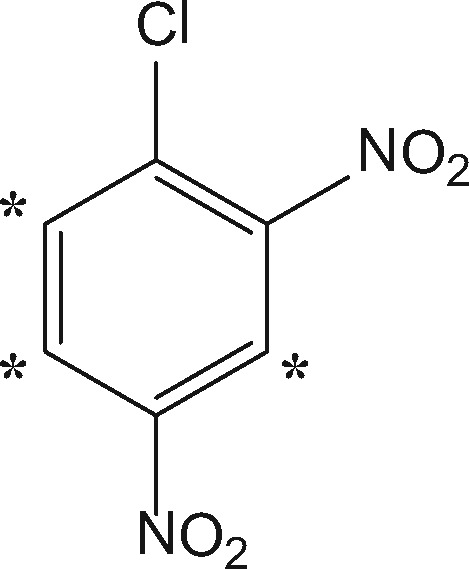 |
Extremec (0.05) | +166.0015d (+169.0195) | SNArd | Cys, His, Lys, Tyre |
| Cinnamaldehyde (CA) | 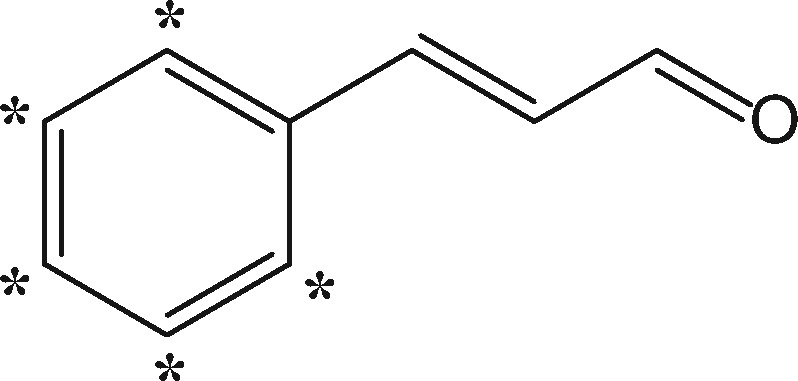 |
Moderatef (3.0) | +114.047 (+119.078) | Schiff baseg | Arg, Lyse |
| +132.0575 (+137.0885) | Michael adduct; acylation | Arg, Cys, His, Lyse | |||
| 6-methylcoumarin (6-MC) | 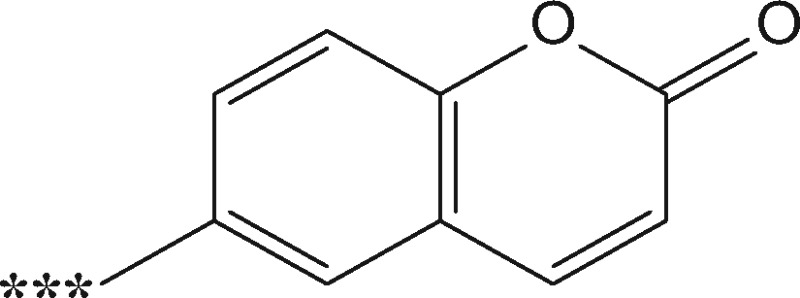 |
Nonsensitizere | +158.0368 (+161.0548) | Michael adduct | Cys, Lyse |
| +160.0525 (+163.0705) | Acylation |
Collection of Human Skin Samples
Full-thickness human skin samples were obtained from mastectomy surgery at Southampton General Hospital with the patients’ signed consent, under the guidelines stated in ethics protocol 07Q170459, snap frozen and stored at −80 °C.
Culturing of Keratinocyte Cells
The adherent keratinocyte cell line (HaCaT) was cultured in Dulbecco’s Modified Eagle Medium, high glucose, (supplemented with 10% heat inactivated fetal calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml Penicillin and 100 µg/ml Streptomycin) (Gibco, ThermoFisher, Paisley, UK), at 37 °C and 5% CO2. Once the cells had reached 70% confluency the media was removed and the cells washed twice with phosphate buffered saline (PBS). Cells were scraped into 10 ml of PBS and centrifuged for 5 min at 300 × g and the cell pellets stored at −80 °C until required.
Processing of Skin Tissue and Cell Line Pellets
Full-thickness skin tissue samples were thawed on ice, washed in Hanks’ Buffered Saline Solution (HBSS, Gibco) and cut into pieces approximately 0.5 × 0.5 cm and placed into reinforced 1.5 ml tubes containing ceramic beads (Matrix D – QBioGene, Cambridge, UK) and 500 µl of lysis buffer (0.1% SDS in 0.1 M TEAB).
Cell line pellets were thawed on ice and transferred in a small volume of HBSS into tubes containing Matrix D and lysis buffer to a final concentration of 0.1% SDS in 0.1 M TEAB as described earlier.
The skin samples and cell pellets were then processed in the same way using a FastPrep macerator (MP Biomedical, Fisher Scientific, Loughborough, UK) for 5 cycles of 45 s; speed setting 6, chilled on ice for 1 min between cycles. Insoluble material was pelleted by centrifugation at 9000 × g for 5 min and discarded and the supernatant stored at −80 °C until required.
Estimation of Protein Concentration
The protein concentrations of the lysates generated were determined using the bicinchoninic acid method (Smith et al., 1985; Wiechelman et al., 1988) using a kit from Sigma-Aldrich.
Protein Modification With Sensitizers
To investigate the differences in protein haptenation with a range of chemicals, stock solutions of sensitizers were prepared in 100% DMSO (for DNCB) or 100% ethanol (for DNCB, CA, MCI and 6-methyl-coumarin) containing 50%, by molar concentration, of unlabeled sensitizer and 50% stable isotope labeled sensitizer (see Table 1). Lysates of keratinocyte cells or ex vivo skin were diluted to a concentration of 1 mg/ml in 0.1 M TEAB (pH 8.0) + 0.1% SDS prior to treatment with a 1:100 molar excess of sensitizer to protein and incubated at 37 °C for 4 weeks. The molarities of the protein lysates were approximated based upon the average molecular weight (66 kDa) of proteins within the samples. Control samples were prepared at the same concentration in 0.1 M TEAB with the addition of 0.2% of the relevant solvent.
