

Abstract
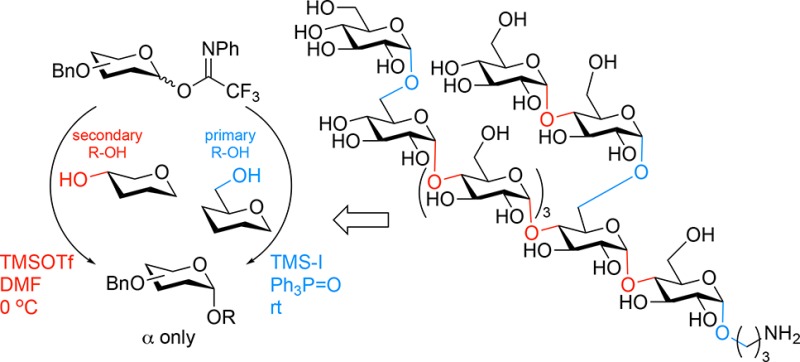
The development of a general glycosylation method that allows for the stereoselective construction of glycosidic linkages is a tremendous challenge. Because of the differences in steric and electronic properties of the building blocks used, the outcome of a glycosylation reaction can vary greatly when switching form one glycosyl donor–acceptor pair to another. We here report a strategy to install cis-glucosidic linkages in a fully stereoselective fashion that is under direct control of the reagents used to activate a single type of donor building block. The activating reagents are tuned to the intrinsic reactivity of the acceptor alcohol to match the reactivity of the glycosylating agent with the reactivity of the incoming nucleophile. A protecting group strategy is introduced that is based on the sole use of benzyl-ether type protecting groups to circumvent changes in reactivity as a result of the protecting groups. For the stereoselective construction of the α-glucosyl linkages to a secondary alcohol, a per-benzylated glusosyl imidate donor is activated with a combination of trimethylsilyltriflate and DMF, while activation of the same imidate donor with trimethylsilyl iodide in the presence of triphenylphosphine oxide allows for the stereoselective cis-glucosylation of primary alcohols. The effectiveness of the strategy is illustrated in the modular synthesis of a Mycobacterium tuberculosis nonasaccharide, composed of an α-(1–4)-oligoglucose backbone bearing different α-glucosyl branches.
Introduction
Despite the tremendous progress that has been made in the synthesis of (complex) oligosaccharides, no general solution exists for the stereoselective construction of challenging glycosidic bonds, such as 1,2-cis and 2-deoxy linkages.1 At the root of this persisting problem is the enormous variation in carbohydrate building blocks and the different mechanistic pathways that can be followed in the union of these.1 Most glycosylation reactions rely on the activation of a glycosyl donor using a (Lewis) acid catalyst to generate a strong electrophile that can either be a covalent species, a close ion pair or a solvent separated ion pair, in which the glycosyl oxocarbenium ion and the counterion are fully dissociated (see Figure 1).2,3 Most often triflate-based activators are used and a multitude of covalently linked anomeric triflates has been described over the last two decades.3 These triflates may engage in a SN2 type substitution reaction, but more often they act as a reservoir for the more reactive glycosyl cation-triflate ion pair, providing reactions with SN1-character. The equilibrium between the covalent species and ion pairs in combination with the reactivity of the incoming nucleophile—the acceptor—determines which pathway(s) will be followed. The reactivity of the donor building block depends on the nature and position of the functional groups on the carbohydrate ring and the different reactivity of donor glycosides has been called upon in reactivity based one-pot chemoselective glycosylation sequences.4 It is also well appreciated—but less well studied—that the reactivity of the acceptor alcohol can differ as a result of the protecting/functional group pattern on the ring and the intrinsic reactivity difference between primary and secondary alcohols often leads to a different stereochemical outcome when glycosylating these acceptors.5 It is a tremendous challenge to design a general glycosylation strategy that accommodates the varying reactivity of different donor–acceptor glycoside combinations and ensures a fully stereoselective glycosylation process.
Figure 1.
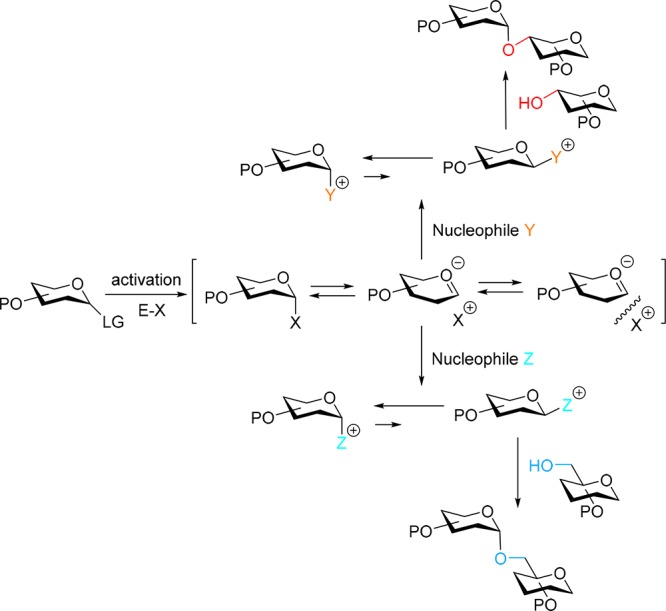
Additive controlled stereoselective glycosylations. The reactivity of the adducts of the different nucleophiles (Y and Z) are tuned to the reactivity of the incoming acceptor alcohol.
