

Abstract
A β-glucosidase (BglA, EC 3.2.1.21) gene from the polycentric anaerobic fungus Orpinomyces PC-2 was cloned and sequenced. The enzyme containing 657 amino acid residues was homologous to certain animal, plant, and bacterial β-glucosidases but lacked significant similarity to those from aerobic fungi. Neither cellulose- nor protein-binding domains were found in BglA. When expressed in Saccharomyces cerevisiae, the enzyme was secreted in two forms with masses of about 110 kDa and also found in two forms associated with the yeast cells. Km and Vmax values of the secreted BglA were 0.762 mM and 8.20 μmol/(min·mg), respectively, with p-nitrophenyl-β-D-glucopyranoside (pNPG) as the substrate and 0.310 mM and 6.45 μmol/(min·mg), respectively, for the hydrolysis of cellobiose. Glucose competitively inhibited the hydrolysis of pNPG with a Ki of 3.6 mM. β-Glucosidase significantly enhanced the conversion of cellulosic materials into glucose by Trichoderma reesei cellulase preparations, demonstrating its potential for use in biofuel and feedstock chemical production.
Index Entries: Cellulose, cellulase, β-glucosidase, Orpinomyces, cellobiase
Introduction
Cellulosic biomass photosynthesized by solar energy from CO2 and H2O is one of the most important renewable energy resources on Earth. Its effective utilization through biologic processes might be an important way to overcome the shortage of foods, feeds, and fuels, which will inevitably be seen in the near future as a consequence of the explosive increase in the human population (1). Degradation of cellulose is accomplished through the synergistic action of endoglucanase (EC 3.2.1.4), exoglucanase or cello-biohydrolase (CBH) (EC 3.2.1.91), and β-glucosidase (EC 3.2.1.21) (2). β-Glucosidase is common among animals, plants, fungi, and bacteria. The enzyme plays an important role in the process of saccharification, catalyzing hydrolysis of β-D-glucosidic linkages of cellobiose and cellooligosac-charides. It alleviates the inhibitory effect of the sugars on exo- and endoglucanases (2,3).
Anaerobic fungi have been isolated from the alimentary tracts of herbivores and other environments (4). These fungi are active in the degradation of plant cell wall polysaccharides and represent a potential source of enzymes against cellulose and hemicelluloses. Among the isolated fungi, Orpinomyces PC-2, a polycentric fungus, produces high levels of cell wall–degrading enzymes (4). Genes coding for several cellulases (5–8), a xylanase (5), a 1,3–1,4-β-D-glucanase (9), and an acetylxylan esterase (10) have been cloned and sequenced. An 85-kDa extracellular β-glucosidase produced by Orpinomyces PC-2 has been purified and characterized (3), but a gene coding for such an enzyme of the fungus has not been described.
In this article, we report the cloning in Escherichia coli and sequencing of an Orpinomyces β-glucosidase cDNA. The enzyme was overexpressed in and secreted from the yeast Saccharomyces cerevisiae. Physiochemical properties of the secreted enzyme were determined after it was purified. Its applicability for cellulose saccharification was also assessed.
Materials and Methods
Strains, Plasmids, and Genes
E. coli TOP10, S. cerevisiae INSC1 (MAT α his 3-D 1 El 2 trp1-289 ura3-52), and the plasmid pYES2 were purchased from Invitrogen (San Diego, CA). pYES2 possesses ampicillin and tetracycline resistance genes for selection in E. coli, a URA3 gene for high-copy-number maintenance and selection in S. cerevisiae INSC1, and a GAL 1 promoter sequence. The bglA cDNA of Orpinomyces sp. PC-2 was cloned and identified by screening a cDNA library (11) as described next.
Screening of Orpinomyces cDNA Library Using Antibodies
The production of antibodies against the different regions of Orpinomyces xylanase A has been described previously (5). Immuno-screening was done following the procedure of a Pico Blue™ Immuno-screening kit (Stratagene). Pure positive plaques were obtained after a secondary screening. λ phages were converted into pBluescript SK– by in vivo excision, and the pBluescript DNA was purified from cultures grown overnight in Luria-Bertani (LB) medium containing 50 μg/mL of ampicillin using a plasmid purification system purchased from Qiagen (Valencia, CA). DNA sequences were determined by automated poly-merase chain reaction (PCR) sequencing (5).