Sample Cleanup and Digestion
Proteins were precipitated using an adapted Bligh Dyer method (Bligh and Dyer, 1959). To 100 µg of modified protein lysate, 4 volumes of methanol were added and the sample vortexed. One volume of chloroform was added to the sample/methanol solution and vortexed before finally adding 3 volumes of water followed by vortexing. The sample was centrifuged at 20 000 × g for 1 min, focusing the proteins between the organic and inorganic phases. The aqueous phase was removed and 4 volumes of ethanol were added, followed by a short vortex. The precipitate was pelleted by centrifugation at 20 000 × g for 2 min, the ethanol removed, and the pellet air-dried.
The pellet was resolubilized in buffer containing 6 M urea, 2 M thiourea and 10 mM HEPES, pH 7.5. Proteins were reduced with dithiothreitol for 1 h at 60 °C, alkylated with 5.5 mM iodoacetamide for 45 min in the dark at room temperature, and then digested for 4 h with the protease Lys-C (Thermo Pierce, Loughborough, UK) (1/50 w/w). Peptides were then diluted 4 times with 20 mM ammonium bicarbonate and further digested using sequencing grade modified trypsin (1/50 w/w, Promega, Southampton, UK) overnight at 37 °C.
Fractionation of Peptides
To increase proteome coverage of the haptenated peptide digests, the samples were separated into 12 fractions based on their isoelectric points. This was performed using the Agilent 3100 OFFGEL Fractionator in combination with 13 cm Immobiline IPG strips, pH 3-10. 100 µg peptide samples were made up to a final volume of 1.4 ml with a 1: 50 solution of IPG buffer, pH 3-10 (GE Life Sciences, Buckinghamshire, UK) diluted in 5% Glycerol. Peptides were focused for 20 kVh at a maximum current of 50 µA and a maximum power of 200 mW.
Each fraction was collected and acidified by adding 10 µl of solvent containing 10% TFA. Each acidified fraction was loaded onto a conditioned C18 reverse-phase Empore Plates (3 M, Maplewood, Minnesota, USA), and washed with 20 µl of 0.5% acetic acid. Peptides were eluted from the tip using 40 µl of 80% acetonitrile + 0.5% acetic acid. Samples were lyophilized using a vacuum concentrator to 6 µl and mixed with 6 µl of 2% acetonitrile + 1% TFA.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analysis
In duplicate, 10 µl of the fractionated sample was loaded onto a reverse phase trap column (Symmetry C18, 5 µm, 180 µm × 20 mm, Waters Corporation, Milford, Massachusetts, USA), at a trapping rate of 5 µl/min and washed for 10 min with buffer A prior to the analytical nanoscale LC separation using a C18 reversed phase column (HSS T3, 1.8 µm, 200 mm × 75 µm, Waters, Wilmslow, UK). The eluted peptides were fractionated over a 90 min linear gradient from 1% acetonitrile + 0.1% formic acid to 60% acetonitrile + 0.1% formic acid, at a flow rate of 300 nl/min. Eluted samples were sprayed directly into a Synapt G2-S MS (Waters Corporation, Wilmslow, UK) operating in the data independent High Definition MS (HDMSE) mode. Data were acquired from 50 to 2000 m/z using alternate low and high collision energy (CE) scans. Low CE was 5 V and elevated CE was ramped from 15 to 40 V. Ion mobility was implemented prior to fragmentation using a wave height of 650 m/s and wave velocity of 40 V. The lockmass Glu[1]-Fibrinopeptide B ((M + 2 H)+2, m/z = 785.8426) was infused at a concentration of 100 fmol/µl at a flow rate of 250 nl/min and acquired every 60 s.
Database Searches
The raw mass spectra were processed using ProteinLynx Global Server (PLGS) 3.0 (Waters Corporation, Wilmslow, UK) to generate reduced charge state and deisotoped precursor and associated product ion peak lists. These peak lists were searched against the UniProt Homo sapiens sequence database (obtained from UniProt 03/2010). A maximum of 2 missed cleavages was allowed for tryptic digestion and the variable modification was set to contain oxidation of methionine, carboxyamidomethylation of cysteine and sensitizer-specific haptenation(s) as detailed in Table 1.
Precursor ion and product ion mass tolerances were calculated automatically during data processing and the allowed protein false discovery rate was set at 4%.
Data Filtering
Following database searching the data was filtered to eliminate falsely identified sensitizer modified peptides. Precursor ion peak pairs were extracted from the monoisotopic deconvoluted spectrum files generated from processing raw data using the data processing software based on the following criteria; ion pairs with a fixed mass difference, (corresponding to the number of stable isotopes incorporated into the labeled sensitizer), with similar ion intensity and according to retention time (with a retention time window of 1 min). Extracted peptide pairs were correlated with modified peptide masses identified after database searching using m/z and retention time. Extracted ion chromatograms of the filtered modified peptides were compared with those from the control samples to filter any remaining false positives. Fragmentation spectra were subsequently manually inspected and the amino acid site of modification determined, where possible.
Calculating Protein Abundance
Protein abundance was calculated based upon the method (Silva et al., 2006) where the sum of the intensity of the 3 most abundant peptides of an enolase digest standard (Waters Corporation, Wilmslow, UK) at a known concentration was used as a response factor to estimate the concentration of each protein in the samples based on the sum of the intensity of their 3 most intense peptide signals.
Calculating Nucleophile Content
The final nucleophile concentration for each protein was calculated as follows:
Protein amount in ng: Estimated protein abundance in ng was calculated as described above, normalized to total protein loaded in each MS run, and then averaged across all MS runs for either HaCaT or skin lysates.
