

SUMMARY
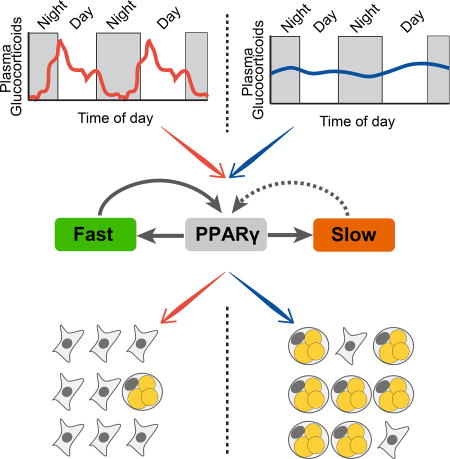
Glucocorticoid and other adipogenic hormones are secreted in mammals in circadian oscillations. Loss of this circadian oscillation pattern correlates with obesity in humans, raising the intriguing question of how hormone secretion dynamics affect adipocyte differentiation. Using live, single-cell imaging of the key adipogenic transcription factors CEBPB and PPARG, endogenously tagged with fluorescent proteins, we show that pulsatile circadian hormone stimuli are rejected by the adipocyte differentiation control system. In striking contrast, equally strong persistent signals trigger maximal differentiation. We identify the mechanism of how hormone oscillations are filtered as a combination of slow and fast positive feedback centered on PPARG. Furthermore, we confirm in mice that flattening of daily glucocorticoid oscillations significantly increases the mass of subcutaneous and visceral fat pads. Together, our study provides a molecular mechanism for why stress, Cushing's disease, and other conditions for which glucocorticoid secretion loses its pulsatility may lead to obesity.
Keywords: Positive feedback, adipogenesis, circadian filtering, hormone oscillations, glucocorticoids, PPARG, CEBPB
eTOC blurb
XXX et al elucidate how adipocytes filter out normal pulsatile circadian hormone stimuli, while strong persistent signals trigger differentiation. Flattening of daily glucocorticoid oscillations in mice results in significantly increases in fat mass, providing a molecular mechanism for Cushing's disease, and other stress-associated obesity conditions, with disrupted pulsatile glucocorticoid secretion.
INTRODUCTION
Slow, ongoing terminal cell differentiation is essential for replacing aging or damaged cells and for maintaining tissue size in all adult mammals. For example, adipocytes (fat cells) and cardiomyocytes renew in humans at rates of approximately 8% and 1% per year, respectively (Bergmann et al., 2009; Spalding et al., 2008). Since terminally-differentiating cells are often derived from large pools of precursor cells, differentiation is expected to be a rare event. In the case of fat cell differentiation, or adipogenesis, for which there are estimates of about one preadipocyte for every five differentiated cells (Tchoukalova et al., 2004), fewer than 1% of preadipocytes are believed to embark on a differentiation path on any given day under normal, homeostatic conditions. How such low rates of differentiation can be reliably maintained is puzzling given that preadipocytes are subjected to daily high increases of differentiation-inducing hormones such as glucocorticoids which are needed in mammals to mobilize energy and increase physical activity, but which also have been shown to strongly accelerate adipogenesis in vitro and in vivo (Campbell et al., 2011; Farmer, 2006; Lee et al., 2014).
Glucocorticoids are secreted in healthy mammals in daily oscillatory patterns (Figure 1A)(Weitzman et al., 1971), as well as in sporadic bursts in response to stress. Levels of other adipogenic hormones such as ghrelin and prolactin, that raise cAMP, and insulin, fluctuate in vivo as well (Carré and Binart, 2014; MacDougald et al., 1995; Thompson et al., 2004). Given that so few preadipocytes differentiate each day despite significant daily increases in adipogenic hormone stimuli, we hypothesized that the regulatory circuit controlling adipocyte differentiation might be filtering out circadian and short hormone stimuli and that differentiation would only start to occur if the duration of the trough between circadian pulses shortens or if the hormone signal remain continuously elevated. Such a temporal filtering of hormone signals is consistent with the observation that, in contrast to short or daily oscillatory signals, more continuous glucocorticoid signals have been linked to increased fat mass (Campbell et al., 2011; Dallman et al., 2000). Shortening of the trough between glucocorticoid pulses, or “flattening of the daily oscillations”, due to irregular feeding or sleep cycles, prolonged treatment with glucocorticoid hormones, chronic stress, or metabolic diseases such as Cushing’s disease have all been shown to closely correlate with increased obesity (Balbo et al., 2010; Dallman et al., 2000; Lee et al., 2014).
Figure 1. Preadipocytes reject circadian and rhythmic glucocorticoid stimuli.

(A) Schematic depicting averaged glucocorticoid time courses in humans (adapted from Weitzman et al, 1971).
(B) Single-cell immunofluorescence assay used to quantitate percent of differentiated cells based on (Park et al., 2012). Preadipocyte cells were fixed and stained with Hoechst to visualize nuclei (blue), BODIPY to visualize lipids (green), and antibodies against PPARG (red) and adiponectin (white). The latter 3 signals are closely correlated in individual cells. Images show mouse OP9 cells. Scale bar, 10 µm. Scatter plot shows that high-PPARG cells display mature fat cell features and accumulate high levels of lipid. Application of an adipogenic stimulus causes cells to split into two populations with low and high PPARG. The middle of the trough between the bimodal PPARG intensity peaks (black dotted line in histogram) was used to separate undifferentiated and differentiated cells. Percent of differentiated cells was assessed at t = 96 hours after applying adipogenic stimulus, using approximately 7,000 cells per technical replicate. See also Figures S1A–S1C.
(C) Different patterns of DMI stimuli were applied to OP9 and 3T3-L1 preadipocyte cells: 48 hours continuous delivery (red) and 3 different pulsatile protocols (blue): 12 hours on/12 hours off, 6 hours on/18 hours off, and 12 hours on/36 hours off. The concentration of DMI stimulus used for a 48-hour continuous pulse was 1µM dexamethasone (dex), 250 µM IBMX, and 1.75 nM insulin. To keep the total amount of stimuli, i.e. area under the curve, constant for all 4 protocols over the 96-hour experimental timeframe, the concentration of dex and IBMX was increased proportionally to compensate for decreases in pulse duration. To end a pulse, the DMI-containing media was aspirated, cells were washed gently three times with fresh media, and the cells were left in media with no glucocorticoids or IBMX, but containing 1.75 nM insulin. The dashed line shows the percent of differentiated cells when no stimulus was applied and cells were placed for the whole 4-day timecourse of differentiation in media with no glucocorticoids or IBMX, but containing 1.75 nM insulin.
(D) Experiments in primary SVF preadipocytes show that the same filtering effects are also observed in primary cells. SVF preadipocytes were plated into 96-well wells and induced to differentiate using the same pulsing protocols from (C). The Total BODIPY Intensity in each well was measured by imaging.
(E) Applying continuous stimuli with durations greater than 12 hours resulted in corresponding increases in the number of differentiated OP9 and 3T3-L1 cells. However, the same total stimuli given in circadian pulses resulted in only minimal differentiation.
(C–E) Barplots represents mean +/− s.e.m. from 3 technical replicates. All data shown are representative of 5 independent experiments.
Adipogenesis occurs over several days by hormone-induced activation of a transcriptional network centered on a core positive feedback loop between PPARG and CEBPA that works together with additional secondary positive feedbacks and regulatory mechanisms (Rosen and Spiegelman, 2014). The positive feedbacks are proposed to control a bistable switch that separates a distinct undifferentiated precursor state from a distinct differentiated state (Ahrends et al., 2014). Nevertheless, while these previous studies provided indirect evidence for bistability using different strengths of continuous stimuli, they did not address the physiologically more relevant question if and how oscillating stimuli of the same total dose control the differentiation process. Furthermore, since these previous studies did not use live cell analysis, they were only able to infer a threshold and bistability of the differentiation process and could not directly show it. Since transcriptional processes are typically variable between cells, investigation of the dynamic processes controlling the switch requires time course analysis in single cells (Loewer and Lahav, 2011).
Here we investigated whether a differentiation system can temporally filter out circadian hormone inputs, a question that to our knowledge has not been addressed by previous studies which typically administered only constant levels of hormone stimuli or single hormone pulses. To test for dynamic control of differentiation, we employed a multi-day, live-cell imaging approach in which we monitored the expression levels of the transcription factors CEBPB and PPARG by endogenously tagging the respective loci in model mouse adipocyte precursor cells (OP9 cells) with fluorescent protein. Strikingly, we demonstrate a temporal control principle whereby preadipocytes reject normal pulsatile, daily hormone inputs due to a combined slow and fast positive feedback circuit that controls the self-amplification of PPARG. The fraction of cells that differentiate only starts to increase when the durations of hormone pulses become extended beyond normal circadian pulse durations.
RESULTS
Circadian and rhythmic hormone stimuli are rejected by the preadipocyte differentiation system
To determine whether and how pulsatile versus continuous hormone stimulation regulate adipogenesis, we used 3T3-L1 and OP9 in vitro models for adipocyte differentiation (Green and Kehinde, 1975; Wolins et al., 2006), as well as primary stromal vascular fraction (SVF) preadipocytes isolated from mice (Ota et al., 2015). To induce differentiation, we applied pulses of an adipogenic hormone cocktail (DMI, see Methods) that mimics glucocorticoids and GPCR-signals that raise cAMP, such as ghrelin and prolactin. These signals have been shown to increase and decrease in circadian or fluctuating patterns in vivo (Carré and Binart, 2014; Thompson et al., 2004), and we used the commonly-used DMI cocktail as a maximal stimulus of glucocorticoids and synergistic hormone signals.
We quantified differentiation by measuring expression of PPARG, the master transcriptional regulator of adipogenesis, whose expression in individual cells correlates closely with lipid accumulation, as well as with expression of GLUT4, adiponectin, and other mature fat cell markers (Tontonoz and Spiegelman, 2008) (Figures 1B, S1A–S1C). To determine the effect of pulsatile versus continuous DMI stimulation on adipogenesis, we used a protocol in which we added and removed DMI in four different pulse patterns while keeping the integrated DMI stimulus constant over four days (Figure 1C, left). Strikingly, rhythmic cycles of DMI stimuli applied to preadipocytes with durations of 12 hours or less caused only minimal differentiation (Figure 1C, blue) while, in contrast, the same total amount of stimulus applied continuously caused robust differentiation (Figure 1C, red). To validate that this is a general phenomenon, we applied the same protocols to primary SVF preadipocytes isolated from mice and measured accumulation of lipids which is a commonly used marker for differentiation in these cells (Ota et al., 2015). Markedly, the primary preadipocytes showed an equally strong failure to differentiate in response to daily 12-hour on/12-hour off DMI stimulation compared to robustly differentiating when the same total integrated DMI stimuli was applied in a continuous manner for 48 hours (Figures 1D and S1D).