DNA Hybridization Screening
A 400-bp DNA fragment of the partial bglA gene sequence obtained by antibody screening was amplified by PCR and labeled with digoxigenin. The labeled fragment was utilized as a hybridization probe and using a Genius kit, the same cDNA library was screened according to the manufacturer’s instructions (Roche, Indianapolis, IN). Positive plaques were converted to pBluescripts, and their inserted DNA sequences were determined as previously described (5).
Construction of Plasmid Cassette
Plasmid pYES2 was digested with SacI and XbaI overnight. The digested plasmid was purified using the Geneclean II kit (Bio 101, La Jolla, CA). On the basis of the nucleotide sequence of the cloned gene, forward (PFBgl, 5′GCCGAGCTCGATGAAGACTCTTACTGTTTTC3′) and reverse (PRBg1, 5′GCTCTAGAGTTAGTTTTGTTCAACATTTTC3′) primers were synthesized. PFBgl corresponded to the first seven amino acids of the open reading frame (ORF) and had a SacI site attached, whereas PRBgl corresponded to the last six amino acids plus a stop codon and had an XbaI site attached. Using PFBgl and PRBgl as primers and plasmid PBgl13 as template, the whole ORF was amplified by PCR. PCR was carried out for 30 cycles of denaturation (1 min at 94°C), annealing (1.5 min at 42°C), and extension (3.5 min at 72°C) on a 480 Thermocycler (Perkin-Elmer, Norwalk, CT). PCR products were purified using a Geneclean kit and digested with SacI and XbaI. Digested DNA fragments were purified and concentrated before they were ligated to the digested pYES2 with T4 ligase.
Transformation of E. coli and Propagation of Plasmids
Ligation reactions were performed using a rapid ligation kit (Roche). E. coli TOP10 transformants were plated onto LB plates containing ampicillin (50 μg/mL). Colonies were picked and grown overnight in LB liquid medium containing ampicillin. Plasmids were purified with a spin column kit from Qiagen. Restriction digestion and nucleotide sequencing were done to verify the presence, orientation, and sequence of the insert.
Transformation of S. cerevisiae
A single colony of yeast strain INVSc1 was grown to an OD600 of 1.3 in YPD medium, pH 6.5, containing 1% (w/v) yeast extract, 1% (w/v) bactopeptone, and 1% (w/v) dextrose. Cells were harvested by centrifuging (4000g, 5 min) at 4°C and washed twice with ice-cold sterile H2O and twice with ice-cold 1 M sterile sorbitol. Then, the cells were resuspended in 0.5 mL of 1 M sorbitol. Approximately 5 μg of plasmid was used to transform 40 μL of prepared yeast cells utilizing an electroporator (Bio-Rad, Hercules, CA). Transformants were grown on DOB medium containing 0.17% (w/v) yeast nitrogen base without amino acids and NH2SO4, 2.0% (w/v) dextrose, 0.08% (w/v) drop-out supplements lacking uracil (Bio101), 2% (w/v) agarose, and 1 M sorbitol. The plates were incubated at 30°C for 3–5 d.
Induction of Gene Expression
Ten putative transformants were chosen for induction experiments. They were cultivated in 10 mL of DOB medium containing 4% (w/v) raffinose. After the OD600 reached 1.0, galactose was added to a final concentration of 2% (w/v). Samples were collected before and periodically after the addition of galactose. Transformant no. 7, which produced the highest level of β-glucosidase activity, was chosen for induction experiments in YPD medium. A single colony of the transformant was used for inoculating 2 mL of DOB medium. After the OD600 reached 0.8, 1.0 mL of the culture was added to 100 mL of YPD-raffinose (4% w/v) medium, and the culture was shaken (250 rpm) at 30°C. Sterile galactose (2.0% w/v) was added to the culture after the OD600 reached 1.0. Samples were collected before and periodically after the addition of galactose. Cells were harvested by centrifuging (5000g, 5 min) at 4°C. All samples were kept at −20°C until analyzed.
Enzyme Assay
β-glucosidase (p-nitrophenyl-β-D-glucosidase) and cellobiase activities were determined by the following standard procedures. With p-nitrophenyl-β-D-glucopyranoside (pNPG) as the substrate, the reaction mixture of 1.2 mL contained 0.3 mL of appropriately diluted enzyme solution; 0.6 mL of 50 mM sodium phosphate buffer, pH 6.0; and 0.3 mL of 12 mM pNPG. The reaction was carried out for 10 min at 40°C and stopped by the addition of 2.4 mL of 1 M Na2CO3. The liberated p-nitrophenol was measured spectrophotometrically at 405 nm (12). Cellobiase activity was determined by using a reaction mixture of 2 mL containing 1 mL of appropriately diluted enzyme solution in 50 mM sodium phosphate buffer, pH 6.0, and 1 mL of 2 mM cellobiose. The reaction was carried out at 40°C for 30 min and was stopped by placing the assay tubes in boiling water for 5 min. Liberated glucose was measured with a glucose determination kit (Sigma, St. Louis, MO) as described in the manufacturer’s instructions. One unit of β-glucosidase or cellobiase activity was defined as the amount of enzyme required to hydrolyze 1 μmol of substrate/min. Specific activity was expressed as units per milligram of protein.