Protein concentration in fmol: (ng protein/MW)*1000; where MW is protein molecular weight in kDa.
Nucleophile concentration: Sum of nucleophilic residues (excluding cysteine residues that are known to form disulfide bridges) * protein concentration (fmol).
RESULTS
Using a previously published dual labeling approach (Parkinson et al., 2014), HaCaT cell and human skin protein lysates were haptenated by DNCB, MCI, cinnamic aldehyde, and 6-MC, with a 100-fold molar excess of chemical to protein, for 4 weeks. By using an artificially high (nonclinical) concentration ratio as well as an extended exposure we hoped to ensure that all possible haptenation reactions would occur at levels that were detectable. This would firstly enable us to gain confidence in detecting these reactions in complex mixtures (i.e. inability to detect any haptenated sites at this level would render the method unsuitable for more clinically relevant experiments with lower concentrations and shorter exposures), and secondly help us to gain baseline data for future experiments where we would want to quantify the levels of haptenation. Haptenated samples were precipitated using chloroform/methanol to remove excess sensitizer prior to digestion with LysC and trypsin. The resulting peptides mixtures were fractionated via OFFGel Fractionation and each fraction was analyzed by MS. Due to the stochastic nature of peptide haptenation, modified peptides are present in low abundance (Figure 1). A ion-mobility assisted data-independent mode of acquisition (HDMSE) in combination with a dual isotope labeling method was used to confidently identify haptenated peptides within these complex lysates.
Figure 1.

Distribution of ion intensity between unmodified and sensitizer modified peptides. Modified peptides are observed at lower ion intensity compared with unmodified peptides within the same sample.
Raw MS data were processed and searched against the UniProt H. sapiens sequence database using PLGS. Modified peptides were confirmed where a peptide signature consisting of 2 peptide isotope clusters of fixed Δ m/z were observed in an MS spectrum. The product ion spectrum of the modified precursor ion was subsequently manually inspected to determine the site of haptenation.
A total of 7208 proteins (≥2 peptides) were identified across the 2 datasets, 6396 proteins in the keratinocyte lysates and 2423 in lysates from the skin tissue. From the total 7208 proteins identified, 400 proteins (5.5%) were modified by at least 1 chemical sensitizer. Analysis of gene ontology terms associated with the proteins identified exhibited a high level of similarity across both datasets with 70% of the proteins assigned in skin also identified in the keratinocyte cell lysates (Supplementary Figure 1). The proportion of modified proteins assigned within each dataset were equally similar demonstrating the utility of the keratinocyte cell line as a useful model for assessing global protein haptenation in skin tissue. In the keratinocyte cell lysates, 213 peptides related to 162 proteins (2.5% of keratinocyte cell proteome) were modified by DNCB; 204 peptides related to 159 proteins (2.5%) by MCI; and 85 peptides related to 71 proteins (1.1%) by CA. In the skin lysates, 66 peptides related to 43 proteins (1.8% of the skin proteome) were modified by DNCB; 41 peptides related to 30 proteins (1.2%) by MCI; and 50 peptides related to 41 proteins (1.7%) by CA. Only a single peptide was modified by 6-MC in the keratinocyte cell lysates and no 6-MC modifications were observed in the skin lysates (see Supplementary Table 1). Despite the high concentration of reactive chemicals used, prolonged incubation time and cell lysis/tissue maceration, the level of haptenation observed was relatively low.
Quantified proteins were ranked in order of abundance from the highest to least abundant to test the hypothesis that high abundant proteins were more likely to be modified because of a greater number of available modifiable sites (Figure 2). Although a large proportion of the haptenated proteins were from proteins present at high abundance, we observed the haptenation of proteins across the abundance range by DNCB, MCI, and CA, demonstrating that protein abundance is not the only single factor driving protein haptenation.
Figure 2.

Graphical representation of all the modified proteins identified with the keratinocyte cell (A) and ex vivo skin (B) lysates, ranked by their abundance which was based on their estimated protein concentration (fmol). The x-axis represents all of the proteins identified in each lysate from most abundant to least; the y-axis shows which of those proteins were modified by each of the 3 chemical sensitizers tested in this study.
The total theoretical nucleophile concentrations within the HaCaT cell line and skin lysates were similar (19.0% of the keratinocyte and 18.6% of the skin proteome containing modifiable nucleophilic residues Lys, Arg, Cys, Tyr, and His). The percentage nucleophile content within each protein identified in both keratinocytes and skin samples was calculated and the proteins ranked according to their nucleophile concentration (high to low). Mapping of sensitizer modified proteins to this ranked list showed a correlation between protein modification and nucleophile concentration within the HaCat cell lysates, but was not observed for the ex vivo skin lysate data (Figure 3).
Figure 3.

Circular representation of the nucleophilic concentration of each protein identified within the keratinocyte (A) and ex vivo skin (B) lysates, from highest concentration moving clockwise to the lowest concentration of nucleophiles. Proteins that were modified by each of the adducts observed in this study are highlighted within these circular representations, to show the distribution of nucleophile concentration amongst the haptenated proteins.
Across the haptenated peptides identified (haptenome), a total of 252 amino acid residues in HaCaT cell lysates were haptenated by DNCB, 210 by MCI, 98 by CA and 1 by the nonsensitizing chemical 6-MC. In human skin lysates, 102 amino acid residues were haptenated by DNCB, 43 by MCI and 57 by CA. No confirmed haptenated amino acids were found for 6-MC. The percentage of haptenated residues observed for each sensitizer adduct as a proportion of the total number of each amino acid residue observed within each proteome were calculated (Figure 4). DNCB and the +99 adduct of MCI both modified the largest number of residues with a preference for lysine. The +113 and +115 adducts of MCI do not show any nucleophile preference, whilst CA appears to modify a higher percentage of arginine and lysine residues. The CA preference for formation of Schiff bases (in particular with arginine) was previously observed when using the model protein HSA (Parkinson et al., 2014). This is particularly interesting since CA reactivity has mainly been studied from the basis that Michael addition reaction with protein thiols is dominant (Roberts et al., 2007). For all chemicals tested, we detected a number of potential further sites of haptenation where the exact site of haptenation could not be confirmed from the fragmentation spectra. These modifications are shown as gray within Figure 4.