Increasing the continuous stimulus durations from 12 to 24, 36, and 48 hours resulted in a progressive increase in differentiated cells (Figure 1E, red). However, once again, when the same total amount of stimulation was applied in circadian cycles, minimal differentiation was observed in OP9 and 3T3-L1 cells (Figure 1E, blue), as well as in primary SVF preadipocytes (Figure S1E). We also performed a number of additional control experiments where we separated the contributions of IBMX and dexamethasone. We combined, for example, IBMX pulses together with continues dexamethasone stimuli (Figure S1F). We conclude from these different pulse protocols that both stimuli have to oscillate in synchrony for effective filtering and low differentiation. Control experiments further confirmed that the doses of applied DMI stimuli were not saturating and that even the application of much stronger doses of DMI failed to induce differentiation when applied in circadian and rhythmic pulses (Figures S2A and S2B). The same filtering and differentiation response to pulsatile versus continuous stimuli was observed whether a synthetic glucocorticoid, dexamethasone, or the physiological glucocorticoid, corticosterone, was used in the hormone stimulus cocktail (Figure S1C). Taken together, these data show that preadipocytes make use of a robust filtering mechanism to maintain low rates of differentiation when exposed to daily hormone pulses. Intriguingly, the near complete filtering of differentiation-inducing stimuli was observed for pulse durations of up to 12 hours (Figure 1E), approximately reflecting the duration of normal circadian glucocorticoid patterns that preadipocytes are expected to experience in vivo (Weitzman et al., 1971) (Figure 1A).
CEBPB mirrors the dynamic changes in pulsatile hormone inputs
To understand at which step in the differentiation signaling pathway input signals are filtered out, we compared the effect of two different stimuli: (i) rosiglitazone which binds to and directly activates PPARG (Tontonoz and Spiegelman, 2008) and (ii) DMI which mimics glucocorticoid stimuli and increases transcription of PPARG (schematic shown in Figure 2A). The stimuli were applied for different durations ranging from 2 to 48 hours. When PPARG was directly activated with rosiglitazone, there was no filtering of input signals, and the fraction of differentiated cells increased proportionally with increasing pulse durations (Figure 2B, blue bars). However, when PPARG was indirectly activated with DMI, preadipocytes showed an increase in differentiation only for pulses longer than 12 hours (Figures 2B and S2D, red bars), arguing that the observed filtering of glucocorticoid input stimuli occurred before or along with PPARG activation.
Figure 2. CEBPB mirrors the dynamics of the hormone input and transmits the pulse durations to PPARG.

(A) Simplified schematic of the canonical adipogenic transcriptional network (Farmer, 2006; Rosen and Spiegelman, 2014).
(B) Stimuli pulses of different durations were applied to OP9 cells. DMI was used to indirectly activate PPARG, and rosiglitazone was used to directly activate PPARG. Data from 3T3-L1 cells is shown in Figure S2D. The rosiglitazone concentration was chosen to induce the same degree of PPARG activity as the DMI stimulus (see Figure S3A).
(C) Suppression of differentiation by siRNA-mediated depletion of CEBPB in OP9 cells.
(B, C) The percent of differentiated cells was measured as in Figure 1B and show mean +/− s.e.m. from 3 technical replicates, representative of 3 independent experiments.
(D) Endogenous CEBPB (LAP* isoform) in OP9 cells was tagged with citrine (YFP) using CRISPR-mediated genome editing. Scale bar, 10 µm.
(E) Single-cell time courses of total citrine-CEBPB nuclear abundance (CEBPB abundance) in response to a 12-hour on / 12-hour off pattern of DMI stimulus (thin lines). The bold line represents the population median and shaded regions represents the 25th to 75th percentile of the population, N > 300 cells. Citrine-CEBPB expression increased with t1/2 ~ 2 hours with stimulus addition and decreased with t1/2 ~ 3 hours with stimulus removal, as estimated by a single exponential decay model.
(F) 3T3-L1 cells were stimulated with circadian pulses (12-hour on / 12-hour off) of DMI and fixed at the respective time points marked with yellow circles (top). Cells were stained with anti-CEBPB antibody and Hoescht as a nuclear marker. Each bar represents the mean +/− s.e.m. of 3 technical replicates, each with approximately 5000 cells, and is representative of 3 independent experiments.
(G) OP9 cells were stimulated with DMI for 24 hours, treated with cyclohexamide or actinomycin-D, fixed at the indicated time points, and immunofluorescence or RNA FISH was performed to measure CEBPB protein levels or count the number of mRNA puncta per cell, respectively. First-order exponential decay curves were fitted to data (t1/2 ~ 3.6 hr and 1 hr, respectively). The shaded region represents the 95th confidence bounds on the fitted coefficients. Values enclosed in parentheses show lower and upper bounds on the estimated half-life derived from the 95% confidence bounds.
Since the transcription factor CEBPB is induced by DMI stimuli and is necessary for PPARG expression (Figures 2A and 2C), we tested whether CEBPB mediates the filtering of hormone pulses. We measured dynamic changes in CEBPB levels by using CRISPR-mediated genome editing to generate an OP9 preadipocyte cell line with Citrine (YFP) fused to the N-terminus of endogenous CEBPB (Figures 2D and S4). Control experiments verified that the tagged endogenous protein was regulated similarly and had similar expression levels and half-life as the untagged endogenous CEBPB protein (Figure S5). To obtain timecourses of CEBPB levels, cells were automatically tracked over several days, and their nuclear YFP fluorescence was recorded. Surprisingly, these live-cell imaging experiments showed that the nuclear abundance of CEBPB was highly dynamic and closely mirrored the hormone input for oscillating stimuli (Figures 2E and S3C). Nuclear abundance of CEBPB has been shown to reflect DNA binding of CEBPB (Siersbæk et al., 2011), arguing that not only the level but also the activity of CEBPB follows the stimulus dynamics. Similar nuclear CEBPB dynamics as in Figure 2E were observed in 3T3-L1 cells stimulated with a pulsatile pattern (Figure 2F), suggesting that fast CEBPB dynamics mirror the external hormone stimulus in different pre-adipocyte models. The observed fast changes in CEBPB levels can be explained by the fast degradation rates of both CEBPB protein and mRNA (Figures 2G and S6).
Live-cell imaging of fluorescently-tagged endogenous PPARG reveals the existence of a threshold where internal positive feedback becomes independent of external stimuli
Because CEBPB dynamics closely mirrored the hormone input stimuli, the filtering must occur downstream of CEBPB, suggesting that PPARG might be involved. We again made use of CRISPR-mediated genome editing to generate an OP9 cell line with citrine (YFP) fused to the N-terminus of endogenous PPARG (Figures 3A and S4). Control experiments verified that the tagged endogenous protein was regulated similarly and had similar expression levels and half-life as the untagged endogenous PPARG protein (Figure S7).
Figure 3. PPARG levels increase slowly and typically do not reach the threshold to differentiate unless hormone stimuli are applied for longer than 24 hours.

(A) Endogenous PPARG2 in OP9 cells was tagged with citrine (YFP) using CRISPR-mediated genome editing. Scale bar, 5 µm.
(B) Identifying a PPARG threshold for differentiation by carrying out live-cell imaging of citrine-PPARG cells induced to differentiate with the standard DMI protocol. When the DMI stimulus is removed at 48 hours, cells either sustain or lose the increase in nuclear PPARG intensity (PPARG abundance). The thin lines show 50 representative single cell traces, and the bold line shows the population mean of 500 cells. Cells that differentiated after 96 hours are marked in red, and cells that stayed in the preadipocyte state are marked in blue. The yellow triangle shows the threshold calculated in Figure 3D.
(C) Timecourses from 419 cells were divided into 5 groups (bins) according to their PPARG level at 48 hours, and the average of each bin was plotted. An initial drop was seen in each averaged timecourse after DMI removal, resulting in a return to the basal state for cells that had lower levels of PPARG and a delayed increase to the maximal state in cells that had higher levels of PPARG.
(D) Scatter plot comparing the level of PPARG in the same cell measured at 48 hours just before DMI is removed and at 96 hours when PPARG levels reach their plateau in differentiated cells. The color of the dots and histograms mark cells that will be differentiated (red) or not differentiated (blue) at 96 hours, respectively. The histograms on top shows the existence of a clear PPARG threshold already at 48 hours before the stimulus is removed (marked with yellow triangle and dotted line) that can distinguish between cells that will go on to differentiate from those that will not.
(E) Live-cell imaging of citrine-PPARG cells transfected with CEBPA-targeting or control siRNA shows that knockdown of CEBPA, a main feedback partner of PPARG, prevents cells from reaching the threshold to differentiate. Cells were induced to differentiate with the standard DMI protocol. Plotted lines are population median traces with shaded regions representing 25th and 75th percentiles of approximately 700 cells per condition.
The role of PPARG in filtering circadian inputs has to be investigated in the context of previous work that showed that PPARG is a critical part of a bistable switch that converts preadipocytes to adipocytes (Park et al., 2012). Bistable switches between two fates are a fundamental part of differentiation processes, and many models predict that a threshold in the expression of a core regulator must exist that decides whether or not a cell will differentiate (Wang et al., 2009). However, the existence of a threshold has never been directly shown since to do so requires live-cell measurement of the presumed regulator in individual cells while being able to remove the differentiation-inducing stimulus and continuing to track individual cells to their final differentiation state. Such live-cell analysis is necessary in order to verify that reaching the threshold level of the presumed regulator indeed results in that cell eventually transitioning irreversibly into the differentiated state.
Our citrine-PPARG cells allowed us now to directly test for the first time whether such a threshold exists in cell differentiation. We continuously imaged and automatically tracked citrine-PPARG preadipocytes over a 4-day time course using a 48-hour continuous DMI stimulation protocol. Markedly, when the DMI stimulus was removed, the population of preadipocytes split into two distinct populations: cells in which PPARG levels caught on and continued to increase with cells reaching the differentiated adipocyte state (red), and cells in the same population in which PPARG level dropped back down with cells staying in the undifferentiated state (blue) (Figure 3B). As shown in Figure 2A, PPARG levels are regulated by both external hormone stimulus and cell-intrinsic positive feedback. This dual regulation of PPARG levels can be more clearly seen in the live-cell traces when cells are binned together according to their level of PPARG at the time of DMI removal (Figure 3C). Markedly, in cells whose PPARG level is above a critical threshold when DMI is removed (red cells), PPARG levels first fall partially down due to the loss of the external hormone stimulus and then increase again as the cell-intrinsic positive feedbacks engage to continue to raise PPARG levels, independently of external stimuli, to the fully differentiated state. In cells whose PPARG level does not increase to this critical threshold (blue cells), the cell-intrinsic positive feedbacks do not engage strongly enough and cells thus fall back to the undifferentiated state. Figure 3D shows measurements extracted from individual cell timecourses which show the PPARG level in each cell at 48 hours before DMI removal and then again in the same cell at 96 hours when the final differentiation state is known. The threshold can then be calculated as the midpoint between the two distributions of the PPARG level for differentiated and undifferentiated cells before DMI removal (Figure 3D). This calculated threshold (marked as a yellow triangle in Figures 3B–D) can predict already at 48 hours, before the stimulus is removed, whether or not a cell will be differentiated 48 hours later.
We also observed in the analysis in Figure 3C that PPARG levels start to increase faster approximately 24 hours after addition of the DMI stimulus and long before the threshold for differentiation is reached. When we knocked down CEBPA, the core positive feedback partner of PPARG, this faster increase in PPARG was absent and cells failed to reach the threshold for differentiation (Figure 3E), arguing that the external hormone stimulus first gradually increases PPARG levels for about 24 hours at which point cell-intrinsic positive feedback to PPARG engages to then allow cells to reach the threshold for differentiation. The threshold level, which is reached after approximately 36–48 hours of continuous external hormone stimulation, is the level of PPARG at which cell-intrinsic positive feedback, initially triggered around 24 hours, has becomes sufficiently strong to sustain the increase in PPARG even in the absence of external hormone stimuli.