Hydrolysis of Avicel cellulose (Sigma) was set up in test tubes with 3.0 mL of solution containing 30 mg of cellulose and 0.25 filter paper activity units of T. reesei cellulase (13). The buffer was 50 mM sodium citrate, pH 5.5. The Orpinomyces β-glucosidase (0.25 U) was added to some of the tubes. The tubes were sealed and shaken (200 rpm) at 37°C for 16 h. Following incubation, insoluble materials were removed by centrifuging (5000g, 15 min), and sugars in the supernatant were separated and quantified using a high-performance liquid chromatography (HPLC) system equipped with a Bio-Rad HPX-87P column and a 1047A RI detector.
Purification of Enzyme
S. cerevisiae culture (7.5 L) harboring pBgl13 was grown in YPD-raffinose medium for 24 h at 30°C. The supernatant was obtained by centrifugation (4000g, 20 min) and concentrated to a volume of approx 155 mL by using an ultrafiltration cell (Amicon, Beverly, MA) equipped with a PM 10 membrane. The buffer was changed to 50 mM sodium phosphate, pH 6.0, and then ammonium sulfate was added to a concentration of 0.8 M. The solution was centrifuged (20,000g, 10 min) at 4°C to remove precipitated material. More than 80% of the activity was found in the supernatant that was loaded on a Phenyl Superose 10/10 (Pharmacia, Piscataway, NJ) column equilibrated with 50 mM sodium phosphate buffer, pH 6.0, containing 0.8 M ammonium sulfate. The major β-glucosidase fraction did not bind to the column. This sample was then concentrated, and the buffer was changed to 20 mM piperazine-HCl, pH 6.0. The solution was applied to a Mono Q 5/5 (Pharmacia) column equilibrated with 20 mM piperazine-HCl, pH 6.0. The enzyme bound to the column. Two peaks of activity were eluted with a linear gradient of NaCl (0 to 1 M). The major fraction was concentrated and changed to 20 mM formic acid buffer, pH 4.0. The sample was applied to a Resource S column (Pharmacia). The enzyme did not adsorb to the column. Final purification was achieved by gel filtration in a Superdex 200 26/60 column (Pharmacia) equilibrated with 20 mM sodium phosphate buffer, pH 6.0, containing 100 mM NaCl. Fractions containing β-glucosidase were stored at −20°C until further analyzed. Procedures for partial purification of the cell-associated β-glucosidases were basically identical to those for the secreted BglA.
N-Terminal Amino Acid Sequencing of Proteins
Proteins were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (14) and transferred to polyvinylidine difluoride membranes in a Mini Trans-Blot cell (Bio-Rad). Protein bands on the membranes were visualized by Coomassie Blue R-250 staining and excised using a razor blade. N-terminal amino acid sequencing of the protein bands was performed on an Applied Biosystems Model 477A gas-phase sequencer equipped with an automatic on-line phenylthiohydantoin analyzer.
Analytical Methods
SDS-PAGE (7.5 and 10%) was carried out in Laemmli’s buffer (14). High-molecular-weight protein standards (Bio-Rad) were used as markers. Electrophoresis was performed in a Mini-Protein II cell and gels were stained with Coomassie brilliant blue R-250 (15). β-Glucosidase activity bands in native gels were visualized by the method of Rutenburg et al. (16) with 6-bromo-naphthyl-β-d-glucopyranoside as substrate.
Characterization of Expressed β-Glucosidases
The carbohydrate content of the purified enzyme was determined using the phenol-sulfuric method of Dubois et al. (17) with mannose as standard. Protein content was measured by the method of Lowry et al. (18) with bovine albumin as standard.