Figure 4.

Percentage of available nucleophilic residues haptenated by adducts of DNCB, MCI, and CA in the keratinocyte (A) and ex vivo skin (B) lysates. Light Gray bars indicate where the exact site of sensitizer modification could not be confirmed from the fragmentation spectra.
Many proteins were found to be modified at more than 1 amino acid residue as well as by more than 1 sensitizer. The 20 proteins found to have the highest number of observed modifications are summarized in Supplementary Table 1, these included Complement C3 with 8 residues modified by DNCB alone; Prelamin-A/C (P02545) with 8 residues modified by DNCB, MCI, and CA; Heat shock cognate 71 (P11142) with 8 residues modified by DNCB and MCI; Pyruvate kinase (P14618) with 10 residues modified with DNCB, MCI and CA; and Serum Albumin (P02768) with 29 residues modified by all 3 chemicals. A summary of all the modified proteins can be found in the Supplementary Table 2.
DISCUSSION
Despite the prevalence of skin allergy, our knowledge about the process of protein haptenation, a key MIE, is limited. Current mechanistic knowledge of haptenation is derived from studies utilizing model peptides or isolated single proteins (Ahlfors et al., 2003; Aleksic et al., 2007, 2008, 2009; Gerberick et al., 2007; Parkinson et al., 2014). Haptenation of a single protein (HSA) has also been shown to stimulate hapten-specific T-cell responses in a number of studies, such as the production of a stable HSA-penicillin G complex (Brander et al., 1995), the occurrence of DNP adducts after the modification with the extreme sensitizer 2, 4-dinitrobenzesulfoinc acid (Dietz et al., 2010) and via p-phenylenediamine modification of cysteine 34 on HSA (Jenkinson et al., 2010). Although useful in understanding the reactivity of a variety of chemicals, these studies do not provide any insights on haptenation within the milieu of the skin proteome. As a first important step towards understanding the complexity of haptenation in a complex protein mixture we have sought to identify sensitizer haptenated peptides in protein lysates of the HaCaT cells and human skin tissue. This was achieved using a novel approach combining isotopic labeling with the data independent MS acquisition method (HDMSE), which successfully pinpointed and identified low abundance haptenated proteins, initially in a single model protein (Parkinson et al, 2014) and now in these complex mixtures.
In total, 7208 proteins were identified in this study, 6396 proteins in keratinocyte cell lysates, and 2423 in lysates from skin tissue. The difference in the numbers of proteins identified likely reflects differences in the efficiency of protein extraction from a monolayer of cells versus whole skin tissue, albeit comparison of the gene ontology terms associated with the identified proteins across the datasets of both sample types are similar (Supplementary Figure 1). Moreover, percentages of proteins that were modified by a sensitizer within each sample type were comparable, and many proteins were found to be modified in both HaCat and ex vivo skin lysates (eg, Keratin’s, Annexin A2, HSP90, Glutathione S transferase, alpha actinin). Although modified samples were analyzed in duplicate, the majority of modifications identified were mainly observed within a single replicate. Detected modified peptides were present at very low abundance with MS measurements close to the lower limits of detection resulting in increased missingness, challenging the repeatability of observed modified peptides across samples.
The underlying concept for the induction of sensitization is that a chemical must be able to covalently react with proteins, either directly or indirectly in skin. Based upon the data obtained in this study, there are clear indications that the previous assumption that only highly abundant proteins are likely to be modified preferentially (Hopkins et al, 2005) may not necessarily be correct. Although the majority of haptenated proteins identified within the keratinocyte cell lysates were highly abundant, the data indicates that low abundant proteins are also haptenated by all 3 of the chemical sensitizers tested. Equally, in skin lysates, where fewer haptenated proteins were identified overall, we found that both high- and low-abundant proteins were modified, indicating specificity in protein haptenation.
To further understand this specificity, and to provide useful parameters for building in silico mathematical models of sensitization, we investigated the relationship between protein modification and proteome nucleophile concentration, ie, whether the numbers of theoretically available reactive sites correlate with their likelihood of modification by a chemical sensitizer. For HaCat cell lysates, our data showed a correlation between the total protein nucleophile concentration and the number of proteins haptenated, whereas for ex vivo skin lysates, this was not evident. This difference is likely to be attributable to the greater proteome coverage obtained for HaCat cell lysates, compared with ex vivo skin. Although we would expect to see more modifications at higher nucleophile concentrations, we also observed modifications of proteins at lower nucleophile concentration, which may reflect differences in the accessibility of nucleophilic residues to modification. Our data demonstrated a specificity for modification of certain nucleophilic residues over others between each of the different sensitizers tested. A similar finding was observed in the direct peptide reactivity assay, although the amino acid specificity in this study differs slightly (Aleksic et al., 2009). For example, after correcting for overall abundance of each residue within each dataset, our data show that DNCB binds predominantly to lysine residues and, to a much lesser extent, available cysteine residues, with very few tyrosine and histidine modifications observed. This is in contrast to the direct peptide reactivity assay, which showed almost 100% depletion of cysteine, lysine and tyrosine containing peptides. Although we believe the observed differences are more likely to be attributed to the effect of secondary and tertiary structure on protein modification as well as an overall high abundance of amines (Parkinson et al, 2014), it is possible that this is the result of an experimental artifact (decreased number of free thiols as a consequence of cysteine oxidation).