An attractive way to modulate the reactivity of a glycosyl donor is through the use of an exogenous nucleophile that can be added to the coupling reaction. These nucleophilic additives or reactivity modulators react with the activated donor to form a new covalent species (see Figure 1).6 Various additives have been probed over the years, including sulfides,7 sulfoxides/sulfinamides,8 phosphine oxides,9 amides and formamides10 and iodide based reagents11 and stereoselective 1,2-cis-glycosylation procedures have been reported based on their use. The most often invoked mechanistic rationale to account for the observed stereoselectivity involves the generation of a stable α-covalent species (often identified and characterized by NMR spectroscopy), that is in equilibrium with its less stable and more reactive β-counterpart (often not detected by NMR), following an in situ anomerisation kinetic scenario as first introduced by Lemieux and co-workers.12 We reasoned that modulation of donor reactivity through external nucleophiles would be very attractive to match the reactivity of acceptor alcohols of different nucleophilicity in order to achieve fully stereoselective glycosylation reactions with both partners. We here report how a single type of donor glycoside can be used for the fully stereoselective glycosylation of both primary and secondary alcohol acceptors. Different additives have been used to accommodate the intrinsic reactivity difference between these two types of alcohols. Key to the success of the strategy is a protecting group strategy that ensures identical reactivity of the parent donor building blocks used, so that the reactivity of the system is under direct control of the activator/additive used. We show the applicability of this approach in the assembly of Mycobacterium tuberculosis (Mtb) derived branched α-glucans. Mtb α-Glucans play an important role in allowing the bacterium to evade the human immune system, but the molecular details behind this process remain obscure.13 To unravel how α-glucans interact with our immune system, well-defined α-glucans fragments will be valuable tools. These structures represent excellent target molecules to test the proposed synthetic strategy, as they only contain 1,2-cis linkages and carry different branches, necessitating flexible building blocks and stereoselective glycosylation methodology for the construction of glycosidic linkages to both primary and secondary alcohol functions.
Results and Discussion
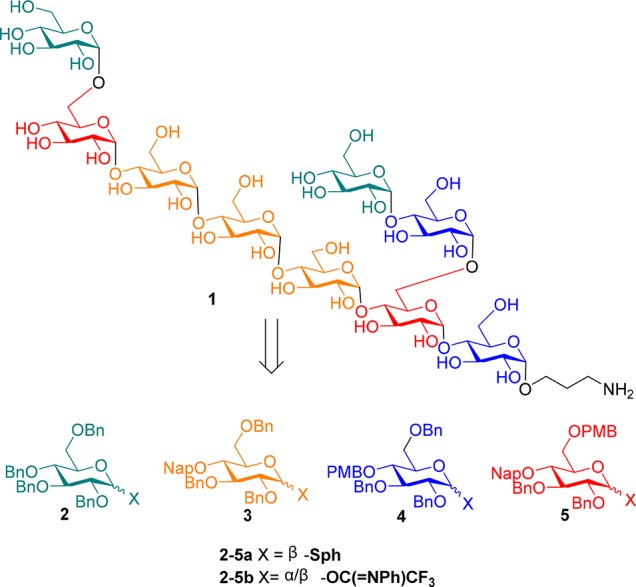
We set out to develop a glycosylation strategy that is under full control of the used reagents and aimed to employ a single donor type, devoid of any stereodirecting protecting groups for the glycosylations of both the primary and secondary alcohol acceptors. We therefore equipped the used donor glycosides and acceptor glycans solely with benzyl type protecting groups. This serves two purposes. First, since all protecting groups on the building blocks are benzyl ethers, the reactivity of the building blocks is as similar as possible. The only factors influencing the relative reactivity of the acceptors are the intrinsic difference between the primary and secondary alcohols and the effect of the growing chain length on the reactivity of the acceptor. Second, global protection of the donor glycoside with benzyl ethers leads to a donor that is as reactive as possible. Previous reports employing nucleophilic additives in glycosylations have shown that this type of glycosylation is generally very slow.6 The reactive intermediates that are generated are relatively stable necessitating long reaction times. The use of acyl type protecting groups would make the system less reactive leading to even longer reaction times. The target α-glucans of this study and the employed building blocks are depicted in Scheme 1. The most complex target, nonasaccharide 1, features a hexa-α-glucan backbone with two different branches. We have selected this target saccharide because its synthesis requires the introduction of all possible structural elements present in naturally occurring α-glucans. To be able to assemble this structure we designed four different building blocks: per-benzylated donor 2, a chain-terminating synthon; donor 3 to build the growing α-1,4-chains; donor 4, to build the branches; and finally, donor 5 to introduce the branches. The triad of benzyl ethers that we aimed to use include benzyl (Bn) ethers for permanent protection, only to removed at the end of the assembly; 2-methylnaphthyl (NAP) ethers that can be selectively removed with respect to the other benzyl ethers under acidic or oxidative conditions and finally the para-methoxybenzyl (PMB) ether that are the most labile of the three benzyl ethers and that can be selectively removed in the presence of the other two using mild acidic conditions, as we recently described.14
Scheme 1. Synthetic Strategy for the Assembly of Mtb α-Glucan 1.
We first directed our attention to the stereoselective construction of the α-1,4-glucosyl linkages. To this end we investigated the condensation of tetra-O-benzyl thioglucoside 2a and tri-O-benzyl-α-O-methyl glucose acceptor 6 using N-iodosuccinimide (NIS) and trimethylsilyl triflate (TMSOTf) activation.15 Following the seminal work of Mong and co-workers10 we explored several amides and formamide additives, including N,N-dimethylformamide (DMF), N-formylpiperidine (NFP), N-formylmorpholine (NFM), N,N-dimethylacetamide (DMA) and tetramethylurea (TMU) as reactivity modulators. We also probed phosphine oxides (triphenylhosphine oxide, Ph3PO) and sulfoxides (benzene sufinyl piperidine, BSP).