The pH optimum was determined by performing an assay with either pNPG or cellobiose as substrate at 40°C in the following buffer systems: 0.1 M sodium acetate (pH 3.8 to 5.6), 0.1 M sodium phosphate (pH 5.8–7.6), and 0.1 M HEPES-NaOH (pH 8.0–8.6). Enzyme stability at different pH values was determined by measuring the residual activity after incubating the enzyme for 24 h at 4°C with these buffers plus glycine-HCl for pH 3.0–3.4 and piperazine-HCl for pH 9.0–10.2. The effect of temperature on β-glucosidase activity was determined by assaying the enzyme at temperatures from 30 to 65°C. To assess the stability of the glycosylated and deglycosylated BglA at various temperatures, enzyme preparations were incubated in 50 mM sodium phosphate buffer, pH 6.0, from 10 min to 8 h at temperatures from 40 to 60°C. During the time course, aliquots were withdrawn and kept on ice. Remaining activity in the samples was determined under standard assay conditions.
Several α- and β-glucosides (1 mM) and polysaccharides (0.5% [w/v]) were tested as substrates for the purified enzyme. p-Nitrophenol (12) and glucose were determined as described by HPLC (see Enzyme Assay). Reducing sugars were determined following the procedure of Miller (19).
To measure kinetic parameters, hydrolysis rates were determined by varying the concentrations of pNPG (0.05–10 mM) and cellobiose (0.04–16 mM). The inhibition by glucose was evaluated with only pNPG (10 mM) as substrate, whereas the inhibitory effect of glucono-1,5-lactone was verified with both pNPG (10 mM) and cellobiose (1.0 mM) as substrates. Km, Vmax, and Ki values were calculated from Lineweaver-Burk plots.
Results and Discussion
Isolation of a BglA cDNA
Previous work (4) revealed that Orpinomyces sp. strain PC-2, a polycentric anaerobic fungus isolated from the rumen of a cow, produces high levels of β-glucosidase in addition to endoglucanase, CBH, and xylanase. A β-glucosidase produced in the culture supernatant of the fungus has been purified and characterized (3). Different hydrolytic enzymes produced by this fungus can function individually or in high-molecular-weight enzyme complexes such as cellulosomes (5–9). The fungal cellulosomes purified from residual solid substrate of the fungal culture contained β-glucosidase activity (20), indicating that β-glucosidases may be components of the cellulosomes produced by the fungus. More recently, the gene coding for a cellulosomal β-glucosidase of the monocentric anaerobic fungus Piromyces E2 was cloned and sequenced (21).
Many of the hydrolytic enzymes of anaerobic fungi sequenced to date contain, in addition to catalytic domains, a noncatalytic docking domain (NCDD) (22), which functions in a fashion similar to dockerins in the cellulosomes of anaerobic bacteria (23), but no sequence similarity has been identified between the fungal NCDDs and the bacterial dockerins (5,24). Polyclonal antibodies raised against the NCDD of Orpinomyces XynA crossreacted with a number of different other polypeptides in the culture media of Orpinomyces and Neocallimastix grown on cellulose (5), suggesting that a number of NCDD-containing enzymes remain to be isolated. To isolate cDNAs coding for NCDD-containing polypeptides, we used the XynA NCDD-specific antibodies to screen an Orpinomyces expression cDNA library (11). Twenty-five positive plaques were isolated after screening 1.0 × 105 plaque-forming units. Sequencing of the inserted cDNAs in the pBluescipts, after being excised from pure positive λ plaques, revealed that several represented different lengths of cDNAs coding for, in addition to xynA (5), celA (6), celB (5), celC (6), and celE (7), three new sequences were isolated. One of the new sequences had 800 bp (pBgl6) and its deduced amino acid sequence was homologous to certain β-glucosidases. Using the cDNA fragment in pBgl6 as a hybridization probe, plaques containing cDNAs (pBgl13) with a complete ORF encoding a putative β-glucosidase (bglA) were isolated from the same cDNA library.
Nucleotide and Deduced Amino Acid Sequences of Orpinomyces sp. Strain PC-2 bgl1
The complete nucleotide sequence of bgl1 cDNA was determined and deposited in GenBank with accession no. AF016864 (submitted on July 31, 1997). The total length of the cDNA is 2435 bp. It contains an ORF of 1974 bp encoding a polypeptide of 657 amino acids with a molecular mass of 75,227 Daltons (BglA). Like cellulase B (5) and cellulase F (8) isolated from the same fungus, a long 3′ noncoding A-T-rich end (423 bp) was observed after the ORF, but no typical long poly (A) stretch was found. The putative translation start codon (ATG) for bgl1 was assigned based on the fact that there were stop codons in all three frames preceding the ORF and there was no ATG codon upstream of the proposed ORF. In addition, the proposed N-terminal region of BglA contains the properties of signal peptides of fungi (25). Furthermore, close examination of the complete amino acid sequence of BglA failed to detect an NCDD sequence, indicating that BglA lacks a dockerin and there is not a component of the Orpinomyces cellulosome. Its cDNA was isolated owing to nonspecific crossreaction between the partial BglA protein and the NCDD-specific antibodies.