The complex reactivity of MCI obtained in this study was in agreement with previously published studies (Alvarez-Sanchez et al., 2003, 2004a,b; Parkinson et al., 2014) in terms of the nucleophile specificity.
Very strong bias for reaction with amines was also observed for CA. As already indicated, experimental Cys oxidation and high abundance of amines may be responsible, but there are additional indications from the literature that may explain this bias in case of CA. Although there is no direct evidence from these experiments, it is plausible that initial Michael addition of CA to thiols may be reversed. This would result first in a thiazolidine type product (making a cross-link between Cys and eg, Lys) followed ultimately by Schiff base adduct formation and release of the thiol originally conjugated to CA. These reactions were observed for similar compounds (α-β unsaturated aldehydes) by several authors (eg, Cai et al., 2009; Esterbauer et al., 1975; Jackson et al., 2016; Randall et al., 2013; Wlodek, 1988). These events are worthy of investigation, however, in light of the complexity of cell/tissue lysates, it would be technically challenging using the current experiments. We have also observed unusual adduct types of CA (+114) with Cys, His, and Tyr, however, we believe that these are an experimental artifact (hemiacetal type products, which lose water in the interaction with the ionized peptide backbone in the electrospray source of the mass spectrometer).
We identified 162 proteins that were haptenated by DNCB, 159 by MCI and 71 for CA. Nonetheless, these account for only approximately 2.5%, 2.5%, and 1.1% of the total protein content in HaCaT cell lysates, respectively. This low proportion of protein haptenation was unexpected, especially for extreme sensitizers such as DNCB and MCI and only further emphasizes the specificity in this initial event. Although the number of modifications was lower than expected, a relationship, albeit weak, between increased levels of protein haptenation and sensitizer potency (as indicated by published in vivo data) was observed. The total number of proteins modified by each of the sensitizers decreased with decreasing sensitizing potency (MCI > DNCB > CA > 6MC). This positive correlation between protein reactivity and the intensity of sensitization reactions is consistent with previous studies (Basketter et al., 1997; Godfrey and Baer, 1971; Roberts and Aptula, 2008; Roberts and Natsch, 2009).
It is important to highlight that these experiments are not directly representative of the protein haptenation that may occur in human skin following topical exposure to a reactive chemical. The data presented here are based upon experiments where the bioavailability aspect was not taken into consideration and is part of generating a baseline haptenome, ie, the skin relevant cells and human skin tissue were lysed prior to contact with study chemicals. However, the protein lysate samples were prepared in buffers containing 0.1% SDS, conditions where proteins are likely to be in their native state, (eg, where trypsin still maintains proteolytic activity), and may sterically influence the availability of nucleophiles for their modification (Gudiksen et al., 2006). It is most likely that the realistic and physiologically relevant haptenation will be a subset of the modifications determined in this baseline study and is an important bridge between previous studies using a single model protein in isolation and the goal of physiologically relevant data.
We have demonstrated the applicability of this approach to provide a robust assessment of global protein haptenation within complex mixtures for a wide range of sensitizers and thereby bringing mechanistic insights into sensitizer reactivity. It is still unclear however whether protein reactivity, selectivity of binding for certain nucleophilic residues, the rate of the protein-binding reaction, or most likely a combination of all 3 provides the best correlate for sensitizer potency (Enoch and Roberts, 2013; Jaworska et al., 2013; Natsch and Gfeller, 2008; Patlewicz et al., 2007).
Having addressed the challenge of method sensitivity, we have the opportunity to investigate these research questions and generate new insights into the types and levels of haptenation closely relevant to human exposure. Further studies bringing a quantitative assessment of protein haptenation to a model cellular system, more relevant to in vivo skin, will be required. This could be achieved by using clinically relevant exposure scenarios in either a hapten-treated 3D cell model or by direct exposure of ex vivo skin tissue. In silico studies that explore and compare the microenvironment of the identified haptenated residues would also be of considerable value, in addition to understanding how the skin proteome responds at the cellular level after sensitizer exposure, particularly in individuals susceptible to skin allergy. However, the findings of this study clearly indicate that haptenation is more than just a statistical process (where all nucleophiles are equally likely to be modified) and that elements such as specificity may play a more important role.
Advancing our knowledge of the skin proteome, the protein targets of modification and the immunogenicity of these covalent protein modifications will ultimately enable us to better interpret reactivity data obtained from studies using model peptides enhancing our understanding of the skin sensitization AOP and its use in quantitative risk assessment. The methodology used within these studies is applicable beyond the investigation of skin sensitization, and has general utility for studying global protein haptenation events across a range of biological research areas, such as identifying drug-haptens in drug allergy.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
SEAC, Unilever plc, study (CH-2208-0181).
Supplementary Material
ACKNOWLEDGMENTS
MCI and 13 C-labeled MCI was obtained from Professor Jean-Pierre Lepoittevin and Dr Elena Gimenez-Arnau, Laboratoire de Dermatochimie, Universite Louis Pasteur, Strasbourg, France. Instrumentation in the Centre for Proteomic Research is supported by the BBSRC (BM/M012387/1) and the Wessex Medical Trust. This work was funded as part of Unilever’s on-going support in developing novel ways of delivering consumer safety.