As shown in Table 1 (entries 2–14), the stereoselectivity of the reactions with additives are better than the condensation reaction without nucleophilic additive (entry 1), barred one: the reaction using Ph3PO. In line with the findings of the Mong laboratory,10 the formamide additives performed best and the use of a larger excess of these additives generally gave better results in terms of stereoselectivity. We observed that the reactions with TMU and the phosphine oxide proceeded slower than the formamide and amide mediated condensations, resulting in diminished yields. This likely reflects the greater stability of the formed covalent intermediates generated with these additives. As shown in Table 1, DMF performed best as additive and this reagent was further studied and we explored the use of imidate donors as these represent a very powerful class of glycosylating agents.16 Where the in situ transformation of thioglycosides into reactive covalent species is commonly applied in glycosylation chemistry, the use of imidate donors for this purpose has not been widely explored. Gratifyingly, the additive controlled condensation of imidate donor 2b and acceptor 6 proceeded in excellent yield and stereoselectivity to provide the desired disaccharide 9 (Table 1, entry 15). To test whether the acid labile naphthyl ether in donor 3b is compatible with the developed reaction conditions, which employ a stoichiometric amount of TfOH, donor 3b was next coupled with acceptor 6. This glycosylation delivered the protected maltoside 10 in similar yield and with comparable stereoselectivity as the condensation of per-benzyl donor 2b and acceptor 6 (Table 1, entry 16), showing that the Nap ether well tolerates the glycosylation conditions.
Table 1. Glycosylations of Perbenzylated Glucose Donors with Secondary Alcohols.

| entry | donor | acceptor | promoter | additives | equiv | product | yield (%) | α:β |
|---|---|---|---|---|---|---|---|---|
| 1 | 2a | 6 | NIS, TMSOTfa | – | – | 9 | 86 | 2:1c |
| 2 | 2a | 6 | NIS, TMSOTfa | DMF | 6 | 9 | 91 | 37:1c |
| 3 | 2a | 6 | NIS, TMSOTfa | 16 | 9 | 83 | >50:1c | |
| 4 | 2a | 6 | NIS, TMSOTfa | NFP | 6 | 9 | 72 | 23:1c |
| 5 | 2a | 6 | NIS, TMSOTfa | 16 | 9 | 69 | >30:1c | |
| 6 | 2a | 6 | NIS, TMSOTfa | NFM | 6 | 9 | 91.5 | 15:1c |
| 7 | 2a | 6 | NIS, TMSOTfa | 16 | 9 | 94 | 19:1c | |
| 8 | 2a | 6 | NIS, TMSOTfa | DMA | 6 | 9 | 83 | 9:1c |
| 9 | 2a | 6 | NIS, TMSOTfa | 16 | 9 | 90 | 19:1c | |
| 10 | 2a | 6 | NIS, TMSOTfa | TMU | 6 | 9 | 32 | 4:1c |
| 11 | 2a | 6 | NIS, TMSOTfa | 16 | 9 | 49 | 3.5:1c | |
| 12 | 2a | 6 | NIS, TMSOTfa | BSP | 3 | 9 | 61 | 3:1c |
| 13 | 2a | 6 | NIS, TMSOTfa | 6 | 9 | 39 | 3:1c | |
| 14 | 2a | 6 | NIS, TMSOTfa | Ph3P=O | 6 | 9 | 60 | 2:1c |
| 15 | 2b | 6 | TfOHb | DMF | 16 | 9 | 94 | >20:1d |
| 16 | 3b | 6 | TfOHb | DMF | 16 | 10 | 91 | >20:1d |
| 17 | 2b | 7 | TfOHb | DMF | 16 | 11 | 85 | >20:1d |
| 18 | 2b | 8 | TfOHb | DMF | 16 | 12 | 90 | >20:1d |
DCM, 0 °C, 24 h.
DCM, −78 to 0 °C, 24 h.
The α:β ratio was determined by chiral HPLC analysis.
The α:β ratio was determined by 1H NMR.
We also briefly explored the scope of the established conditions with two other secondary carbohydrate alcohols. Acceptors 7 and 8, could be glucosylated at the C-3 and C-2 OH, respectively, in good yield and with excellent stereoselectivity (Table 1, entries 17 and 18).
To probe the robustness of the established methodology, we next set out to generate a longer α-glucan, as depicted in Scheme 2. To this end, the Nap-protecting group was removed from maltoside 10 using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to furnish acceptor 13. Compound 13 was glycosylated with donor 3b using the DMF-conditions to give the desired trisaccharide 14 in 81% yield. Repetition of the deprotection and glycosylation reactions then provided the tetrasaccharide 16, pentasaccharide 18 and hexasaccharide 20. All through this reaction sequence the yields and stereoselectivity of the condensations did not erode showcasing the reliability of the methodology.
Scheme 2. Synthesis of α-(1,4)-Hexasaccharide 21.

(a) DMF, TfOH, DCM, −78 to 0 °C, 10: 82%; 14: 81%; 16: 82%; 18: 80%; 20: 79%. (b) DDQ, DCM/H2O, 13: 78%; 15: 78%; 17: 84%; 19: 81%. (c) Pd(OH)2/C, H2, THF:H2O:t-BuOH, 3.5 atm, 80%.