The G+C content of the entire cDNA and the ORF of bgl1 were 36 and 42.3%, respectively, and that of the 5′ and 3′ noncoding region was only 9.1%. Low G+C contents have also been found in other cDNAs of Orpinomyces (5–9).
Homology of BglA with Other β-Glucosidases
The deduced amino acid sequence of BglA was compared with other protein sequences in the SWISS PROT and GP databanks. Comparison using the Bestfit program revealed that BglA had significant levels of identity with β-glucosidases from Piromyces E2 (Cel1A, 72.0%) (26), Cavia porcellus (pig, 41.2%) (27), Costus speciosus (40%) (28), Clostridium thermocellum (40.2%) (29), Bacillus circulans (41.7%) (30), Thermoanaerobacter sp. (40.6%) (31), and Thermotoga maritima (40.7%) (32). According to Henrissat and Bairoch (33) and http://afmb.cnrs-mrs.fr/~cazy/CAZY, these enzymes belong to family 1 glycosyl hydrolases. No significant identity (<20%) was found with the family 3 β-glucosidases from T. reesei and Aspergillus aculeatus. Comparison of the homologous β-glucosidases of the family revealed that the β-glucosidases of Orpinomyces and Piromyces have about 50 more amino acid residues after their signal peptides than those of the other organisms. These 50 amino acid regions are probably not critical for catalysis because the recombinant polypeptides of Orpinomyces BglA with this region partially truncated remained catalytically active. These regions between the Orpinomyces and Piromyces enzymes are the least homologous to each other (Fig. 1). Furthermore, the short repeated sequences within the region of the Piromyces enzyme (26) are absent in the Orpinomyces enzyme. Glu-250 and Glu-523 of the Orpinomyces BglA are conserved between all the enzymes, and these two residues in the Bacillus polymyxa β-glucosidase were found to be directly involved in catalysis (34). Gln82, His 260, Tyr 433, Glu-523, and Tyr 607, which are also conserved, have been identified as determinant residues for the recognition of substrates (34).
Fig. 1.

Comparison of the amino-terminal regions of Orpinomyces BglA (AF016864) and Piromyces CelA1 (26) β-glucosidases. Vertical lines indicate identical matches, and single and double dots indicate different levels of conservation.
Expression of bgl1 in and Secretion of BglA from S. cerevisiae
No β-glucosidase activity was detected in E. coli culture harboring the complete bglA cDNA. This finding is in agreement with no positive colonies detected when using 4-methylumbelliferyl-β-D-glucoside, a fluorescent substrate of β-glucosidases, as a screening substrate. Lack of functional expression in E. coli might be related to differences between anaerobic fungi and E. coli with respect to posttranslational modifications such as glycosylation and folding. We attempted to express the gene in S. cerevisiae, since several heterologous genes coding for hydrolytic enzymes have been expressed in various strains of the yeast. These include genes coding for two endoglucanases (35), two CBHs (36), and one β-glucosidase from T. reesei (37); a xylanase from Aureobasidium pullulans (38); an α-amylase from wheat (39); and others. Recently, a cellulase gene cassette encoding the Butyrivibrio fibrisolvens endo-β-1,4-glucanase (END1), the Phanerochaete chrysosporium CBH (CBH1), the Ruminococcus flavefaciens cellodextrinase (CEL1), and the Endomyces fibrilizer cellobiase (Bgl1) was successfully expressed in a laboratory strain of S. cerevisiae (40).
After transformation, 10 transformants were grown in synthetic dropout supplemented medium without uracil, using raffinose as growth substrate and galactose as inducer (see Materials and Methods). β-Glucosidase activity was determined for the cells and in culture medium. All activity was found to be associated with cells, and no activity was found in the culture medium for all the transformants. It has been reported that culture conditions can strongly affect the secretion of enzymes from S. cerevisiae. For example, the secretion of a wheat α-amylase from S. cerevisiae into the medium was efficient only in a rich medium but barely detectable in a minimal medium (39). The secretion of the Orpinomyces BglA from S. cerevisiae was similar (Fig. 2). A substantial percentage (40%) of the total β-glucosidase activity was found in the culture medium after 24 h of growth in rich medium. The levels of activity in cell-associated and culture medium fractions stayed almost constant during the cultivation period (96 h). The growth rates between the transformants using plasmids with and without bgl1 inserted were almost identical, indicating that BglA and its gene did not affect the physiology of the yeast. A higher percentage of a T. reesei β-glucosidase, when expressed in S. cerevisiae, was found in the culture medium (37).