The core adipogenic transcription factors PPARG and CEBPA rapidly degrade which keeps PPARG levels below the threshold to differentiate for daily oscillatory stimuli
Given the existence of a PPARG threshold, it was plausible that daily hormone stimuli fail to trigger differentiation due to a failure of PPARG levels to reach the threshold. Such a mechanism is plausible due to the very slow PPARG increase we observed in Figures 3B and 3C. It takes PPARG approximately 24–48 hours after continuous stimulation to reach the irreversible threshold for differentiation, meaning that shorter stimuli pulses may fail to trigger differentiation. Indeed, live-cell imaging experiments showed that repetitive 12-hour on / 12-hour off stimuli resulted in only minimal increases in PPARG levels in most cells (Figures 4A and 4B), and thus prevented differentiation compared to when an equally-strong continuous stimulus was applied (Figure 3B). Interestingly, when the stimulus was off during pulsatile conditions, PPARG levels not only stopped to increase but also dropped (blue traces in Figures 4A and 4C). The drops between pulses prevent PPARG levels from gradually increasing to the threshold to trigger differentiation for repetitive pulse protocols. The rapid changes in PPARG levels can be explained by the short half-lives of PPARG protein and mRNA, approximately 1 and 0.8 hour respectively (Figure 4D).
Figure 4. The core adipogenic transcription transcription factors PPARG and CEBPA rapidly degrade which keeps PPARG below the threshold to differentiate in response to oscillatory circadian hormone inputs.

(A) Single-cell timecourses showing the dynamics of citrine-PPARG expression when adipogenic DMI stimuli are applied in a pulsatile 12-hour on/12-hour off manner over 4 days. Cells were marked as differentiated (red) and undifferentiated (blue) based on whether their final citrine-PPARG values was above or below the threshold for differentiation defined in Figure 3.
(B) Comparison of the percent of differentiated cells resulting from continuous (Figure 3B) versus circadian (Figure 4A) stimuli. Error bars show mean +/− s.e.m. from 6 technical replicates, representing 3 independent experiments.
(C) Zoom-in of the undifferentiated cells (blue traces) in (A). If PPARG levels do not reach the threshold to differentiate, PPARG levels drop back down to precursor levels each time the DMI stimulus is removed. Plot shows data when pulses of DMI stimulus (blue) or control pulses consisting of media with no glucocorticoids or IBMX stimulus (grey) were applied in parallel to different wells of citrine-PPARG cells plated in 96-well wells and imaged over time. Dark lines represent the population median traces and shaded regions represent the 25th and 75th percentiles of approximately 700 cells per condition.
(D) Half-life analysis of PPARG protein and mRNA as in Figure 2G.
(E) Plot of the times required to reach steady state for molecules with different half-lives and first order decay.
(F) A slow-degrading regulator converts input stimuli with increasing pulse durations into increasing output signal amplitudes whereas a fast-degrading regulator outputs only a single maximal amplitude. Model output in response to different input pulse protocols is shown on the right and assumes exponential increase and decay of proteins with t1/2 = 3 and 34 hrs, respectively.
(G) Half-life analysis of CEBPA protein and mRNA as in Figure 2G.
The fast degradation times of CEBPB and PPARG and the failure of PPARG levels to sufficiently increase under pulsatile conditions can explain the filtering seen in response to daily hormone pulses. However, the fast dynamics also raised the puzzling question of how PPARG levels can slowly build over days to result in differentiation when cells are subjected to continuous adipogenic stimuli. It has previously been shown that 2 days of stimulation are needed to raise PPARG levels to be able to convert a large fraction of preadipocytes to the point where they stay on a differentiation path even when the stimulus is removed (Chawla et al., 1994). As shown in Figure 3B, it takes most cells 24–48 hours to build PPARG levels to the threshold to differentiate. Since the time for a protein to accumulate to a steady-state level depends only on its degradation rate when the synthesis rate is not changing (Rosenfeld et al., 2002), a regulatory circuit built from only fast-degrading proteins such as PPARG and CEBPB would be unable to gradually increase PPARG expression levels for days since proteins with a 1 hour half-life will have already reached more than 99% of their maximal level within 12 hours of stimulation (Figure 4E, red line). However, a slow-degrading regulator of PPARG would be able to slowly buildup up and increase PPARG levels for days for continuous stimuli (Figure 4E, blue line).
The existence of a slow-degrading PPARG feedback partner would also explain another observed phenomenon: that the adipogenic differentiation system responds very differently to oscillating versus continuous stimuli (Figures 1C–E, 3B–C, 4A–C), filtering out stimuli in the first case and regulating increasing fractions of the cell population to differentiate in the second case. A system with only fast-degrading regulators would only have one response since it would rise rapidly to the same expression level for both oscillating or continuous stimuli and would either trigger differentiation or not for both types of stimuli without a delay (Figure 4F, top). In contrast, a slow-degrading PPARG regulator could rise to different steady-state amplitudes in response to different input pulse durations and thus would be able to keep its steady-state level below a threshold for oscillating stimuli while also being able to rise above the threshold and trigger differentiation for continuous stimulation (Figure 4F, bottom, solid red and blue lines). In other words, the existence of a slow-degrading PPARG regulator would allow a system to selectively prevent differentiation for pulsatile stimulation as long as the trough between pulses is sufficiently long (~12 hours) which allows for sufficient degradation of the slow regulator between pulses. The dashed lines in Figure 4F show how steady-state levels increase for daily pulses of 18-hour and become lower for daily pulses of 6-hour duration, further demonstrating how the existence of slow-degrading regulators in the adipogenic transcriptional architecture would enable adipocyte precursor cells to convert daily hormone stimuli of different pulse durations into different longterm steady-state amplitudes of expression of the regulator above and below a threshold. In summary, a slow feedback regulator of PPARG is needed both to increase PPARG levels slowly to the threshold to differentiate (Figures 3B–C, 4E) and to allow PPARG levels to rise to different steady-state levels for oscillating versus continuous stimuli (Figure 4F). Since a transcriptional positive feedback loop between PPARG and CEBPA has been shown to be particularly critical for differentiation (Rosen and Spiegelman, 2014) (Figure 2A), an obvious candidate for such a slow regulator is CEBPA. However, CEBPA also has short mRNA and protein half-lives of approximately 1 and 3 hours, respectively, and cannot be the required slow co-regulator (Figure 4G).
Identification of a slow feedback regulator of PPARG that can mediate the slow buildup in PPARG levels to the threshold to differentiate
Since their mRNA and protein all degrade quickly even in the presence of continuous stimuli, the three core regulators CEBPA, CEBPB, and PPARG (Figure 2A) cannot be alone responsible for the observed slow buildup of PPARG. A plausible alternative mechanism to explain the delayed PPARG increase would be if additional positive feedback regulators of PPARG had a much longer life time. Several such positive regulators of PPARG have been identified (Ahrends et al., 2014). At least two have been shown to have long protein half-lives, FLNA and CEPBZ (Schwanhäusser et al., 2011), and one has been shown to have a long mRNA half-life, FABP4 (Spangenberg et al., 2013). To understand how a slow regulator might work in a differentiation system, we focused on FABP4 since previous work showed that it had a particularly strong effect on differentiation of OP9 cells compared to FLNA or CEBPZ (Ahrends et al., 2014).
FABP4 has a relatively fast protein half-life (Figure 5A), but had been shown to have a long mRNA half-life (Spangenberg et al., 2013). Using different methods, we confirmed that FABP4 mRNA has a long half-life of between 14 to 34 hours (Figures 5B–5C, S6) and that the increase in PPARG level is closely correlated with an increase in FABP4 in individual cells (Figure 5D). Previous studies had established FABP4 is a downstream target of PPARG (Hotamisligil and Bernlohr, 2015) that can positively regulate PPARG expression and adipogenesis (Ahrends et al., 2014; Ayers et al., 2007). Indeed, siRNA-mediated knockdown of FABP4 suppressed adipogenesis (Figure 5E), and overexpression of FABP4 increased adipogenesis in the absence of adipogenic stimulus in OP9 cells (Figure 5F). Furthermore, identical to the effect of CEBPA knockdown, siRNA-mediated depletion of FABP4 and overexpressing FABP4 resulted in a slowing-down (Figure 5G) and acceleration (Figure 5H) of the normally-observed PPARG increase in response to DMI. Together with previous studies by other groups (Adida and Spener, 2006; Ayers et al., 2007), our results support that FABP4 is in a positive-feedback relationship with PPARG in OP9 cells. Furthermore, the timecourses in Figures 5G and 5H support that slow-degrading FABP4 can mediate the slow buildup in PPARG levels. While there are likely additional slow regulators of PPARG expression, our study argues that FABP4 has a critical role as a slow positive feedback partner that mediates the slow PPARG dynamics in response to oscillatory versus continuous stimuli.
Figure 5. FABP4 is an example of a slow-degrading PPARG regulator that can mediate a slow increase in PPARG expression during adipogenesis.

(A) Half-life analysis of FABP4 protein as in Figure 2G.
(B) Half-life of FABP4 mRNA obtained using mRNA FISH as in Figure 2G. CEBPA mRNA FISH images are shown for comparison. Scale bars are 50 µm. See also Figure S6.
(C) Measurements using actinomycin D and RT-PCR show that the half-life of FABP4 mRNA is much longer than RPL18 mRNA, a known long-lived control. The half-life of CEBPA versus RPL18 mRNA is shown for comparison. First-order exponential decay curves were fitted to data. The shaded region represents the 95th confidence bounds. See also Figure S6.
(D) Single cell immunofluorescence analysis showing the mean nuclear PPARG and the mean cytosolic FABP4 values in each cell stimulated with DMI for 24 hours or 48 hours and fixed at 96 hours. Each scatter plot represents approximately 5000 cells and is colored based on cell density.
(E) Suppression of differentiation by siRNA-mediated depletion of FABP4 and CEBPA in OP9 cells.
(F) Overexpression of FABP4 increases differentiation even without DMI stimulation. OP9 cells were transfected with constructs expressing either mcherry(control) or mcherry-FABP4 and left in normal growth media for 5 days. No adipogenic stimulus was added.
(E–F) Percent of differentiated cells was quantitated as in Figure 1B. Barplots show mean +/− s.e.m. from 3 technical replicates, representative of 3 independent experiments.
(G–H) Live-cell imaging of citrine-PPARG cells transfected in (G) with FABP4-targeted or control siRNAs or in (H) with mcherry-FABP4 or control(mcherry). Cells were induced to differentiate using the standard DMI protocol. Plotted lines are population median traces with shaded regions representing 25th and 75th percentiles of approximately 700 cells per condition.