We then turned our attention to the condensations of the primary acceptor. For this we used model acceptor 22 in combination with thioglycoside 2a and the panel of additives described above. The higher reactivity of the primary alcohol 22 with respect to its secondary alcohol counterpart 6 leads to significant erosion of the stereoselectivity, when identical condensation conditions are used (Table 2, entries 1–5). We were surprised to see that the condensation using the phosphine oxide actually led to the formation of more β- than α-linked product. Cognizant of the work of Mukaiyama and co-workers on the use of phosphine oxides in highly stereoselective condensation reactions of perbenzylated glucosyl pyranosyl iodides,9 we switched to the use of imidate donor 2b and transformed it in situ into the anomeric iodide using trimethylsilyl iodide (TMSI).17 Under these conditions, the disaccharide 23 was formed with moderate α-selectivity and in rather poor yield (Table 2, entry 6). The addition of 1.2 equiv of diphenylmethyl phosphine oxide, as advocated by Mukaiyama and co-workers, led to a significant improvement of the stereoselectivity (α/β = 6:1, Table 2, entry 7). As we planned to use this activation system in combination with a donor glucoside bearing a relative acid labile PMB group we explored these conditions for the coupling of donor 4b and acceptor 22. We increased the amount of phosphine oxide to ensure that sufficient Lewis-basic reagent was present to protect the labile PMB. As displayed in Table 2, entry 8, the desired disaccharide 24 was obtained in good yield with improved stereoselectivity and the PMB group proved to be completely stable to the conditions used. Increasing the amount of the phosphine oxide additive to 6 equiv led to a further increase in stereoselectivity (Table 2, entry 9). More phosphine oxide did not further improve the stereoselectivity (Table 2, entries 10 and 11). Triphenyl phosphine oxide performed equally well as an additive and the use of 6 equiv of this reagent proved optimal for the condensation of donor 4b and acceptor 22 (Table 2, entry 12).
Table 2. Glycosylations of Primary Alcohol 22.
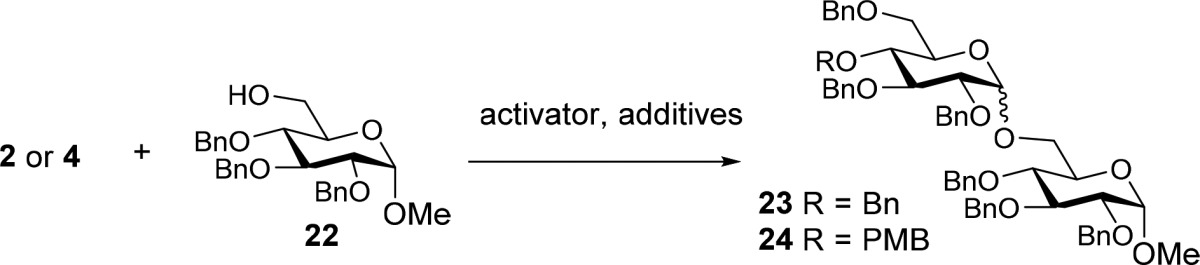
| entry | donor | promoter | additive | equiv | T (°C) | product | time (h) | yield (%) | α:βa |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2a | NIS, TMSOTf | DMF | 16 | 0 | 23 | 12 | 90 | 2.7:1 |
| 2 | 2a | NIS, TMSOTf | NFM | 16 | 0 | 23 | 24 | 83 | 2.1:1 |
| 3 | 2a | NIS, TMSOTf | DMA | 16 | 0 | 23 | 24 | 69 | 1:1.3 |
| 4 | 2a | NIS, TMSOTf | TMU | 6 | 0 | 23 | 24 | 82 | 1:1.1 |
| 5 | 2a | NIS, TMSOTf | Ph3P=O | 6 | 0 | 23 | 24 | 70 | 1:3 |
| 6 | 2b | TMSI | – | – | rt | 23 | 24 | 41 | 2:1 |
| 7 | 2b | TMSI | Ph2(Me)P=O | 1.2 | rt | 23 | 24 | 46 | 6:1 |
| 8 | 4b | TMSI | Ph2(Me)P=O | 3 | rt | 24 | 24 | 82 | 16:1 |
| 9 | 4b | TMSI | Ph2(Me)P=O | 6 | rt | 24 | 24 | 76 | 20:1 |
| 10 | 4b | TMSI | Ph2(Me)P=O | 10 | rt | 24 | 24 | 73 | 20:1 |
| 11 | 4b | TMSI | Ph2(Me)P=O | 16 | rt | 24 | 24 | 70 | 20:1 |
| 12 | 4b | TMSI | Ph3P=O | 6 | rt | 24 | 24 | 78 | >20:1 |
| 13 | 2b | TMSOTf | Ph2(Me)P=O | 6 | rt | 23 | 24 | 84 | 3:2 |
The α:β ratio was determined by 1H NMR.
To explore the necessity of the intermediate iodide, we explored the activation of imidate donor 2b with TMSOTf instead of TMSI, in the presence of 6 equiv phosphineoxide (Table 2, entry 13). This led to formation of the diglucoside 23 in good yield, but very poor selectivity, indicating that the anomeric iodide plays an important role in the coupling mechanism. To shed further light on the reactive intermediates formed with the TMSI-phosphine oxide reagent combination we studied the activation of donor 2b by NMR spectroscopy. When donor 2b was activated with TMSI in CDCl3 in the absence of a phosphine oxide additive, a mixture of two products was formed. The products were tentatively assigned as α-iodide 26 (Scheme 3, H-1: δ = 6.82 ppm; C-1: δ = 81.04 ppm; see SI for NMR spectra) and its β-counterpart 25 (H-1: δ = 5.68 ppm; C-1: δ = 61.42 ppm).18 In time (±45 min), the β-iodide 25 isomerized into its more stable α-congener 26. Alternatively, treatment of a mixture of donor 2b and Ph2(Me)PO in CDCl3 with TMSI, showed a clean conversion of the imidate into the anomeric α-iodide 26. The β-iodide 25 was not observed, nor could we detect the presence of any anomeric phosphonium species. Given the importance of the phosphine oxide for the stereoselectivity of the reaction (compare Table 2, entries 6 and 9) we suggest that the anomeric α-iodide serves a reservoir for the more reactive β-phosphonium iodide, which is the actual glycosylating species (see Scheme 3). The phosphine oxide also catalyzes the transformation of the β-iodide 25 into α-iodide 26.