Fig 2.

β-glucosidase production of S. cerevisiae cultures grown in raffinose-YPD medium after galactose induction. An aliquot of an overnight culture grown in DOB medium was used to inoculate raffinose-YPD medium. After growth to an OD600 of 1.0, sterile galactose was added. Samples were withdrawn at the time points shown. The OD600 of control culture (●), OD600 (○), and β-glucosidase activity of cell extract (□) and culture medium (■) of transformant no. 7 were determined. Culture conditions, preparation of the samples, and enzyme assays were described in Materials and Methods. The control culture corresponded to the yeast containing the pYES2 without any insert.
Purification and N-Terminal Processing of BglA Produced by S. cerevisiae
A summary of purification of the Orpinomyces BglA secreted by S. cerevisiae culture is given in Table 1. The enzyme was purified about 28-fold to homogeneity with a specific activity of 18.8 U/mg and a yield of about 1%. Multiple peaks of activity were observed during the purification steps, but only the major activity peak was used for further purification, indicating that BglA may be secreted into culture medium with multiple forms owing to proteolysis or different levels of glycosylation.
Table 1.
Summary of Purification of Recombinant BglA from Culture Medium of S. cerevisiae
| Purification step | Total protein (mg) | Total units (μmol/min)a | Specific activity (μmol/[min·mg]) | Yield (%) |
|---|---|---|---|---|
| Culture filtrate | 480.00 | 326.8 | 0.68 | 100.0 |
| Concentrated supernatant | 165.30 | 319.7 | 1.93 | 97.8 |
| Phenyl Sepharose | 17.10 | 101.4 | 5.90 | 31.0 |
| Mono Q | 5.20 | 66.0 | 12.80 | 20.2 |
| Resource S | 2.23 | 38.3 | 17.20 | 11.7 |
| Superdex 200 | 0.17 | 3.2 | 18.80 | 1.0 |
Activities were measured with pNPG as substrate.
Two cell-associated forms (BglA1 and BglA2) of BglA were also partially purified from the cell-free extract of yeast cells using Phenyl Sepharose, Mono Q, and Superdex 200. The sizes of BglA1 (first band in lanes 4 and 7 of Fig. 3) and BglA2 (first band in lanes 5 and 8 of Fig. 3) were estimated to be about 65 kDa on SDS-PAGE/zymogram analysis (Fig. 3).
Fig. 3.
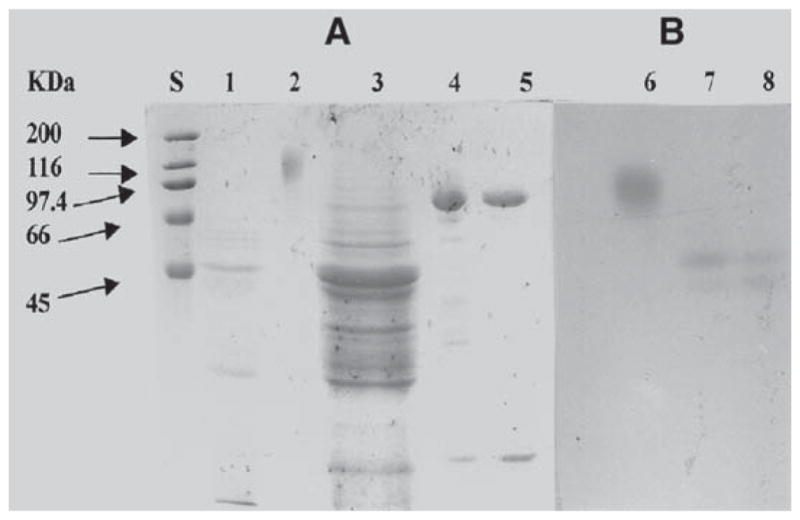
SDS-PAGE (10%)/zymogram analysis of secreted and cell-associated forms of BglA. (A) SDS-PAGE stained with Coomassie brilliant blue R-250; (B) β-glucosidase zymogram gel. Lane S, protein molecular mass standards; lane 1, crude culture supernatant (10 μg); lane 2, purified secreted BglA (2 μg); lane 3, crude cell extract (60 μg); lane 4, partially purified BglA1 (2 μg); lane 5, partially purified BglA2 (2 μg); lane 6, purified secreted BglA (2 μg); lane 7, BglA1 (2 μg); lane 8, BglA2 (2 μg).