REFERENCES
- Aeby P., Ashikaga T., Bessou-Touya S., Schepky A., Gerberick F., Kern P., Marrec-Fairley M., Maxwell G., Ovigne J. M., Sakaguchi H. et al. , (2010). Identifying and characterizing chemical skin sensitizers without animal testing: ‘research and method development program. Toxicol. In Vitro 24, 1465–1473. [DOI] [PubMed] [Google Scholar]
- Ahlfors S. R., Sterner O., Hansson C. (2003). Reactivity of contact allergenic haptens to amino acid residues in a model carrier peptide, and characterization of formed peptide-hapten adducts. Skin Pharmacol. Appl. Skin Physiol. 16, 59–68.http://dx.doi.org/10.1159/000068288 [DOI] [PubMed] [Google Scholar]
- Aleksic M., Pease C. K., Basketter D. A., Panico M., Morris H. R., Dell A. (2007). Investigating protein haptenation mechanisms of skin sensitisers using human serum albumin as a model protein. Toxicol. In Vitro 21, 723–733. [DOI] [PubMed] [Google Scholar]
- Aleksic M., Pease C. K., Basketter D. A., Panico M., Morris H. R., Dell A. (2008). Mass spectrometric identification of covalent adducts of the skin allergen 2, 4-dinitro-1-chlorobenzene and model skin proteins. Toxicol. In Vitro 22, 1169–1176. [DOI] [PubMed] [Google Scholar]
- Aleksic M., Thain E., Roger D., Saib O., Davies M., Li J., Aptula A., Zazzeroni R. (2009). Reactivity profiling: Covalent modification of single nucleophile peptides for skin sensitization risk assessment. Toxicol. Sci. 108, 401–411. [DOI] [PubMed] [Google Scholar]
- Alvarez-Sanchez R., Basketter D., Pease C., Lepoittevin J. P. (2003). Studies of chemical selectivity of hapten, reactivity, and skin sensitization potency. 3. Synthesis and studies on the reactivity toward model nucleophiles of the 13C-labeled skin sensitizers, 5-chloro-2-methylisothiazol-3-one (MCI) and 2-methylisothiazol-3-one (MI). Chem. Res. Toxicol. 16, 627–636. [DOI] [PubMed] [Google Scholar]
- Alvarez-Sanchez R., Basketter D., Pease C., Lepoittevin J. P. (2004a). Covalent binding of the 13C-labeled skin sensitizers 5-chloro-2-methylisothiazol-3-one (MCI) and 2-methylisothiazol-3-one (MI) to a model peptide and glutathione. Bioorg. Med. Chem. Lett 14, 365–368. [DOI] [PubMed] [Google Scholar]
- Alvarez-Sanchez R., Divkovic M., Basketter D., Pease C., Panico M., Dell A., Morris H., Lepoittevin J. P. (2004b). Effect of glutathione on the covalent binding of the 13C-labeled skin sensitizer 5-chloro-2-methylisothiazol-3-one to human serum albumin: Identification of adducts by nuclear magnetic resonance, matrix-assisted laser desorption/ionization mass spectrometry, and nanoelectrospray tandem mass spectrometry. Chem. Res. Toxicol. 17, 1280–1288. [DOI] [PubMed] [Google Scholar]
- Ashby J., Basketter D. A., Paton D., Kimber I. (1995). Structure activity relationships in skin sensitization using the murine local lymph node assay. Toxicology 103, 177–194.http://dx.doi.org/10.1016/0300-483X(95)03132-Y [DOI] [PubMed] [Google Scholar]
- Basketter D., Dooms-Goossens A., Karlberg A. T., Lepoittevin J. P. (1995). The chemistry of contact allergy: Why is a molecule allergenic? Contact Dermatitis 32, 65–73. [DOI] [PubMed] [Google Scholar]
- Basketter D. A., Dearman R. J., Hilton J., Kimber I. (1997). Dinitrohalobenzenes: Evaluation of relative skin sensitization potential using the local lymph node assay. Contact Dermatitis 36, 97–100.http://dx.doi.org/10.1111/j.1600-0536.1997.tb00421.x [DOI] [PubMed] [Google Scholar]
- Basketter D. A., Gerberick G. F., Kimber I. (2001). Measurement of allergenic potency using the local lymph node assay. Trends Pharmacol. Sci. 22, 264–265.http://dx.doi.org/10.1016/S0165-6147(00)01704-1 [DOI] [PubMed] [Google Scholar]
- Bligh E. G., Dyer W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917.http://dx.doi.org/10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- Brander C., Mauri-Hellweg D., Bettens F., Rolli H., Goldman M., Pichler W. J. (1995). Heterogenous T cell responses to beta-lactam-modified self-structures are observed in penicillin-allergic individuals. J. Immunol 155, 2670–2678. [PubMed] [Google Scholar]
- Burden N., Sewell F., Andersen M. E., Boobis A., Chipman J. K., Cronin M. T. D., Hutchinson T. H., Kimber I., Whelan M. (2015). Adverse Outcome Pathways can drive non-animal approaches for safety assessment. J. Appl. Toxicol. 35, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J., Bhatnagar A., Pierce W. M. Jr, (2009). Protein modification by acrolein: Formation and stability of cysteine adducts. Chem. Res. Toxicol. 22, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipinda I., Hettick J. M., Siegel P. D. (2011). Haptenation: Chemical reactivity and protein binding. J. Allergy 2011, 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codreanu S. G., Zhang B., Sobecki S. M., Billheimer D. D., Liebler D. C. (2009). Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell Proteomics 8, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C. C., Choi J., Malakowsky C. A., Talent J. M., Dai R., Marshall P., Gracy R. W. (2001). Identification of protein carbonyls after two-dimensional electrophoresis. Proteomics 1, 829–834. [DOI] [PubMed] [Google Scholar]
- Dietz L., Esser P. R., Schmucker S. S., Goette I., Richter A., Schnolzer M., Martin S. F., Thierse H. J. (2010). Tracking human contact allergens: From mass spectrometric indetification of peptide-bound reactive small chemicals to chemical-specific naïve human T-cell priming. Toxicol. Sci. 117, 336–347. [DOI] [PubMed] [Google Scholar]
- Elahi E. N., Wright Z. M., Hinselwood D., Hotchkiss S. A., Basketter D. A., Pease C. (2004). Protein binding and metabolism influence the relative skin sensitization potential of cinnamic compounds. Chem. Res. Toxicol. 17, 301–310. [DOI] [PubMed] [Google Scholar]
- Enoch S. J., Roberts D. W. (2013). Predicting Skin Sensitization Potency for Michael Acceptors in the LLNA Using Quantum Mechanics Calculations. Chem. Res. Toxicol. 26, 767–774.http://dx.doi.org/10.1021/tx4000655 [DOI] [PubMed] [Google Scholar]