Scheme 3. Proposed Mechanism for the Activation and Glyosylation of Imidate Donors with TMSI and Phosphineoxides.

Having identified the required reagents and conditions to stereoselectively construct cis-glucosidic linkages with both primary and secondary acceptor glucosyl alcohols we turned our attention to assemble nonaglucoside 1 as outlined in Scheme 4.19 First 3-azidopropanol was condensed with donor 4b using the Ph3PO mediated glycosylation conditions to deliver monosaccharide 27 in 91% yield and 11:1 α/β selectivity (Scheme 4). The spacer-equipped 27 was then treated with a catalytic amount of HCl in a mixture of hexafluoro-isopropanol (HFIP) and dichloromethane (DCM) to remove the PMB protecting group and liberate the C-4-alcohol. At this stage pure α-linked 28 was obtained after silica gel chromatography. Acceptor 28 was then reacted with “branching” donor 5b using the DMF-mediated glycosylation conditions to provide disaccharide 30 in 81% yield and excellent α-selectivity.20,21 The PMB ether in disaccharide 30, was chemoselectively removed using the aforementioned HCl/HFIP conditions. Of note, the Nap-ether at the C-4′ position was completely stable under these acidic conditions. Disaccharide 31 was then elongated at its C-6′-OH with C-4-PMB-donor 4b using the TMSI-Ph3PO reagent combination to stereoselectively provide the trisaccharide 32. Liberation of the C-4″-OH, again using HCl/HFIP, then set the stage for the elongation of the branching arm with perbenzyl donor 2b under the aegis of TfOH and DMF. Having completed the first arm, we continued to grow the α-(1,4)-backbone. To this end the Nap ether was oxidatively removed and the resulting secondary alcohol coupled to C-4-Nap donor 3b with TfOH-DMF to give pentasaccharide 36. Reiteration of this deprotection-coupling cycle let to hexasaccharide 38 and heptasaccharide 40 in a completely stereoselective fashion. To introduce the second α-(1,6)-arm the C-4-OH was unmasked and the heptasaccharide acceptor 41 was coupled to branching glucoside 5b to deliver octamer 42. Liberation of the primary alcohol was then followed by the final TMSI-Ph3PO-condensation leading to the fully protected nonasaccharide 44. Global deprotection of the nonasaccharide was accomplished in a single hydrogenation event to complete the total synthesis of branched α-glucan 1.
Scheme 4. Stereoselective Synthesis of Branched Alpha Nonasaccharide 1.

(a) TMSI, Ph3P=O, DCM, rt, 27: 91%, α:β = 11:1; 32: 68%; 44: 67%. (b) 0.2 M HCl/HFIP, DCM/HFIP, 28: 85%; 31: 85%; 33: 83%; 43: 88%. (c) TMSOTf, DMF, 0 °C, 30: 81%. (d) TfOH, DMF, 0 °C, 34: 81%; 36: 91%; 38: 93%; 40: 91%; 42: 80%. (e) DDQ, DCM/H2O, 35: 84%; 37: 75%; 39: 66%; 41: 70%. (f) Pd(OH)2/C, H2 (3.5 atm), THF/H2O/t-BuOH, 61%.
Conclusion
In conclusion, we have described a strategy to assemble α-glucans in a fully stereoselective manner, using a single type of donor, relying solely on the activating agents and additives to control the stereoselectivity of the glycosylation reactions. The reactivity of the donor building blocks was matched to the intrinsically different reactivity of primary and secondary alcohols through the use of different activator/additive combinations (TfOH or TMSOTf/DMF and TMSI/Ph3PO). To keep the reactivity of all donor synthons on par, we introduced the triad of benzyl, 2-methylnaphthyl and para-methoxybenzyl ethers, as a set of semiorthogonal protecting groups that can be used to differentiate the hydroxyl groups on the building blocks that need permanent protection, that have to be extended to form the glycan backbone or removed to introduce branching.22 The applicability of the strategy has been illustrated by the fully stereoselective assembly of an Mtb nonasaccharide α-glycan, bearing two different branches. Matching the reactivity of both donor and acceptor through the use of external nucleophiles as reactivity moderators presents an important step toward the generation of a general glycosylation reaction. Increasing our insight into the reactivity of donor and acceptor building blocks in combination with the development of tailor-made additives, covering a broad range of reactivity, will likely allow the fine-tuning of many glycosylation systems in the future. Besides the stereoselective construction of cis-glycosidic linkages in a reagent controlled manner, it should be feasible to conceive nucleophilic additives that allow for the stereoselective formation of trans-glycosidic bonds using the same donor/acceptor pair, further broadening the scope of the methodology.
Experimental Section
Standard Procedure for Glycosylation of Secondary Alcohols with Thiodonors (2a–5a) (Procedure A)
The donor (1.0 equiv, coevaporated with toluene) was dissolved in dry DCM under nitrogen and stirred over fresh flame-dried molecular sieves 3Å, after which DMF (16 equiv) was added to the solution. The solution was cooled to 0 °C, after which NIS (1.0 equiv) and TMSOTf (1.0 equiv) were added. After 1 h, the preactivation was complete as indicated by TLC analysis. Then acceptor (0.7 equiv) was added to the solution. The reaction was stirred at 0 °C until TLC analysis showed complete conversion of the acceptor. The reaction mixture was diluted and the reaction was quenched with saturated Na2S2O3. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The products were purified by size exclusion and silica gel column chromatography.
Standard Procedure for Glycosylation of Secondary Alcohols with Imidate Donors (2b–5b) (Procedure B)
The donor (1.0 equiv, coevaporated with toluene) was dissolved in dry DCM under nitrogen and stirred over fresh flame-dried molecular sieves 3Å, after which DMF (16 equiv) was added to the solution. The solution was cooled to −78 °C, after which TfOH (1.0 equiv) was added. After 30 min, the preactivation was complete as indicated by TLC analysis. Acceptor (0.7 equiv) was added to the solution and the mixture was placed in an ice bath. The reaction was stirred at 0 °C until TLC analysis showed complete conversion of the acceptor. The reaction was quenched with Et3N, filtered and concentrated in vacuo. The products were purified by size exclusion and silica gel column chromatography.