The purified BglA, BglA1, and BglA2 were all subjected to N-terminal amino acid sequencing. The secreted BglA had an N-terminal sequence of KKCIVKSDAA, which matched amino acid residues 17–26 (Fig. 1), demonstrating that amino acid residues 1–16 were cleaved during secretion. Thus, the first 16 amino acid residues might serve as a signal peptide in both Orpinomyces and S. cerevisiae. Removal of 16 amino acid residues at the N-terminus resulted in 641 amino acid residues with a calculated mass of 73,608 Daltons for the mature BglA. The signal peptide had a basic amino acid (Lys) as the second N-terminal residue, followed by a hydrophobic amino acid region containing in some points nonhydrophobic residues. This is in agreement with the work of Ngsee et al. (41), who, using site-directed mutagenesis of the signal sequence of yeast invertase gene, suc 2, demonstrated that the essential feature of a signal peptide for yeast is a hydrophobic core of 6–15 amino acids. The core region can be interrupted to a certain extent by nonhydrophobic residues. The purified recombinant BglA yielded a broad band with an average size of about 110 kDa on SDS-PAGE (Fig. 3), which was larger than that calculated for the deduced mature enzyme. According to Orlean et al. (42), only one N-glycosylation site, Asn-X-Ser/Thr, corresponding to amino acid residues 280–282 was found on the entire BglA sequence. However, the size of the purified enzyme after treatment with N-glycosidase F, an enzyme specifically removing N-glycosylation, shifted to two sharp bands with very similar sizes (87 and 92 kDa) on SDS-PAGE (Fig. 4). N-terminal amino acid sequencing revealed that these two bands had amino acid sequences at their N-termini identical to those of the secreted BglA. These results indicate that about 20% (w/w) of N-glycosylation is added to BglA during secretion from S. cerevisiae and that the size difference between the two similar bands after the N-glycosidase F treatment is probably owing to O-glyco-sylation. The β-glucosidase purified from the culture supernatant of the same fungus had a mass of 85 kDa including 8.5% (w/w) carbohydrate (3). Assuming that the native β-glucosidase (3) and the secreted BglA reported here are products of the same gene, bgl1 of Orpinomyces PC-2, glycosylation (hyperglycosylation) by S. cerevisiae is much greater than by Orpinomyces. Hyperglycosylation was also found for T. reesei endoglucanases (35), CBHs (36), and β-glucosidase (37) expressed in and secreted from S. cerevisiae.
Fig. 4.
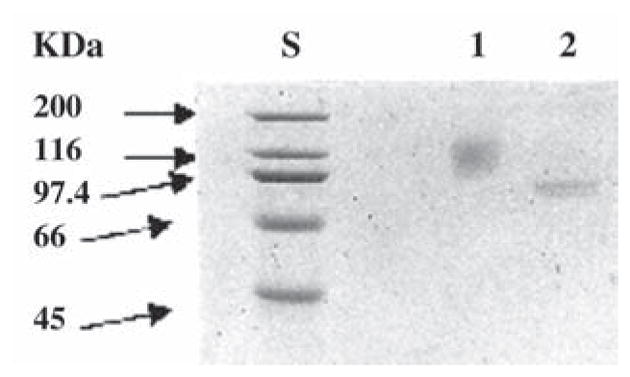
SDS-PAGE analysis of BglA treated with N-glycosidase F. Lane S, protein molecular mass standards; lane 1, purified secreted BglA (2.4 μg); lane 2, purified secreted BglA (2.4 μg) treated with N-glycosidase F.
The N-terminal sequence for BglA1 was APEDSGVES, which matched amino acid residues 40–48 (Fig. 1), and that of BglA2, GEDDELLDLS, corresponded to amino acid residues 49–58 (Fig. 1). Thus, the cleavages resulted in two truncated forms (BglA1 and BglA2) of BglA (Fig 3). These results suggest that BglA1 and BglA2 were cleaved at the wrong sites and subsequently trapped during transport in the secretory pathway. The fact that these two truncated forms retained catalytic function indicates that the sequences of the Orpinomyces BglA and the Piromyces Cel1A up to amino acid residue 48 are not critical for catalysis. This may be why this region is absent in the homologous bacterial β-glucosidases.