Model calculations show that transcriptional circuits with slow and fast positive feedback can filter periodic stimuli and regulate fractional differentiation
Our results so far suggest that the ability to both reject single and repetitive pulses of stimuli and to regulate increasing fractions of cells in the population to differentiate for continuous stimuli of increasing durations requires a regulatory circuit with fast and slow positive feedback (Figure 6A). To validate this requirement, we carried out simulations using an ordinary differentiation model to predict abundance changes of PPARG driven by the action of combined fast and slow positive feedback (see Methods). In our model, the time constants of the fast and slow feedbacks were set to 3 and 34 hours, respectively, which corresponds to typical values found for the CEBPA and FABP4 feedbacks to PPARG (Figures 4G and 5B). For a 48-hour continuously-applied stimulus, model calculations showed that a slow feedback partner would mediate a delayed buildup of PPARG past the threshold where differentiation is triggered (Figure 6B). For an oscillating stimulus, the model shows that the concentrations of a slow feedback partner would level off to a much lower steady-state than that reached by continuous stimulus, thereby preventing PPARG from reaching the threshold for differentiation (Figure 6C). Variations of the model parameters suggest that the transcriptional circuit depicted in Figure 6A is able to filter out circadian input pulses as long as the slow feedback partner has a half-life longer than 12 hours and the fast feedback partners a half-life much shorter than 12 hours.
Figure 6. A signaling circuit with a fast and slow positive feedback can trigger differentiation for continuous stimuli and reject daily oscillations.

(A) Schematic of the dual feedback loop circuit that rejects circadian inputs but locks cells in the differentiated state for continuous stimuli.
(B) Quantitative simulations when a continuous 48-hour stimulus is applied show that the slow positive feedback regulator mediates a slow buildup of PPARG to the threshold for differentiation. The threshold is marked with the dotted line.
(C) Same model simulations as in (B) but with an input stimulus applied in circadian 12-hour on / 12-hour off oscillations. The slow regulator builds to a steady-state below the threshold in cells stimulated with circadian hormonal oscillations which keeps levels of its feedback partner PPARG permanently low and below the PPARG threshold where differentiation is triggered.
(D) Addition of cell-to-cell variability (30% lognormal noise) to a model for a continuous 48-hour stimulus results in high rates of differentiation, but now with variable delays.
(E) In contrast, addition of cell-to-cell variability to the model with daily oscillating stimuli now allows PPARG levels in a few cells to reach the threshold and result in differentiation.
(F) Barplot summarizing 400 simulations with continuous stimulus was applied for 48 hours and 400 stimulations when an oscillating 12-hr on / 12-hr off stimulus was applied. Cells were categorized as differentiated if the PPARG level in that cell rose above the threshold and high PPARG was maintained even when input stimulus was removed. Model results are similar to results from the fixed and live-cell experiments shown in Figures 1C–D, 3B, and 4A–B.
(B–E) Each trace represents a different simulated cell.
The model so far could explain the experimentally-observed rejection of single and repetitive pulses of differentiation stimuli. However, our experiments showed that still a few cells could differentiate for pulsatile stimuli (Figures 1C and 4A). In addition, pulse durations of longer than approximately 12 hours resulted in an increasing fraction of the cell population differentiating (Figure 2B) which also cannot be explained by the above model. We next added noise to our model in order to determine whether cell-to-cell variation (noise) in the slow and fast positive feedback circuit is sufficient to explain why stimulated cells cross the threshold at different times and why periodic stimuli generate low differentiation rates and not zero differentiation. The rationale for the addition of the noise is based on our finding that cell-to-cell variation in PPARG expression enables control of low rates of adipogenesis in a population of precursor cells (Ahrends et al., 2014). Our experimental data in Figures 3B and 3C had shown that cells differentiated at different times when a continuous stimulus was applied. Indeed, adding lognormal stochastic variation to each simulation resulted in individual cells reaching the differentiation threshold with variable delays for continuous stimulation (Figure 6D), reproducing our experimental data in Figure 3B and 3C. Adding the same stochastic noise to the simulations in which differentiation was induced by daily 12-hour on/12-hour off pulses of stimuli showed that cells differentiated only rarely (Figure 6E), again reproducing our experimental data (Figures 4A). Differentiation outcome statistics of oscillating and continuous simulations are shown in Figure 6F. Thus, a circuit with fast and slow positive feedback, together with stochastic variation in PPARG signaling, is sufficient to explain variable delays in cells reaching the threshold and also recapitulates the low differentiation rates observed experimentally for daily oscillations of DMI stimuli.
Flattening the circadian glucocorticoid oscillations in mice results in a striking increase in fat mass
While previous studies had shown that increased glucocorticoid levels can increase fat mass in rodents and humans (Campbell et al., 2011; Lee et al., 2014), our goal here was to test if and how oscillating versus continuous levels of glucocorticoid stimuli affect fat mass and adipogenesis. To generate flattened, sustained levels of Corticosterone (Cort), we implanted eight-week old C57BL/6J male mice with pellets that released Cort continuously over 21 days (Scheme in Figure 7A and data in Figure 7B). The Cort dose released per day from the pellet was chosen based on previous studies using Cort pellets in mice (Hodes et al., 2012) such that mean Cort levels would not exceed normal mean physiological levels. As shown in Figure 7B, we confirmed that animals with sham pellets showed the normal diurnal Cort oscillations with a nadir between 08:00–11:00 and peak levels between 17:00–20:00. Mice implanted with Cort pellets showed significant elevated Cort levels in the nadir of the diurnal pattern, and also showed lower peak Cort levels, effectively flattening the level of circulating glucocorticoids without significantly changing the total amount of circulating glucocorticoids in the mice compared to mice implanted with sham pellets. Flattening of circadian glucocorticoid oscillations by implanted Cort pellets has also been demonstrated in rats (Campbell et al., 2011; Dallman et al., 2000). Notably, due to the nocturnal waking of mice, the timing of circadian glucocorticoid oscillations in mice is shifted, peaking at approximately 19:00 (7PM) instead of at approximately 7AM as in humans.
Figure 7. Animals implanted with Corticosterone (Cort) pellets that caused sustained, flattened elevations in Cort showed significant increases in adipose tissue mass compared to animals with normal circadian Cort osillations.

(A) Schematic of expected flattening (blue curve) of glucocorticoid oscillations in animals with Cort pellet implantation.
(B) Timecourse of average Cort levels in mice implanted with Cort pellets compared to mice implanted with Sham pellets. N=3, mean +/− s.e.m.
(C) Timecourses showing weight of mice implanted with Cort pellets compared to mice implanted with Sham pellets. N=6, mean +/− s.e.m. Unpaired t-test (Sham pellet vs. Cort pellet): * p < 0.05, ** p < 0.01. Chow intake during the 26 days per mouse was 73.9 and 70.9 grams for Sham-pellet and Cort-pellet mice, respectively (see Table S7).
(D) Schematic of daily injection protocol to selectively increase peak amplitudes of Cort in animals.
(E) Peak amplitudes of Cort are greater than 40-fold higher for mice injected daily with Cort compared to mice Sham-injected daily with phosphate buffer solution (PBS). N=3, mean +/− s.e.m.
(F) Timecourses showing weight of mice injected daily with Cort compared to Sham-injected daily with PBS. N=6, mean +/− s.e.m. Chow intake during the 26 days per mouse was 68.2 and 74.0 grams for PBS-injected and Cort-injected mice, respectively (see Table S7).
(G and H) The weights of both the Epididymal (visceral) and Inguinal (subcutaneous) fat depots were significantly increased for the Cort pellet group but not for the other 3 groups of mice. N=6, mean +/− s.d. Unpaired t-test: * p < 0.05, ** p < 0.01, *** p < 0.001.
(I) A custom cell contour image analysis program written in MATLAB was used to automatically measure adipocyte size from H & E stained fat tissue slices. Left, example of original H & E image. Right, segmented image showing identified adipocytes in green. Cells touching borders or not meeting shape criteria were excluded.
(J–K) Barplots of the average cell volume (J) and number of cells (K) in the Epididymal and Inguinal adipose fat depots in mice treated with Sham or Cort pellets for 21 days. The number of cells were derived by dividing total fat depot weight by the average cell volume. Each bar represents ~10,000 adipocytes analyzed from 90 images per condition (6 mice with either implanted Sham or Cort pellets × 3 tissue sections per mouse of either Epididymal or Inguinal fat × 5 images per section). Error bars show s.e.m. based on 90 images.
Confirming previous results in which Cort pellets were implanted in rats (Campbell et al., 2011), the mice we had implanted with Cort pellets initially showed a small decrease in body weight before increasing their weight over the next 3 weeks at a faster rate compared to animals with Sham pellets and ending up weighing approximately 5% more than Sham animals on day 26 (Figure 7C). Since our in vitro data had shown that daily glucocorticoid pulses with durations less than 12 hours do not result in differentiation independent of their amplitude within a 12-fold amplitude range (Figure S2B), we next tested whether this was also the case in vivo. We generated a 40-fold increase in daily peak amplitude of glucocorticoids compared to Sham-injected mice by injecting Cort into mice at 17:00 (5PM) every day for 21 days (Figures 7D and 7E). Strikingly, despite the greatly increased daily peak levels - and thus total dose - of Cort, we did not observe a significant difference in weight between the Cort- and sham-injected animals (Figure 7F), arguing that peak amplitude and total dose of glucocorticoids can change over wide ranges and still not cause an increase in weight as long as the increase in glucocorticoids occurs during a short time window and leaves a sufficiently long time period each day with low Cort. Together, these results raise the question whether the weight gain we observed in the Cort pellet experiments is a reflection of an increase in fat mass.
Markedly, when we dissected animals in the four groups after 26 days, we found that the animals with implanted Cort pellets - and flattened glucocorticorticoid levels - had significantly greater amounts of both inguinal (subcutaneous) and epidydimal (visceral) adipose mass compared to the mice with sham pellets and normal circadian glucocorticoid oscillations (Figures 7G and 7H). Furthermore, despite experiencing daily 40-fold greater peak levels of glucocorticoids for 21 days, the Cort-injected animals showed indistinguishable increases in inguinal and epididymal adipose mass compared to sham-injected animals. A summary of the Cort pellet and injection experiments is presented in Table S7.
To further characterize the adipose tissues in Cort versus Sham-treated mice, we prepared paraffin sections from the inguinal and epididymal fat pads and carried out routine hematoxylin and eosin (H & E) histology staining. We used automated image analysis to compare histology images for the two conditions: Cort-pellet or Sham-pellet mice, and for both types of fat depots: inguinal and epididymal (Figure 7I). We quantified cell volumes since previous studies showed that glucocorticoid-mediated increases in fat pad size are associated with both increased number of cells, as well as with increased cell volume (Lee et al., 2014; Rebuffe-Scrive et al., 1992). In both inguinal and epididymal fat pads, animals implanted with Cort pellets had significantly larger adipocytes compared to animals implanted with sham pellets (Figures 7J and 7K). We then used the cell volume measurement and the respective fat pad weights to derive an estimated number of cells in the fat pads. Epididymal, but not inguinal, fat pads showed a significantly higher number of adipocytes, consistent with previous results in rats (Campbell et al., 2011), and suggesting that Cort pellet treatment, which flattens daily glucocorticoid oscillations, results in increased adipogenesis as well as an increase in adipocyte volume.