- Esterbauer H., Ertl A., Scholz N. (1976). The reaction of cysteine with α,β-unsaturated aldehydes. Tetrahedon 32, 285–289. [Google Scholar]
- Ezendam J., Braakhuis H. M., Vandebriel R. J. (2016). State of the art in non-animal approaches for skin sensitization testing: From individual test methods towards testing strategies. Arch. Toxicol 90, 1–23. [DOI] [PubMed] [Google Scholar]
- Gerberick G. F., Vassallo J. D., Bailey R. E., Chaney J. G., Morrall S. W., Lepoittevin J. P. (2004). Development of a peptide reactivity assay for screening contact allergens. Toxicol. Sci. 81, 332–343. [DOI] [PubMed] [Google Scholar]
- Gerberick G. F., Vassallo J. D., Foertsch L. M., Price B. B., Chaney J. G., Lepoittevin J. P. (2007). Quantification of chemical peptide reactivity for screening contact allergens: A classification tree model approach. Toxicol. Sci. 97, 417–427. [DOI] [PubMed] [Google Scholar]
- Godfrey H. P., Baer H. (1971). The effect of physical and chemical properties of the sensitizing substance on the induction and elicitation of delayed contact sensitivity. J. Immunol. 106, 431–441. [PubMed] [Google Scholar]
- Gudiksen K. L., Gitlin I., Whitesides G. M. (2006). Differentiation of proteins based on characteristic patterns of association and denaturation in solutions of SDS. Proc. Natl. Acad. Sci. U.S.A. 103, 7968–7972.http://dx.doi.org/10.1073/pnas.0602816103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F., Sekhar K. R., Freeman M. L., Liebler D. C. (2005). Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 280, 31768–31775. [DOI] [PubMed] [Google Scholar]
- Hopkins J. E., Naisbitt D. J., Kitteringham N. R., Dearman R. J., Kimber I., Park B. K. (2005). Selective haptenation of cellular or extracellular protein by chemical allergens: Association with cytokine polarization. Chem. Res. Toxicol. 18, 375–381. [DOI] [PubMed] [Google Scholar]
- Jackson P. A., Widen J. C., Harki D. A., Brummond K. M. (2017). Covalent Modifiers: A chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael Addition reactions. J. Med. Chem. 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T., Marnett L. J. (2010). Systems analysis of protein modification and cellular responses induced by electrophile stress. Accounts Chem. Res. 43, 673–683.http://dx.doi.org/10.1021/ar900286y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworska J., Dancik Y., Kern P., Gerberick F., Natsch A. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: From theory to practice. J. Appl. Toxicol. 33, 1353–1364. [DOI] [PubMed] [Google Scholar]
- Jenkinson C., Jenkins R. E., Aleksic M., Pirmohamed M., Naisbitt D. J., Park B. K. (2010). Characterisation of p-phenylenediamine-albumin binding sites and T-cell repsonses to hapten-modified protein. J. Invest. Dermatol. 130, 732–742. [DOI] [PubMed] [Google Scholar]
- Karlberg A.-T., Bergström M. A., Börje A., Luthman K., Nilsson J. L. G. (2008). Allergic Contact Dermatitis - Formation, structural requirements and reactivity of skin sensitizers. Chem. Res. Toxicol. 21, 53–69. [DOI] [PubMed] [Google Scholar]
- Kimber I., Basketter D. A., Gerberick G. F., Ryan C. A., Dearman R. J. (2011). Chemical allergy: Translating biology into hazard characterization. Toxicol. Sci. 120, S238–S268. [DOI] [PubMed] [Google Scholar]
- Kimber I., Dearman R. J. (2003). What makes a chemical an allergen? Ann. Allergy Asthma Immunol. 90, 28–31.http://dx.doi.org/10.1016/S1081-1206(10)61645-6 [DOI] [PubMed] [Google Scholar]
- Koppes S. A., Engebretsen K. A., Agner T., Angelova-Fischer I., Berents T., Brandner J., Brans R., Clausen M.-L., Hummler E., Jakasa I. et al. , (2017). Current knowledge on biomarkers for contact sensitization and allergic contact dermatitis. Contact Dermatits 77, 1–16. [DOI] [PubMed] [Google Scholar]
- Landsteiner K., Jacobs J. (1935). Studies on the sensitization of animals with simple chemical compounds. J. Exp. Med. 61, 643–656.http://dx.doi.org/10.1084/jem.61.5.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepoittevin J. P. (2006). The chemistry of skin allergy. Altex 23(Suppl), 234–238. [Google Scholar]
- Loveless S. E., Ladies G. S., Gerberick G. F., Ryan C. A., Basketter D. A., Scholes E. W., House R. V., Hilton J., Dearman R. J., Kimber I. (1996). Further evaluation of the local lymph node assay in the final phase of an international collaborative trial. Toxicology 108, 141–152. [DOI] [PubMed] [Google Scholar]
- MacKay C., Davies M., Summerfield V., Maxwell G. (2013). From pathways to people: Applying the adverse outcome pathway (AOP) for skin sensitization to risk assessment. Altex 30, 473–486. [DOI] [PubMed] [Google Scholar]
- Majeti V. A., Suskind R. R. (1977). Mechanism of cinnamaldehyde sensitization. Contact Dermatitis 3, 16–18.http://dx.doi.org/10.1111/j.1600-0536.1977.tb03581.x [DOI] [PubMed] [Google Scholar]
- Martin S. F. (2015). Immunological mechanisms in allergic contact dermatitis. Curr. Opin. Aleergy Clin. Immunol. 15, 124–130.http://dx.doi.org/10.1097/ACI.0000000000000142 [DOI] [PubMed] [Google Scholar]
- Maxwell G., Mackay C. (2008). Application of a systems biology approach to skin allergy risk assessment. Altern. Lab. Anim. 36, 521–556. [DOI] [PubMed] [Google Scholar]
- Maxwell G., MacKay C., Cubberley R., Davies M., Gellatly N., Glavin S., Gouin T., Jacquoilleot S., Moore C., Pendlington R. et al. , (2014). Applying the skin sensitisation adverse outcome pathway (AOP) to quantitative risk assessment. Toxicol. In Vitro 28, 8–12. [DOI] [PubMed] [Google Scholar]
- Mello C. F., Sultana R., Piroddi M., Cai J., Pierce W. M., Klein J. B., Butterfield D. A. (2007). Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience 147, 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsch A., Gfeller H. (2008). LC-MS-based characterization of the peptide reactivity of chemicals to improve the in vitro prediction of the skin sensitization potential. Toxicol. Sci. 106, 464–478.http://dx.doi.org/10.1093/toxsci/kfn194 [DOI] [PubMed] [Google Scholar]
- OECD. (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence, Eds., Testing and Assessment. No 168. ENV/JM/MONO(2012)10/PART1 ed.