Standard Procedure for the Glycosylation of Primary Alcohols (Procedure C)
A mixture of donor (1.0 equiv), acceptor (0.7 equiv) (donors and acceptors coevaporated with toluene three times), Ph3P=O (6 equiv) in dry DCM were stirred over fresh flame-dried molecular sieves 3Å under nitrogen. Then TMSI (1.0 equiv) was added slowly in the mixture. The reaction was stirred at room temperature until TLC analysis indicated the reaction to be complete. The solution was diluted and the reaction quenched with saturated Na2S2O3. The organic phase was washed with water and brine, dried with anhydrous MgSO4, filtered and concentrated in vacuo. The products were purified by size exclusion and silica gel column chromatography.
Acknowledgments
This work was supported by the Chinese Scolarship Council (CSC grant to L.W.) and the European Research Council (ERC-CoG-726072-“GLYCONTROL”, to J.D.C.C.)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b00669.
Full experimental procedures and characterization of prepared compounds, NMR spectra of all new compounds, NMR analysis of the TMSI-activation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Nigudkar S. S.; Demchenko A. V. Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci. 2015, 6 (5), 2687–2704. 10.1039/C5SC00280J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Demchenko A. V.Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH Verlag GmbH & Co. KGaA, 2008. [Google Scholar]; c Bennett C. S.Selective Glycosylations: Synthetic Methods and Catalysts; Wiley VCH Verlag GmbH, 2017. [Google Scholar]; d Christina A. E.; van der Marel G. A.; Codée J. D. C.. Recent Developments in the Construction of cis-Glycosidic Linkages. In Modern Synthetic Methods in Carbohydrate Chemistry; Werz D. B., Vidal S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, 2013; pp 97–124. [Google Scholar]; e Huang M.; Garrett G. E.; Birlirakis N.; Bohé L.; Pratt D. A.; Crich D. Dissecting the mechanisms of a class of chemical glycosylation using primary 13C kinetic isotope effects. Nat. Chem. 2012, 4 (8), 663–667. 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bohé L.; Crich D. A propos of glycosyl cations and the mechanism of chemical glycosylation. C. R. Chim. 2011, 14, 3–16. 10.1016/j.crci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bohé L.; Crich D. A propos of glycosyl cations and the mechanism of chemical glycosylation; the current state of the art. Carbohydr. Res. 2015, 403, 48–59. 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Walvoort M. T. C.; Dinkelaar J.; van den Bos L. J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. The impact of oxacarbenium ion conformers on the stereochemical outcome of glycosylations. Carbohydr. Res. 2010, 345, 1252–1263. 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- a Crich D. Mechanism of a Chemical Glycosylation Reaction. Acc. Chem. Res. 2010, 43, 1144–1153. 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]; b Crich D.; Cai Chemistry of 4,6-O-Benzylidene-d-glycopyranosyl Triflates: Contrasting Behavior between the Gluco and Manno Series. J. Org. Chem. 1999, 64, 4926–4930. 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]; c Frihed T. G.; Bols M.; Pedersen C. M. Mechanisms of Glycosylation Reactions Studied by Low-Temperature Nuclear Magnetic Resonance. Chem. Rev. 2015, 115, 4963–5013. 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]; d Dhakal B.; Bohé L.; Crich D. Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J. Org. Chem. 2017, 82, 9263–9269. 10.1021/acs.joc.7b01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Andrews C. W.; Rodebaugh R.; Fraser-Reid B. A Solvation-Assisted Model for Estimating Anomeric Reactivity. Predicted versus Observed Trends in Hydrolysis of n-Pentenyl Glycosides. J. Org. Chem. 1996, 61, 5280–5289. 10.1021/jo9601223. [DOI] [Google Scholar]; b Douglas N. L.; Ley S. V.; Lücking U.; Warriner S. L. Tuning glycoside reactivity: New tool for efficient oligosaccharide synthesis. J. Chem. Soc., Perkin Trans. 1 1998, 51–66. 10.1039/a705275h. [DOI] [Google Scholar]; c Zhang Z.; Ollmann I. R.; Ye X.-S.; Wischnat R.; Baasov T.; Wong C.-H. Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc. 1999, 121, 734–753. 10.1021/ja982232s. [DOI] [Google Scholar]; d Fraser-Reid B.; López J. C.. Armed–Disarmed Effects in Carbohydrate Chemistry: History, Synthetic and Mechanistic Studies. In Reactivity Tuning in Oligosaccharide Assembly; Fraser-Reid B., Cristóbal López J., Eds.; Springer: Berlin Heidelberg, 2011; pp 1–29. [DOI] [PubMed] [Google Scholar]; e Codée J. D. C.; Litjens R. E. J. N.; Van den Bos L. J.; Overkleeft H. S.; Van der Marel Thioglycosides in sequential glycosylation strategies. Chem. Soc. Rev. 2005, 34, 769–782. 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]