DISCUSSION
Our study shows that different preadipocyte model systems (i.e., primary SVF, 3T3-L1, and OP9 cells) fail to differentiate in response to 12-hour on / 12-hour off pulses of hormone stimuli that correspond to the timing of circadian hormone secretions observed in humans and mice. In striking contrast, the same preadipocytes differentiate at gradually increasing rates if the duration of the daily pulses increases from 12 to 18 to 24 hours. Because an increase in pulse duration leads to a shorter trough, or off-period, during each stimulation cycle, our study shows that cells increasingly fail to reset their PPARG level down as pulse durations get longer, thereby increasing the probability that PPARG levels will reach above the threshold and cause cells to continue on the path to the differentiated state. We validated these findings in mice and showed that flattening the circadian glucocorticoid oscillations while keeping the overall circulating glucocorticoid concentrations the same over 21 days caused fat mass to double compared to mice with normal circadian oscillations. Furthermore, when we raised the peak of the glucocorticoid oscillations 40-fold by injecting corticosterone into mice daily for 21 days, there was no increase in fat mass compared to sham (PBS)-injected mice, providing strong validation that as long as glucocorticoid increases occur within the correct circadian timeperiods, there is only minimal adipogenesis and that it is not increased levels of glucocorticoids, but rather losing the nadirs or "off-periods" that leads to increased fat mass. It should be noted that our quantitation of adipogenesis in vivo could be improved by using, for example, BRDU-injection or the Adipochaser mouse to label the adipocytes generated in a specific time window (Jeffery et al., 2015; Wang et al., 2013).
We were surprised to find in our live-cell analysis of endogenous CEBPB that the expression of CEBPB increases and decreases along with the daily hormone pulses, reaching each time quickly a maximal level and then dropping again back to basal levels after the hormone stimulus is removed. The closely mirrored relationship between CEBPB levels in cells and the external pulse pattern argues that filtering has to occur downstream of CEBPB. We further showed that the life-time of the mRNA and protein of PPARG, CEBPB and CEBPA, are all relatively short lived, and all drop back down rapidly when the input stimulus is removed, which can explain how cells can reset back to their basal state after each external hormone pulse as long as the PPARG level in the cell has stayed below the threshold for differentiation. However, these findings did not explain how continuous stimuli can trigger a gradual buildup in PPARG which motivated us to search for slow-acting PPARG regulators. As one such slow-degrading co-factor, we identified FABP4, whose mRNA has a long half-life of between 14 to 34 hours (Figures 5B–5C, S6). We showed that FABP4 expression can accelerate PPARG induction and that it is also needed to mediate the slow 24 to 48-hour increase in PPARG levels up to the threshold where differentiation is triggered. This argues for a control principle for differentiation whereby a combined fast and slow positive feedback generates a differentiation system with a delayed and irreversible threshold that can filter out short pulses and circadian hormone stimuli.
FABP4 is a fatty-acid binding protein that is a downstream target of PPARG and is highly upregulated during adipogenesis (Hotamisligil and Bernlohr, 2015). Our live-cell analysis of the differentiation decision showed that FABP4 has a critical role in activating PPARG and in controlling the irreversible switch from preadipocyte to adipocyte differentiation. Our results are in line with other studies that showed that FABP4 upregulates PPARG expression and activity, likely by transporting fatty-acid ligands through the cytosol and across the nuclear membrane to PPARG in the nucleus (Adida and Spener, 2006; Ayers et al., 2007).
As was found previously in rats (Campbell et al., 2011), we found a lack of adipogenesis in the subcutaneous fat of Cort-treated male animals. Interestingly, this finding is in line with previous studies highlighting the inability of the subcutaneous fat depot to expand by adipocyte hyperplasia in high fat diet (HFD) fed male mice (Wang et al., 2013). However, it is possible that subcutaneous adipogenesis might have been observed if female mice had been used in our study. The subcutaneous fat depot of female mice has been shown to be capable of expanding through adipocyte hyperplasia in HFD fed mice (Jeffery et al., 2016). Likely the subcutaneous microenvironment, or niche, is strongly regulating adipogenesis in vivo.
Since preadipocytes differentiate at low rates in humans and mice (Spalding et al., 2008; Wang et al., 2013) and since a flattening of daily glucocorticoid oscillations leads to increased fat mass and obesity (Campbell et al., 2011; Dallman et al., 2000; Lee et al., 2014), our live-cell measurements provide a molecular basis for strategies to alter the daily timing of glucocorticoid pulses that might be beneficial therapeutically to influence differentiation rates. Specifically, it is suggestive that the dramatic suppression of differentiation for circadian hormone pulse patterns and the much higher rates of differentiation that we found for more sustained hormone signals may provide an explanation as to why conditions such as Cushing's disease, chronic stress, or long-term glucocorticoid treatments that disrupt normal circadian patterns of hormone oscillations also result in increased obesity. Finally, the molecular filtering mechanism for differentiation we uncovered for adipocytes provides support for the development of temporal therapeutic regimens aimed at changing adipogenic or other hormone pulse durations to control differentiation of precursor cells.
In conclusion, our study introduces a temporal control mechanism for adipogenesis that allows precursor cells to reject normal, daily, oscillating hormone inputs. We show that a dual fast and slow positive feedback system centered on PPARG has the marked characteristic to remain unresponsive to circadian and rhythmic hormone pulses as long as the duration of the trough between pulses remains longer than 12 hours. However, when pulse duration increases and the trough duration becomes shorter, cells convert the duration of the daily pulses into an increasing probability for differentiation until the rate of differentiation becomes maximal for continuous stimulation. Our findings are likely of relevance for many, if not most, differentiation systems since oscillating hormone stimuli are a near universal stimulus pattern in mammalian physiology.
Limitations of study
Our work made use of different in vitro models to determine how oscillating versus constant hormone stimuli control adipocyte differentiation. Future work will be needed to determine the precise timing requirements by which hormones that increase cAMP, glucocorticoids, and potentially other signaling pathways control adipogenesis in different physiological settings and whether and how the hormones work synergistically. Furthermore, we and others have found that FABP4 has key functions not only in mature adipocytes but also during adipogenesis to increase PPARG expression, and the molecular mechanisms of this regulation will need to be further explored. Finally, further studies will be needed to work out in more detail how oscillating versus constant glucocorticoid signaling regulates preadipocytes and mature adipocytes to better explain how the two cell populations contribute to the large increase in fat mass we identified here for constant glucocorticoid stimulus
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author, Dr. Mary N. Teruel (mteruel@stanford.edu).
EXPERIMENTAL MODEL DETAILS
OP9 mouse stromal cell line (Wolins et al., 2006)
3T3-L1 mouse preadipocyte cell line (Green and Kehinde, 1975)
3T3-F442A mouse preadipocyte cell (Green and Kehinde, 1975)
C57/Bl6 male mice (source: Jackson Labs)
METHOD DETAILS
Cell culture and differentiation
Primary SVF preadipocytes were cultured according to previously published protocols (Ota et al., 2015). Briefly, stromal vascular cells (SVC) were isolated from inguinal subcutaneous WAT from 6–8 week old male C57BL/6J mice. The tissue was rinsed in PBS, minced, and digested with 1mg/ml collagenase type D (Roche 11088866001) and 1mg/ml Dispase II (Sigma-Aldrich D4693) in PBS with 1mM CaCl2 for 40 minutes in a 37C shaking water bath. The digest was filtered to exclude large debris and separated into the adipocyte (supernatant) and SVC fractions (pellet) by spinning cells at 300 RCF for 3 minutes. The SVC fraction, which contains preadipocytes, was washed once and then plated in warm culture medium (DMEM with 10% FBS + 100U/mL pen/strep + 2.5ug/ml amphotericin B) for 2 hours and then washed with culture media to remove cell debris and non-adherent cells, thus enriching for preadipocytes which adhere well to the culture surface. Cells were further grown in culture for one to two passages before plating cells for experiments.
OP9 and 3T3-L1 cells were cultured according to previously published protocols (Ahrends et al., 2014; Park et al., 2012; Wolins et al., 2006). OP9 cells were cultured in 20% Fetal Bovine Serum (FBS) in growth media consisting of MEM-α (Invitrogen, # 12561) and 100 units/mL Penicillin, 100µg/mL Streptomycin, and 292 µg/mL L-glutamate (Invitrogen, # 10378-016). 3T3-L1 cells were cultured in 10% bovine calf serum in growth media consisting of DMEM, 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin.
To induce differentiation of SVF preadipocytes, OP9 cells, and 3T3-L1 cells, a standard DMI protocol was used: Confluent cells were treated with a differentiation medium containing a commonly used DMI (dexamethasone / IBMX / insulin) stimulus to initiate adipogenesis. DMI consists of dexamethasone (dex), a synthetic glucocorticoid; 3-isobutyl-1-methylxanthine (IBMX), an inhibitor of phosphodiesterase that increases cAMP levels; and insulin. Applying the DMI stimulus consisted of replacing the media on the cells with growth media plus 10% FBS, 250 µM IBMX (Sigma Cat # 7018), 1 µM dexamethasone (Sigma Cat #D1756), and 1.75 nM insulin (Sigma Cat # I6634) (DMI Stimulus I). Two days after initiating differentiation, the media was removed and was replaced with growth media plus 10% FBS and 1.75 nM insulin for two more days. As noted in some experiments, corticosterone (Sigma Cat #174) was used instead of dexamethasone. Also, for other experiments as noted, rosiglitazone (Cayman, USA) was added to the media to result in a final concentration of 1 to 10 µM.
Unless otherwise noted, the concentration of DMI stimulus used for a 48-hour continuous pulse was 1µM dexamethasone (dex), 250 µM IBMX, and 1.75 nM insulin. In some experiments - for example Figure 1C - to keep the total amount of stimuli, i.e. area under the curve, constant for all 4 protocols over the 96-hour experimental timeframe, the concentration of dex and IBMX was increased proportionally to compensate for decreases in pulse duration. To apply pulses of dexamethasone, corticosterone, or other stimuli, the current media on the cells was gently removed.. To remove all traces of the previous stimulus, the cells were then washed by adding fresh growth media to the cells and removing it three times. Then the new stimulus was applied to the washed cells. Typically to end a pulse, media with no glucocorticoids or IBMX, but containing 1.75 nM insulin. The doses of dex used were not saturating (Figures S2A and S2B), and using corticosterone instead of dex in the stimulus showed the same filtering effects (Figure S2C).
Immunofluorescence Staining
OP9, 3T3-L1, and SVF preadipocyte cells were fixed with 3% paraformaldehyde in PBS for 30 min. Then the cells were gently washed 3× with PBS. Permeabilization was carried out with 0.1 % Triton X-100 in PBS for 15 minutes on ice, followed by blocking with 5% bovine serum albumin (Sigma #7906). The cells were stained with DAPI (1:20000), anti-PPARG (1:1000 Santa Cruz Biotech #sc-7273), anti-PPARG2 (Abcam ab45036), anti-CEBPB (1:1000, Santa Cruz Biotech #sc-150), anti-CEBPA (1:1000, Santa Cruz Biotech #sc61), or anti-FABP4 (1:200, Abcam, #ab13979). Alexa Fluor-514 (#A31558), 594 (#A11032) and 647 (#A31571) (1:1000, Invitrogen) were used as secondary antibodies.
In cases in which BODIPY was used to stain lipids, permeablization was carried out more gently using 0.05% saponin (Sigma #47036), to better preserve the lipid structure, followed by blocking with 5% bovine serum albumin (Sigma #7906). The cells were then stained with the antibodies listed above, plus BODIPY 493/503 (1µg/ml, Molecular Probes #D-3922).