- Parkinson E., Boyd P., Aleksic M., Cubberley R., O’Connor D., Skipp P. (2014). Stable isotope labeling method for the investigation of protein haptenation by electrophilic skin sensitizers. Toxicol. Sci. 142, 239–249. [DOI] [PubMed] [Google Scholar]
- Patlewicz G., Aptula A. O., Uriarte E., Roberts D. W., Kern P. S., Gerberick G. F., Kimber I., Dearman R. J., Ryan C. A., Basketter D. A. (2007). An evaluation of selected global (Q)SARs/expert systems for the prediction of skin sensitisation potential. SAR QSAR. Environ. Res. 18, 515–541. [DOI] [PubMed] [Google Scholar]
- Pickard C., Louafi F., McGuire C., Lowings K., Kumar P., Cooper H., Dearman R. J., Cumberbatch M., Kimber I., Healy E. et al. , (2009). The cutaneous biochemical redox barrier: A component of the innate immune defenses against sensitization by highly reactive environmental xenobiotics. J. Immunol. 183, 7576–7584.http://dx.doi.org/10.4049/jimmunol.0901064 [DOI] [PubMed] [Google Scholar]
- Randall M. J., Hristova M., van der Vliet A. (2013). Protein alkylation by the α,β-unsaturated aldehyde acrolein. A reversible mechanism of electrophile signaling?. FEBS Lett. 587, 3808–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D. W., Aptula A. O. (2008). Determinants of skin sensitisation potential. J. Appl. Toxicol. 28, 377–387.http://dx.doi.org/10.1002/jat.1289 [DOI] [PubMed] [Google Scholar]
- Roberts D. W., Aptula A. O., Patlewicz G. (2007). Electrophilic chemistry related to skin sensitization. reaction mechanistic applicability domain classification for a published data set of 106 chemicals tested in the mouse local lymph node assay. Chem. Res. Toxicol. 20, 44–60.http://dx.doi.org/10.1021/tx060121y [DOI] [PubMed] [Google Scholar]
- Roberts D. W., Natsch A. (2009). High throughput kinetic profiling approach for covalent binding to peptides: Application to skin sensitization potency of Michael acceptor electrophiles. Chem. Res. Toxicol. 22, 592–603.http://dx.doi.org/10.1021/tx800431x [DOI] [PubMed] [Google Scholar]
- Sanderson P. N., Simpson W., Cubberley R., Aleksic M., Gutsell S., Russell P. J. (2016). Mechanistic understanding of molecular initiating events (MIEs) using NMR spectroscopy. Toxicol. Res. 5, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn C. T., Fritz K. S., Shearn A. H., Saba L. M., Mercer K. E., Engi B., Galligan J. J., Zimniak P., Orlicky D. J., Ronis M. J. et al. , (2016). Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice. Redox. Biol. 7, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006). Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell Proteomics 5, 144–156. [DOI] [PubMed] [Google Scholar]
- Simonsson C., Andersson S. I., Stenfeldt A.-L., Bergstrom J., Bauer B., Jonsson C. A., Ericson M. B., Broo K. S (2011). Caged fluorescent haptens reveal the generation of cryptic epitopes in allergic contact dermatitis. J. Investig. Dermatol. 131, 1486–1493. [DOI] [PubMed] [Google Scholar]
- Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985). Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Spiess P. C., Deng B., Hondal R. J., Matthews D. E., van der Vliet A. (2011). Proteomic profiling of acrolein adducts in human lung epithelial cells. J. Proteomics 74, 2380–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland J., Zang Q., Kleinstreuer N., Paris M., Lehmann D. M., Choksi N., Matheson J., Jacobs A., Lowit A., Allen D. et al. , (2016). Integrated decision strategies for skin sensitization hazard. J. Appl. Toxicol. 36, 1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyssen J. P., Linneberg A., Menne T., Johansen J. D. (2007). The epidemiology of contact allergy in the general population–prevalence and main findings. Contact Dermatitis 57, 287–299. [DOI] [PubMed] [Google Scholar]
- Vinken M. (2013). The adverse outcome pathway concept: A pragmatic tool in toxicology. Toxicology 312, 158–165.http://dx.doi.org/10.1016/j.tox.2013.08.011 [DOI] [PubMed] [Google Scholar]
- Wiechelman K. J., Braun R. D., Fitzpatrick J. D. (1988). Investigation of the bicinchoninic acid protein assay - Identification of the groups responsible for color formation. Anal. Biochem. 175, 231–237. [DOI] [PubMed] [Google Scholar]
- Wlodek L. (1988). The reaction of sulfhydryl groups with carbonyl compounds. Acta Biochim. Pol. 35, 307–317. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.