- a Van der Vorm S.; Hansen T.; Overkleeft H. S.; Van der Marel G. A.; Codee J. D. C. The influence of acceptor nucleophilicity on the glycosylation reaction mechanism. Chem. Sci. 2017, 8, 1867–1875. 10.1039/C6SC04638J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hagen B.; Ali S.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Mapping the Reactivity and Selectivity of 2-Azidofucosyl Donors for the Assembly of N-Acetylfucosamine-Containing Bacterial Oligosaccharides. J. Org. Chem. 2017, 82, 848–868. 10.1021/acs.joc.6b02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang L.; Qin Q.; Ye X.-S. Preactivation: An Alternative Strategy in Stereoselective Glycosylation and Oligosaccharide Synthesis. Asian J. Org. Chem. 2013, 2, 30–49. 10.1002/ajoc.201200136. [DOI] [Google Scholar]; b Mulani S. K.; Hung W.-C.; Ingle A. B.; Shiau K.-S.; Mong K.-K. T. Modulating glycosylation with exogenous nucleophiles: an overview. Org. Biomol. Chem. 2014, 12, 1184–1197. 10.1039/c3ob42129e. [DOI] [PubMed] [Google Scholar]
- a Park J.; Kawatkar S.; Kim J.-H.; Boons G.-J. Stereoselective Glycosylations of 2-Azido-2-deoxy-glucosides Using Intermediate Sulfonium Ions. Org. Lett. 2007, 9, 1959–1962. 10.1021/ol070513b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nokami T.; Shibuya A.; Manabe S.; Iyo Y.; Yoshida J. α- and β-Glycosyl Sulfonium Ions: Generation and Reactivity. Chem. - Eur. J. 2009, 15, 2252–2255. 10.1002/chem.200802293. [DOI] [PubMed] [Google Scholar]
- a Garcia B. A.; Gin D. Y. Dehydrative Glycosylation with Activated Diphenyl Sulfonium Reagents. Scope, Mode of C(1)-Hemiacetal Activation, and Detection of Reactive Glycosyl Intermediates. J. Am. Chem. Soc. 2000, 122, 4269–4279. 10.1021/ja993595a. [DOI] [Google Scholar]; b Crich D.; Li W. Efficient Glycosidation of a Phenyl Thiosialoside Donor with Diphenyl Sulfoxide and Triflic Anhydride in Dichloromethane. Org. Lett. 2006, 8, 959–962. 10.1021/ol060030s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kobayashi Y.; Mukaiyama T. Highly α-Selective Glycosylation with Glycosyl Acetate via Glycosyl Phosphonium Iodide. Chem. Lett. 2004, 33, 874–875. 10.1246/cl.2004.874. [DOI] [Google Scholar]; b Mukaiyama T.; Kobayashi Y. Highly α-Selective Synthesis of Disaccharide Using Glycosyl Bromide by the Promotion of Phosphine Oxide. Chem. Lett. 2004, 33, 10–11. 10.1246/cl.2004.10. [DOI] [Google Scholar]
- a Koto S.; Morishima N.; Zen S. A Stereoselective α-Glucosylation by Use of a Mixture of 4-Nitrobenzenesulfonyl Chloride, Silver Trifluoromethane Sulfonate, N,N-dimethylacetamide and Trietheylamine. Carbohydr. Res. 1984, 130, 73–83. 10.1016/0008-6215(84)85271-4. [DOI] [Google Scholar]; b Bock K.; Guzman J. F. B.; Refn S. Synthesis and Properties of 1,1,3,3-Tetramethyl-2-(2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl)uranium Triflate. Carbohydr. Res. 1992, 232, 353–357. 10.1016/0008-6215(92)80067-B. [DOI] [Google Scholar]; c Lu S.-R.; Lai Y.-H.; Chen J.-H.; Liu C.-Y.; Mong K.-K. T. Dimethylformamide: An Unusual Glycosylation Modulator. Angew. Chem., Int. Ed. 2011, 50, 7315–7320. 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]; d Ingle A. B.; Chao C.-S.; Hung W.-C.; Mong K.-K. T. Tuning Reactivity of Glycosyl Imidinium Intermediate for 2-Azido-2-deoxyglycosyl Donors in α-Glycosidic Bond Formation. Org. Lett. 2013, 15, 5290–5293. 10.1021/ol402519c. [DOI] [PubMed] [Google Scholar]
- a Lam S. N.; Gervay-Hague J. Solution-Phase Hexasaccharide Synthesis Using Glucosyl Iodides. Org. Lett. 2002, 4, 2039–2042. 10.1021/ol025887d. [DOI] [PubMed] [Google Scholar]; b Lam S. N.; Gervay-Hague J. Solution- and solid-phase oligosaccharide synthesis using glucosyl iodides: a comparative study. Carbohydr. Res. 2002, 337, 1953–1965. 10.1016/S0008-6215(02)00227-6. [DOI] [PubMed] [Google Scholar]; c Chu A. A.; Nguyen S. H.; Sisel J. A.; Minciunescu A.; Bennett C. S. Selective Synthesis of 1,2-cis-α-Glycosides without Directing Groups. Application to Iterative Oligosaccharide Synthesis. Org. Lett. 2013, 15, 2566–2569. 10.1021/ol401095k. [DOI] [PubMed] [Google Scholar]
- a Lemieux R. U.; Hendriks K. B.; Stick R. V.; James K. Halide ion catalyzed glycosidation reactions. Syntheses of.alpha.-linked disaccharides. J. Am. Chem. Soc. 1975, 97, 4056–4062. 10.1021/ja00847a032. [DOI] [Google Scholar]; b Lemieux R. U.; Driguez H. Chemical synthesis of 2-acetamido-2-deoxy-4-O-(α-l-fucopyranosyl)-3-O-(β-d-galactopyranosyl)-d-glucose. Lewis a blood-group antigenic determinant. J. Am. Chem. Soc. 1975, 97, 4063–4068. 10.1021/ja00847a033. [DOI] [PubMed] [Google Scholar]