Defining differentiation
We have previously shown that the transition from a proliferating preadipocyte precursor cell into a mature, non-proliferating adipocyte capable of accumulating lipid occurs via a bistable switch from low PPARG expression in the cell to high PPARG expression (Ahrends et al., 2014; Park et al., 2012). As shown in Figure 1B, a histogram of PPARG expression for a population of differentiating adipocytes shows two peaks of PPARG expression - low and high. The high state predicts the subsequent lipid droplet formation. We thus define a cell as being differentiated if its level of PPARG placed the cell in the high PPARG-expressing peak of the cell population which correlates with high lipid accumulation and markers of mature adipocytes such as adiponectin and GLUT4 (Figures 1B, S1A, and S1B).
siRNA transfection
siRNA was purchased from the respective manufacturers listed in the Key Resources table. OP9 cells were transfected with the respective siRNA or control (YFP) siRNA 24 hours before the start of an experiment by reverse transfection using Lipofectamine RNAiMax (Invitrogen #13778150), following the manufacturer’s protocol. OP9 cells were induced to differentiate by the standard DMI protocol. The knockdown efficiency of the CEBPB siRNA was over 60%, and the knockdown efficiencies of the FABP4 and CEBPA siRNAs were both over 75% (Figure S3B). For the fixed-cell siRNA experiments in Figure 2C and 5E, differentiation was assessed at 96 hours as in Figure 1B.
Measuring protein decay rates using cyclohexamide
To obtain protein decay rates, 10,000 OP9 cells were seeded in 96-well plates) one plate for each timepoint. Cells were induced to differentiatiate with DMI for 24 hours. Cyclohexamide was added at a final concentration of 30 µM. Cells were fixed and stained at different times after addition of cyclohexamide, and immunofluorescence was used to quantify protein concentration following the protocol shown in Figure 1B. Half-lives were obtained by fitting first order exponential decay curves to the data.
Measuring mRNA decay rates using Actinomycin D and RNA FISH
To carry out an mRNA decay timecourse with RNA FISH are the readout, 10,000 OP9 cells were seeded in 96-well glass plates (Greiner), one plate for each timepoint. Cells were induced to differentiate with DMI for 24 hours. Actinomycin D (Cat #A1410, Sigma) was added 5 minutes before the first timepoint (t=0 hours) at a concentration of 5 µg/ml. Cells were fixed with 4% formaldehyde for 15 min, stored in 75% ethanol at 4°C. RNA FISH w as performed using the Affymetrix Quantigene ViewRNA ISH cell assay (Affymetrix eBioscience, San Diego, CA). FISH probes for mouse PPARG, CEBPA, CEBPB, and FABP4 were purchased from Affymetrix eBioscience. Cells were rehydrated in PBS for 10 minutes and then permeabilized with a detergent solution provided by the assay kit for 5 min at room temperature. The cells were incubated with diluted FISH probes (1:25) at 40°C for 3 hrs. Cells were then hybridized with the three different probes provided by the assay kit: preamplification probe, amplification probe, and label probe which were carried out by incubating cells with a probe for 30 min at 40°C. Finally, cells w ere incubated with Hoechst (1:10,000 in PBS) for 5 min, washed three times with PBS, and left in PBS for imaging.
Measuring mRNA decay rates using Actinomycin D and RT-PCR
To carry out an mRNA decay timecourse using RT-PCR as the readout, OP9 cells were seeded in 6-well tissue-culture plates, one plate for each timepoint. Cells were induced to differentiate with DMI for 24 hours. Actinomycin D (Cat #A1410, Sigma) was added 5 minutes before the first timepoint (t=0 hours) at a concentration of 5 ug/ml. Cells were collected at the respective timepoints, and RT-PCR was performed using the primers listed in Table S6.
Measuring mRNA decay rates using 5-Ethnyluridine (EU) pulse-labeling
Analysis of mRNA half-lives was performed by EU pulse-labeling of RNA using the Click-iT Nascent RNA Capture Kit (ThermoFisher). OP9 cells were seeded in 10 cm tissue culture dishes, grown to confluence, and then induced to differentiate with a modified DMI cocktail protocol. Briefly, cells were stimulated with the typical DMI cocktail for 24 hours. Then half the volume of differentiation media in each plate was removed and combined. To avoid stimulation by fresh media, 5EU was directly added to the combined differentiation media to a final concentration of 200 µM. The remaining half of differentiation media in each plate was then aspirated and replaced with media containing 5EU and incubated for another 24 hours. At 48 hours, all plates were washed 3 times with fresh culture media and replaced with 5EU-free culture media containing only insulin. Cells were harvested at the indicated time points and total RNA was isolated using TRIzol reagent (ThermoFisher). EU-labeled RNAs were biotinylated and captured on streptavidin coated magnetic beads using the Click-iT Nascent RNA Capture Kit (ThermoFisher), following the manufacturers protocol. RT-qPCR was performed on the captured RNA to assess mRNA half-lives.
Image Acquisition
Images were acquired on an ImageXpress MicroXL automated epifluorescence microscope (Molecular Devices; Sunnyvale, CA, USA) using 10× Plan Fluor objective and a 2560 × 2160 pixel Andor Zyla 5.5 sCMOS camera with a 16-bit readout. A camera bin of 1 was used for fixed cell imaging, and a camera bin of 2 was used for live cell imaging.
For fixed-cell imaging, cells were plated in 96-well, optically clear, polystyrene plates (Costar #3904) For live-cell imaging, 7,000 – 8,000 OP9 cells were plated >6–12 hours prior to imaging in full growth media in 96-well optically clear, glass-bottom plates (either Greiner Sensoplate or Invitro Scientific).
Living cells were imaged in FluoroBrite DMEM media (Invitrogen) to reduce background fluorescence. Before image acquisition, the full growth media was switched to media consisting of FluoroBrite DMEM with 10% FBS and 1% Penicillin/Streptomycin. Time-lapse imaging was performed in 200 µl of media per well. Cells were imaged in a humidified 37degC chamber at 5% CO2. Images were taken every 10 min in CFP, YFP, RFP channels (depending on the experiment). Total light exposure time was kept less than 600 ms for each time point. Two, non-overlapping sites were imaged per well.
Image Processing and Analysis
Segmentation
Cells were segmented for their nuclei based on either Hoechst staining (fixed-cell imaging) or H2B-Turquoise (live-cell imaging). Nuclear segmentation was performed as follows: A Laplacian of Gaussian filter was applied to the nuclear image to identify the edges of cell nuclei. Pixels within the nuclei edges were identified as cell nuclei and pixels outside the nuclei edges were identified as background. To split cells in contact with their nearest neighbor(s), a custom segmentation algorithm was implemented to detect and bridge concave inflections in the perimeter of each object (hereafter referred to as the ‘deflection bridging algorithm’).
Signal Measurement
Each channel global background subtraction was used to measure all immunofluorescence and fluorescent protein intensities as follows: the nuclear mask was dilated by 50 m and the background for the image was calculated as the mode pixel intensity of all non-masked pixels. Nuclear immunofluorescence and nuclear fluorescent protein signals were calculated as median nuclear intensity. In the case of CEBPB, the sum nuclear intensity was used instead because it exhibited a punctate pattern due to its centromeric localization in the nucleus.
Tracking
The deflection-bridging algorithm was implemented on every object in the first imaging frame, and then only adaptively in subsequent frames. This was accomplished by iteratively tracking cells in each frame, detecting probable merge events (as discussed below), and selectively implementing the deflection-bridging algorithm on putative merged objects. This method reduced the probability of over-segmentation, increased processing speed, and improved tracking fidelity. Tracking of cells between frames was implemented by screening the nearest future neighbor for consistency in total H2B-Turquoise fluorescence. This ‘conservation of total fluorescence’ was further exploited to detect merges or splits, which allowed recovery of overlapping traces. Mitosis events (called at anaphase) were called when the total H2B fluorescence of the two nearest future neighbors of a given cell were both between 45–55% of the total H2B fluorescence of the past cell. Frame-to-frame jitters were accounted for by registering sequential H2B-Turquoise images by cross-correlation.
Workflow used to generate single OP9 cell colonies with endogenously tagged CEBPB and PPARG (Figure S3)
A) Construction of DNA plasmids
A.1) Construction and design of the donor template
CRISPR-mediated genome editing was used to tag the N-terminus of endogenous PPARG and CEBPB with Citrine, a bright version of the yellow fluorescent protein. We tagged the PPARG2 isoform since it is the PPARG isoform that is highly expressed in adipose tissue and that controls fat cell differentiation. CEBPB has three isoforms LAP*, LAP, and LIP, and we chose to tag the longest isoform LAP* at the N-terminus. The DNA repair template to promote homology directed repair (HDR)-mediated insertion of the fluorescent protein (FP) was constructed by inserting the cDNA of FP-3xGly flanked by two 800 bp homology arms into the entry vector backbone pENTR1a (Addgene Plasmid #17398). The repair template was assembled such that after HDR, the FP is inserted in frame at the N-terminus and separated with a 3xGly linker from PPARG and CEBPB, respectively. The pENTR1a backbone vector was digested with EcoRΙ-HF and BamHΙ-HF (NEB), and assembled together with three DNA fragments coding for homology arm 1, Citrine or mKate2, and homology arm 2 using Gibson assembly. The homology arm fragments were PCR amplified from OP9 genomic DNA with primers introducing a 15–20 bp overhang used for Gibson assembly. Similarly, the FP was PCR amplified from a DNA template using primers introducing the linker and an overhang for Gibson assembly. The sequences of the assembled donor vector constructs were verified by sequencing.
A.2) Construction of the Cas9 plasmids
To carry out CRISPR genome editing to make Citrine-CEBPB, we used Plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330) (Addgene plasmid # 42230) to deliver the SpCas9 protein and guide RNA (Cong et al., 2013). A 20 nucleotide “targeting” sequence was inserted into the guide sequence insertion site of the pX330 plasmid to produce guide RNAs directed to the N-terminal of CEBPB. Targeting sequences were designed using the web tool, crispr.mit.edu. From the predicted target sites, the sequences with the highest scores and in proximity of the ATG were selected. To carry out CRISPR genome editing to make Citrine-PPARG, we initially used the same single guide RNA strategy to insert citrine at the N-terminus of PPARG. However, we found that the majority of PPARG clones generated using SpCas9 had undesired off-target integrations of Citrine into the OP9 genome. To reduce off-target integration events we switched to using the “double nickase” system which uses two different guide RNAs that create adjacent and opposing nicks in the DNA at the site of insertion (Ran et al., 2013). We found that using the double-nickase system greatly improved the specificity of Cas9-mediated double strand breaks at the PPARG locus. To carry out double-nickase genome-editing, two different targeting sequences directed to the PPARG locus were designed as described above and inserted into the guide RNA site of two plasmids, pX335-U6-Chimeric_BB-CBh-hSpCas9n (pX335) (Addgene plasmid # 42335) encoding the SpCas9 D10A nickase. Oligonucleotide duplexes encoding each desired targeting sequence were ligated into the BbsΙ cut sites of px330 or px335, respectively. All constructs were validated by sequencing.
B) Transfection
To endogenously tag PPARG with Citrine, 1 µg of each of the two pX335 guide RNA/SpCas9n constructs and 5 µg of the Citrine donor template were transfected into 1 million OP9 cells using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. To endogenously tag CEBPB with Citrine, 2 µg of the desired pX330 guide RNA/SpCas9 construct and 5 µg the Citrine donor template were transfected into 1 million OP9 cells using Lipofectamine 2000.
C) Clone selection by single-cell FACS
Seven days post-transfection, single cells expressing Citrine were sorted into separate wells of 96-well culture plates and allowed to grow. We chose to wait 7 days post transfection to avoid false positive fluorescent signal originating from the un-integrated donor DNA plasmid.
D) Stimulus Response Test
Once the single-cell colonies grew to 50% confluency, each colony was passaged into wells on two different 96-well plates. One plate was used to expand the colonies, and the other half was imaged using a Molecular Devices MicroXL fluorescence imaging system to select for clones with correct localization of the Citrine signal and the appropriate response to stimuli. Before imaging, PPARG clones were stimulated with Rosiglitazone for 24 hours to induce expression of the Citrine-PPARG, and CEBPB clones were stimulated for 24 hours with DMI to induce expression of Citrine-CEBPB.
E) Differentiation capacity test
Clones that expressed Citrine were further characterized for their differentiation capacity using the standard four-day adipocyte differentiation protocol detailed under “Cell Culture and Differentiation.” Clones that acquired mature adipocyte morphology and accumulated lipid droplets in response to DMI treatment were expanded and subjected to further validation steps.
F) Further validation
F.1) Validation of citrine-CEBPB clones
We first performed genomic PCR with different primer sets to look at the genotype and verify correct insertion of Citrine into the CEBPB locus. The first PCR was performed using primers annealing to regions flanking the site where Citrine is inserted (Supp Table 3, Figure S5A). Using this set of primers, CEBPB tagged clones were shown to be heterozygous, and the PCR products were subjected to sequencing. The second set of PCRs was performed using primers annealing to regions around 5’ and 3’ ends of the inserted Citrine. (Supp Table 4, Figure S5B). Both reactions resulted in a single band, indicating that Citrine was correctly inserted into the 5’ end of the CEBPB locus in all clones.
Next, Western blot analysis of citrine-CEBPB clones was performed using anti-GFP antibody and anti-CEBPB to verify protein expression and to check for the correct molecular weight of the tagged protein (Figures S5C–D). The size compatible with the correct predicted molecular weight of citrine-protein fusion were shown for each clone. The anti GFP blot shows the expression of tagged-CEBPB and did not detect any free GFP. The anti CEBPB blot shows the expression of the three CEBPB isoforms (LAP*, LAP, LIP) in OP9 cells. In the citrine-CEBPB clones, the LAP* isoform is shown to be tagged with citrine as expected.
Next, Southern blot analysis was performed to confirm locus-specific knock-in using a probe directed towards citrine. All examined clones showed the presence of a specific copy of citrine within the genome, as evidenced by the detection of a single band of expected size (1.5kb) in the Southern blot (Figure S5E).
Finally, immunohistochemistry analysis was used to validate correct nuclear localization and differentiation capacity of the clones (Figure S5F–H). Colocalization of the citrine fluorescence signal with the immunohistochemistry signal of the untagged proteins throughout four days of differentiation, was used to choose clones for live cell imaging. The selected clone CEBPB-3 differentiated well and showed similar expression of citrine-CEBPB and untagged CEBPB over the time course of differentiation (Figure S5F). Thus, clone CEBPB-3 was subsequently used for all further experiments.
F.2) Validation of citrine-PPARG clones
The same steps used for CEBPB validation were used to validate the PPARG clones (Figure S4). Since it passed all the validation criteria described above, the PPARG-2 clone was used for all the time course measurements in the current manuscript.
T7 assay
The cutting efficiency of Cas9 with the designed guide RNAs was determined using a T7 Endonuclease assay. The assembled plasmids (pX330 or pX335) were transfected together into OP9 cells with a construct containing mCherry in the Clontech C1-vector (mcherry-C1). The mCherry-C1 served as a co-transfection marker for cells that are likely transfected with pX330 or pX335 plasmids. The constructs were transfected at a 1:10 ratio of gRNA-pX330 or gRNA-pX335 to mCherry-C1 into OP9 cells using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Seventy-two hours post-transfection, the mCherry-C1 expressing cells were sorted by FACS, grown up using our standard cell culture protocol, and genomic DNA was extracted using a Qiagen DNeasy Blood and Tissue kit. Approximately 600 bp of the genomic DNA (~200 and 400 bp around the predicted cut site) was amplified by PCR and used for a T7 assay that was carried out using Surveyer® mutation kit hybridization conditions (IDT, cat # 706020). Briefly, 200ng DNA was hybridized in a thermocycler to form heteroduplexes of cut and uncut DNA strands, incubated with T7 endonuclease I (New England Biolabs, Cat #M0302S) for 1 hour at 37 °C to cleave mismatched heteroduplexes, run on a 10% TBE polyacrylamide gel (Invitrogen) and stained with ethidium bromide. The fraction of strands that were cleaved by T7 endonuclease, as visualized by the gel, is representative of the cutting efficiency of the CRISPR constructs. For each site of interest, the CRISPR construct that showed the highest cutting efficiency was used to direct insertion of Citrine.
Western Blot Analysis
CRISPR-tagged clones and wtOP9 cells were stimulated for 48h with Rosiglitazone (Citrine-PPARG and FABP4-mKate2 clones) or for 12h with DMI (Citrine-CEBPB clones) to increase protein levels for western blot analysis of PPARG and CEBPB, respectively. The presence and integrity of Citrine-tagged proteins was verified with an antibody directed to the tagged protein and by immunoblotting for GFP. Cell pellets of citrine-PPARG clones were lysed on ice with nuclear extraction kit (Abcam), according to the manufacturer’s protocol, and quantified using a BCA assay (Thermo Scientific). For citrine-CEBPB clones, cells were lysed on ice in RIPA lysis buffer (Millipore, CA) in the presence of protease inhibitors (cOmplete™, Mini, EDTA-free, Sigma Aldrich). All samples were boiled for 10 min at 96 °C in NuPAGE LDS sample buffer containing 1× reducing agent (Invitrogen). Lysates were separated by SDS–PAGE using the XCell SureLock Electrophoresis Cell on NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels (Invitrogen). Proteins were transferred onto polyvinylidene fluoride membranes (Thermo Scientific) using the XCell II Blot Module (Invitrogen). Membranes were blocked by incubating with TBS with 0.1% Tween-20 containing 5% non-fat milk for 2h at room temperature or overnight at 4 °C for anti-GFP. Membranes were subsequently incubat ed overnight at 4 °C with primary antibodies at a dilution of 1:1,000 (anti-PPARG, Santa Cruz Biotech #sc-7273) and 1:2,000 (anti-CEBPB antibody, Santa Cruz Biotech #sc-7962) in blocking buffer or for 1h at RT at a dilution of 1:2,500 (anti-GFP abcam #ab290). Membranes were washed 3 times for 5 min using wash buffer (TBS with 0.1% Tween-20), and further incubated in 1:5,000 horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse and anti-rabbit (Cell Signalling #7076 and #7074) for 1 h at room temperature. After another set of three washes, antibody-bound proteins were visualized on film using supersignal™ West Femto chemiluminescence substrate (Thermo Scientific).
Southern blot analysis
To check whether Citrine and mKate2 was integrated at any unspecific loci, a southern blot was performed with a 32P-labeled probe directed towards Citrine. Two probes about 500 bp was amplified by standard PCR from a Citrine plasmid and gel purified. The labeled probe was prepared with the RadPrime DNA kit (Invitrogen) in the presence of dCTP [α-32P] (Perkin Elmer) and a clean-up step was performed using illustra™ Microspin™ G-25 Columns (GE Healthcare), all according to the manufacturer’s guidelines. Genomic DNA was extracted using a Qiagen DNeasy Blood and Tissue kit, and 7 µg DNA was digested overnight. Digestion enzymes were chosen such that they cut around Citrine and generate fragments of ~1.8 kb, ~1.5kb and 1.9 kb for PPARG and CEBPB, respectively. Digested samples were separated with 1% agarose gel electrophoresis, and the gel was incubated 1× 20-min in 0.25M HCl followed by 2× 15-min in 0.5M NaOH/1.5M NaCl to denature the dsDNA. Subsequently, 2× 15-min washes were performed in transfer buffer (1M NH4OAc) to neutralize the gel. The DNA fragments were transferred via capillary forces from the gel to an Amersham Hybond-N+ membrane (GE Healthcare). Afterwards, the DNA was cross-linked to the membrane using ultraviolet light (0.3J/cm2; UVStratalinker 1800, Stratagene, La Jolla, CA). After 1h of prehybridization of the membrane at 65 °C in a Te chne hybridizer HB-1D in hybridization buffer (0.5M sodium phosphate buffer pH 7.2, 7% SDS), the probe was hybridized overnight at 65 °C in the presence of salmon sperm DNA (100 µg/ml) to avoid unspecific binding of the probe. After hybridization, the blots were washed at hybridization temperature for 2× 15-min in each of the following buffers in sequential order: 0.3× SSC, 0.1% SDS, 0.1× SSC, 0.1% SDS 0.1× SSC, 1.5% SDS. The probe was visualized using a storage phosphor screen and Typhoon™ imager (GE Healthcare).
Description of Model
Stim represent the stimulus. Assume Stim ~ CEBPB
In the first equation, Stim and CEBPA are added together because CEBPB and CEBPA bind to the same DNA sequences and can replace each other at binding sites.
Cooperativity of 4 for (Stim+CEBPA) because CEBPB and CEBPA have to dimerize in order to function and there are multiple CEBPB/CEBPA binding sites on the PPARG promoter.
Z represents that FABP4 needs to activate PPARG in order for PPARG to have transcriptional activity on target genes like FABP4 and CEBPA.
FABP4’s activation of PPARG is limited such that it can only increase 6-fold (max. Z = 1.2 * PPARG).
Cooperativity of 2 in the second and third equations because there are multiple binding sites for PPARG on the CEBPA and FABP4 promoters
Degradation rates correspond to 1 hour for PPARG, 3.5 hours for CEBPA, and 30 hours for FABP4.
Lognormal noise (with mean=0, standard dev=30%) randomly to each simulation shown in Figures 7G and 7H through a noise term before the PPARG term in the equation calculating dCEBPA/dt. A noise term was added only to one equation for simplicity. We have established in previous work that adding a larger noise to a single parameter is similar to adding smaller noise terms to each parameter in different equations (Ahrends et al., 2014).
Mice
Seven-week-old C57BL/6J male mice were purchased from Jackson Laboratory (cat. 000664). Mice were housed on a 12h light/dark cycle (lights on at 7:00 hours) in the animal facility at Stanford University. All animal care and experimentation was conducted in accordance with current NIH and Stanford University Institutional Animal Care and Use Committee guidelines. Mice were housed in the animal facility for 7 days prior the start of experiments.