- a Geurtsen J.; Chedammi S.; Mesters J.; Cot M.; Driessen N. N.; Sambou T.; Kakutani R.; Ummels R.; Maaskant J.; Takata H.; Baba O.; Terashima T.; Bovin N.; Vandenbroucke-Grauls C. M. J. E.; Nigou J.; Puzo G.; Lemassu A.; Daffe M.; Appelmelk B. J. Identification of Mycobacterial α-Glucan As a Novel Ligand for DC-SIGN: Involvement of Mycobacterial Capsular Polysaccharides in Host Immune Modulation. J. Immunol. 2009, 183, 5221–5231. 10.4049/jimmunol.0900768. [DOI] [PubMed] [Google Scholar]; b Sambou T.; Dinadayala P.; Stadthagen G.; Barilone N.; Bordat Y.; Constant P.; Levillain F.; Neyrolles O.; Gicquel B.; Lemassu A.; Daffe M.; Jackson M. Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol. Microbiol. 2008, 70, 762–774. 10.1111/j.1365-2958.2008.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gagliardi M. C.; Lemassu A.; Teloni R.; Mariotti S.; Sargentini V.; Pardini M.; Daffe M.; Nisini R. Cell wall-associated alpha-glucan is instrumental for Mycobacterium tuberculosisto block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell. Microbiol. 2007, 9, 2081–2092. 10.1111/j.1462-5822.2007.00940.x. [DOI] [PubMed] [Google Scholar]
- Volbeda A. G.; Kistemaker H. A. V.; Overkleeft H. S.; Van der Marel G. A.; Filippov D. V.; Codée J. D. C. Chemoselective Cleavage of p-Methoxybenzyl and 2-Naphthylmethyl Ethers Using a Catalytic Amount of HCl in Hexafluoro-2-propanol. J. Org. Chem. 2015, 80, 8796–8806. 10.1021/acs.joc.5b01030. [DOI] [PubMed] [Google Scholar]
- We also explored a preactivation protocol using diphenylsulfoxide/triflic anhydride to activate the thioglycoside donor followed by addition of the additive and subsequent addition of the acceptor. This generally gave similar results in terms of stereoselectivity, but diminished yields.
- a Jung K.-H.; Müller M.; Schmidt R. R. Intramolecular O-Glycoside Bond Formation. Chem. Rev. 2000, 100, 4423–4442. 10.1021/cr990307k. [DOI] [PubMed] [Google Scholar]; b Yu B.; Tao H. Glycosyl trifluoroacetimidates. Part 1: Preparation and application as new glycosyl donors. Tetrahedron Lett. 2001, 42, 2405–2407. 10.1016/S0040-4039(01)00157-5. [DOI] [Google Scholar]
- During the course of this research Mong and co-workers also showed that tetrabutylammonium iodide can be used to couple primary alcohols in a stereoselective fashion:Hu J.-H.; Feng A.-F. W.; Chang B.-Y.; Lin C.-H.; Mong K.-K. T. A flexible 1,2-cis α-glycosylation strategy based on in situ adduct transformation. Org. Biomol. Chem. 2017, 15, 5345–5356. 10.1039/C7OB00839B. [DOI] [PubMed] [Google Scholar]
- a Hsieh H.-W.; Davis R. A.; Hoch J. A.; Gervay-Hague J. Two-Step Functionalization of Oligosaccharides Using Glycosyl Iodide and Trimethylene Oxide and Its Applications to Multivalent Glycoconjugates. Chem. - Eur. J. 2014, 20, 6444–6454. 10.1002/chem.201400024. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gervay-Hague J. Taming the Reactivity of Glycosyl Iodides To Achieve Stereoselective Glycosidation. Acc. Chem. Res. 2016, 49, 35–47. 10.1021/acs.accounts.5b00357. [DOI] [PubMed] [Google Scholar]
- For a selection of previous syntheses of α-glucans, see:; a Yasomanee J. P.; Demchenko A. V. Hydrogen Bond Mediated Aglycone Delivery: Synthesis of Linear and Branched α-Glucans. Angew. Chem., Int. Ed. 2014, 53, 10453–10456. 10.1002/anie.201405084. [DOI] [PubMed] [Google Scholar]; b Fang T.; Mo K.-F.; Boons G.-J. Stereoselective Assembly of Complex Oligosaccharides Using Anomeric Sulfonium Ions as Glycosyl Donors. J. Am. Chem. Soc. 2012, 134, 7545–7552. 10.1021/ja3018187. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hahm H. S.; Hurevich M.; Seeberger P. H. Automated assembly of oligosaccharides containing multiple cis-glycosidic linkages. Nat. Commun. 2016, 7, 12482. 10.1038/ncomms12482. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Komarova B. S.; Orekhova M. V.; Tsvetkov Y. E.; Beau R.; Aimanianda V.; Latgé J.-P.; Nifantiev N. Synthesis of a Pentasaccharide and Neoglycoconjugates Related to Fungal α-(1–3)-Glucan and Their Use in the Generation of Antibodies to Trace Aspergillus fumigatus Cell Wall. Chem. - Eur. J. 2015, 21, 1029–1035. 10.1002/chem.201404770. [DOI] [PubMed] [Google Scholar]; e For a review on approaches employing long range participation, see:Komarova B. S.; Tsvetkov Y. E.; Nifantiev N. E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]; f Also see ref (1a)
- 2,3-Di-O-benzyl-4-O-naphthylmethyl-1,6-anhydroglucose (29) was obtained as a side product in this reaction. Likely this product is formed by attack of the C-6-O-PMB ether on the activated anomeric center and subsequent loss of the PMB cation. Loss of PMB groups from the product was not observed.
- Purification of the oligosaccharides generated in the glycosylation reactions was accomplished using only size exclusion chromatography.
- For the development of halo-benzyl ethers as a set of orthogonal protecting groups, see:Plante O. J.; Buchwald S. L.; Seeberger P. H. Halobenzyl Ethers as Protecting Groupls for Organic Synthesis. J. Am. Chem. Soc. 2000, 122, 7148–7149. 10.1021/ja0008665. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.