Corticosterone administration experiment
Mice (n=24) were divided equally into four groups. The first group of 6 mice was implanted with a corticosterone releasing pellet, the second group with a placebo pellet, the third group was injected with corticosterone, and the fourth group was injected with phosphate buffer solution (PBS). For pellet implantation, mice were anesthetized via inhalation of isoflurane. Placebo and corticosterone pellets (5mg, 21-day release; Innovative Research of America, Sarasota, FL, USA) were implanted subcutaneously with a trochar. Mice weighed an average of 24.2 ± 1.4 g, which results in a daily dose of 9 mg/kg/day. For injections, corticosterone complexed with 2-hydroxypropyl-β-cyclodextrin (C174, Sigma) was dissolved in PBS and injected subcutaneously once daily at 5PM for 21 days with the same corticosterone dose (9 mg/kg/day) as released by the corticosterone pellets per day.
Body weight and food intake were monitored in all mice for 26 days. After 26 days, mice were anesthetized with isoflurane and sacrificed by cervical dislocation. The epididymal and inguinal fat depots were surgically removed and weighed, followed by standard preparation of paraffin sections and hematoxylin and eosin (H & E) staining.
Measurement of corticosterone in blood serum
In addition to the 24 mice used above, 12 mice were divided into four groups, treated in parallel to the 24 mice as described above in the "Corticosterone administration experiment" section, and used to obtain blood serum corticosterone measurements. Eighteen days after pellet implantation or daily corticosterone/PBS injections, blood was taken at multiple time points over a 15 h time period. At the first timepoint, blood was taken by nicking the tail vain. Blood samples collected at following timepoints were taken by removal of the crust formed after first blood withdrawal. The blood was allowed to clot by leaving it undisturbed at room temperature for 45 minutes. The clot was removed by centrifuging at 2000 × g for 15 minutes. The corticosterone concentration in the blood serum was determined using the Enzyme Immunoassay kit (K014-H1, Arbor Assays, Michigan, USA) following the manufacturer's instructions.
Mouse Statistics
All data are represented as mean ± SD or mean ± SEM and analyzed by ANOVA followed by Students t test. N indicates the number of animals per group. Results were considered significant if p < 0.05.
Measuring amount of hyperplasia (adipogenesis) and hypertrophy (increase in cell size) in fat tissue
To compare hyperplasia and hypertrophy in the fat of mice subjected to continuous or oscillatory Cort-stimulus conditions, we carried out H&E staining on adipose tissue slices from the mice, imaged the slices, and used image analysis software to count the number of adipocytes using metrics that included a minimum size threshold to exclude debris, over-segmented objects, and to not exclude smaller cells; a maximum size threshold to exclude very large objects that for example occurred if the membrane between two cells broke and creating an object twice as large as a typical fat cell; as well as an eccentricity metric to take into account roundness and shape of the counted objects. One advantage of our automated approach is that we were able to obtain good statistics by being able to analyze many slides and sites. Approximately 10,000 cells were identified for each fat depot type and stimulus condition and input into the calculation to obtain mean cell volume for that fat depot type and condition. Since the density of fat is approximately 1 g/ml, the mass of the fat pad is approximately equal to the volume. To obtain the number of fat cells per fat pad, we then divided the mass (weight) of the fat pad by the median cell volume.
However, we realize that our analysis of in vivo adipogenesis could be improved. For example, we could use established protocols (Jeffery et al., 2015) in which BRDU was injected into mice to pulse-label proliferating cells. After a specified amount of time to allow proliferating preadipocytes to become adipocytes, two approaches could be used to measure adipogenesis: 1) isolate mature adipocytes could be isolated from fat tissue by collagenase digestion followed by centrifugation. Nuclei could then be extracted from these mature adipocytes and FACS-sorted to count how many adipocytes have BRDU; or 2) fat tissue slices could be obtained and stained for caveolin which marks mature adipocytes. Then one could count how many cells stained for caveolin have BRDU-labeled nuclei. Another approach would be to use Adipochaser mice (Wang et al., 2013) or an adipocyte-specific, tamoxifen-inducible Adiponectin-cre Estrogen Receptor mouse model (Jeffery et al., 2015) which would for inducible pulse-labeling of all pre-existing mature adipocytes.
DATA AND SOFTWARE AVAILABILITY
MATLAB analysis scripts used in this paper are available from Dr. Mary N. Teruel.
Supplementary Material
HIGHLIGHTS.
Circadian adipogenic stimuli are rejected by the adipocyte differentiation system.
This circadian filtering requires fast and slow positive feedback to PPARG.
Loss of circadian glucocorticoid oscillations in mice increases fat mass.
Acknowledgments
This work was supported by National Institutes of Health RO1-DK101743, RO1-DK106241, P50-GM107615 and a Stanford BioX Seed Grant (to M.N.T.), T32-NIH T2HG00044 and 1F31DK112570-01A1 (to M.L.Z.), AHA 16POST2700015 (to D.H.), and DFG Research Fellowship TH2156/1-1 (to S.T.). We thank Sean Collins (UC Davis), James Ferrell, Tobias Meyer, Fredric Kraemer, Connie Phong (Stanford University), and members of the Teruel Lab for discussions and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
Z.B., M.L.Z., S.T., D.H., K.T., and M.N.T. conceived experiments. Z.B., M.L.Z., S.T., D.H., K.T., S.V., and M.N.T. performed experiments and analyzed data. M.L.Z. and M.C. wrote image analysis scripts. Z.B., M.L.Z., and M.N.T wrote the paper with input from all authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Adida A, Spener F. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. Biochim. Biophys. Acta. 2006;1761:172–181. doi: 10.1016/j.bbalip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Ahrends R, Ota A, Kovary KM, Kudo T, Park BO, Teruel MN. Controlling low rates of cell differentiation through noise and ultrahigh feedback. Science. 2014;344:1384–1389. doi: 10.1126/science.1252079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers SD, Nedrow KL, Gillilan RE, Noy N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry. 2007;46:6744–6752. doi: 10.1021/bi700047a. [DOI] [PubMed] [Google Scholar]
- Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamopituitary-adrenal axis activity. Int. J. Endocrinol. 2010;2010:759234. doi: 10.1155/2010/759234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Peckett AJ, D’souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol. 2011;300:C198–209. doi: 10.1152/ajpcell.00045.2010. [DOI] [PubMed] [Google Scholar]
- Carré N, Binart N. Prolactin and adipose tissue. Biochimie. 2014;97:16–21. doi: 10.1016/j.biochi.2013.09.023. [DOI] [PubMed] [Google Scholar]
- Chawla A, Schwarz EJ, Dimaculangan DD, Lazar M. Peroxisome Proliferation-Activated Receptor (PPAR) gamma : Adipose-Predominant Expression And Induction Early In Adipocyte Differentiation. Endocrinology. 1994;135:798–800. doi: 10.1210/endo.135.2.8033830. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Bhatnagar S, Bell ME, Strack AM. Bottomed out: metabolic significance of the circadian trough in glucocorticoid concentrations. Int. J. Obes. 2000;24:S40–S46. doi: 10.1038/sj.ijo.0801276. [DOI] [PubMed] [Google Scholar]
- Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, Kehinde O. An established preadipose cell line and its differentation in culture II. Factors affecting the adipose conversion. Cell. 1975;5:19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- Hodes GE, Brookshire BR, Hill-Smith TE, Teegarden SL, Berton O, Lucki I. Strain differences in the effects of chronic corticosterone exposure in the hippocampus. Neuroscience. 2012;222:269–280. doi: 10.1016/j.neuroscience.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015;11:592–605. doi: 10.1038/nrendo.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat. Cell Biol. 2015;17:376–385. doi: 10.1038/ncb3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra-Peña R, Church CD, Colman L, Berry R, Rodeheffer MS. The Adipose Tissue Microenvironment Regulates Depot-Specific Adipogenesis in Obesity. Cell Metab. 2016;24:142–150. doi: 10.1016/j.cmet.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta. 2014;1842:473–481. doi: 10.1016/j.bbadis.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Lahav G. We are all individuals: Causes and consequences of non-genetic heterogeneity in mammalian cells. Curr. Opin. Genet. Dev. 2011;21:753–758. doi: 10.1016/j.gde.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougald O, Cornelius P, Liu R, Lane M. Insulin regulates transcription of the CCAAT/enhancer binding protein (C/EBP) alpha, beta, and delta genes in fully-differentiated 3T3-L1 adipocytes. J. Biol. Chem. 1995;270:647–654. doi: 10.1074/jbc.270.2.647. [DOI] [PubMed] [Google Scholar]
- Ota A, Kovary KM, Wu OH, Ahrends R, Shen W, Costa MJ, Feldman BJ, Kraemer FB, Teruel MN. Using SRM-MS to quantify nuclear protein abundance differences between adipose tissue depots of insulin-resistant mice. J. Lipid Res. 2015;56:1068–1078. doi: 10.1194/jlr.D056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BO, Ahrends R, Teruel MN. Consecutive Positive Feedback Loops Create a Bistable Switch that Controls Preadipocyte-to-Adipocyte Conversion. Cell Rep. 2012;2:976–990. doi: 10.1016/j.celrep.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebuffe-Scrive M, Walsh UA, McEwen B, Rodin J. Effect of chronic stress and exogenous glucocorticoids on regional fat distribution and metabolism. Physiol. Behav. 1992;52:583–590. doi: 10.1016/0031-9384(92)90351-2. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, Alon U. Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol. 2002;323:785–793. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Siersbæk R, Nielsen R, John S, Sung M-H, Baek S, Loft A, Hager GL, Mandrup S. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J. 2011;30:1459–1472. doi: 10.1038/emboj.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz Ba, Bergmann O, Blomqvist L, Hoffstedt J, Näslund E, Britton T, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- Spangenberg L, Shigunov P, Abud APR, Cofre AR, Stimamiglio MA, Kuligovski C, Zych J, Schittini AV, Costa ADT, Rebelatto CK, et al. Polysome profiling shows extensive posttranscriptional regulation during human adipocyte stem cell differentiation into adipocytes. Stem Cell Res. 2013;11:902–912. doi: 10.1016/j.scr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Tchoukalova YD, Sarr MG, Jensen MD. Measuring committed preadipocytes in human adipose tissue from severely obese patients by using adipocyte fatty acid binding protein. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R1132–40. doi: 10.1152/ajpregu.00337.2004. [DOI] [PubMed] [Google Scholar]
- Thompson NM, Gill DAS, Davies R, Loveridge N, Houston PA, Robinson ICAF, Wells T. Ghrelin and ges-octanoyl ghrelin promote adipogenesis directly in vivo by a mechanism independent of Type 1a growth hormone secretagogue receptor. Endocrinology. 2004;145:234–242. doi: 10.1210/en.2003-0899. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu. Rev. Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Wang L, Walker BL, Iannaccone S, Bhatt D, Kennedy PJ, Tse WT. Bistable switches control memory and plasticity in cellular differentiation. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6638–6643. doi: 10.1073/pnas.0806137106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman ED, Fukushima D, Nogeire C, Roffwarg H, Gallagher TF, Hellman L. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J. Clin. Endocrinol. Metab. 1971;33:14–22. doi: 10.1210/jcem-33-1-14. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Park C, Choi K, Bickel PE. OP9 mouse stromal cells rapidly differentiate into adipocytes: characterization of a useful new model of adipogenesis. J. Lipid Res. 2006;47:450–460. doi: 10.1194/jlr.D500037-JLR200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.