Catalytic Properties
The activity of the purified secreted BglA against pNPG and cellobiose was determined from pH 3.8 to 8.6 at 40°C. The optimum pH with both substrates was found to be between 5.5 and 7.5 (Table 2) with detectable levels of activity from pH 4.0 to 8.0. The enzyme was stable for at least 24 h between pH 3.4 and 10.2 at 4°C. Hydrolysis of pNPG and cellobiose by BglA, determined in 50 mM sodium phosphate buffer (pH 6.0), was most active at 50°C (Table 2) when assayed for 10 min. Enzyme activity decreased rapidly above 55°C and the enzyme lost its activity at 65°C. The enzyme maintained 100% of its activity for 8 h at 40 and 50°C. Inactivation of BglA occurred slowly at 55°C, with 50% of the enzyme activity remaining after 8 h of incubation. At 60°C the enzyme was quickly inactivated. The optimum pH and temperature ranges of the recombinant BglA are similar to those reported for the native β-glucosidases of Orpinomyces sp. strain PC-2 (3), Neocallimastix frontalis (43), and Piromyces sp. strain E2 (44).
Table 2.
Some Properties of Purified Recombinant BglA of Orpinomyces Produced in S. cerevisiae
| Molecular mass | |
| Deduced | 75,227 Daltons |
| Before deglycosylation | 110,000 Daltons |
| After glycosidase F treatment | 87 and 97 kDa |
| Optimum pH at 40°C | 5.5–7.5 |
| Optimum Temperature at pH 6.0 | 55°C |
| Km | |
| pNPG | 0.762 mM |
| Cellobiose | 0.310 mM |
| Vmax | |
| pNPG | 8.20 μmol/(min·mg) |
| Cellobiose | 6.20 μmol/(min·mg) |
| KI of glucose | 3.6 mM |
Km, Ki, and Vmax values for the secreted BglA were obtained from Lineweaver-Burk plots (Table 2). The Km value with pNPG as substrate at 40°C and pH 6.0 was 0.762 mM, higher than that (0.35 mM; [3]) reported for the native β-glucosidase of the same fungus. However, the Km value with cellobiose as substrate in the range of 0.04–1.0 mM was 0.31 mM, which is very similar to the Km (0.25 mM) for the native β-glucosidase. These values are within the range of Km values reported for several β-glucosidases of anaerobic fungi (43–46). Comparison of the Km values for β-glucosidases from various sources indicates that those from anaerobic fungi have lower Km values than those from bacteria or aerobic fungi. The differences in Km values between the Orpinomyces native β-glucosidase and the recombinant BglA could be owing to the different levels of glycosylation. Ward (47) has reported that when a site for N-glycosylation was introduced, chymosin had resulted in lower specific activity. The effect on specific activity was considered to be probably a consequence of active site change by the glycosylation (47,48).
Hydrolysis rate decreased as cellobiose concentration was increased higher than 2.0 mM (Fig. 5). This phenomenon is caused by substrate inhibition by cellobiose and transglycosylation for the formation of cellooligosaccharides, as described for a cellobiase of N. frontalis (46). A close comparison of the same plots between the Orpinomyces BglA (Fig. 5) and the Neocallimastix cellobiase (Fig. 4, of ref. 46) shows that the Orpinomyces and the Neocallimastix enzymes had their highest activity when cellobiose concentrations were 1.0 and 0.25 mM, respectively, suggesting that the former tolerates higher cellobiose concentrations.
Fig. 5.

Lineweaver-Burk plot of the Orpinomyces BglA on hydrolysis of cellobiose (0.04–16 mM). The release of glucose was measured. Reciprocal initial velocities (mg/U) are plotted against the reciprocal concentrations of substrates (1 mM). The inset is a plot of initial velocities (U/mg) against the log cellobiose concentrations (mM).
Glucose and glucono-1,5-lactone competitively inhibited BglA with a Ki of 3.6 and 0.05 mM, respectively. These numbers are lower than those for the native Orpinomyces β-glucosidase, and the reason for this could be again owing to difference in glycosylation.
BglA is specific for aryl-β-glucoside bonds and was not able to hydro-lyze alkyl-β-glucoside bonds or α-1,4-glucoside bonds. The enzyme rapidly hydrolyzed sophorose (β-1,2-glucobiose), laminaribiose (β-1,3-glucobiose), and cellobiose (β-1,4-glucobiose) but lacked activity on gentibiose (β-1,6-glucobiose), methyl-β-glucoside, p-nitrophenyl-β-xyloside (pNPX), salicin, maltose, sucrose, lactose, xylan, microcrystalline cellulose, or carboxym-ethyl cellulose. A low level of activity was found against pNPX when the enzyme level in the assays was increased 20 times. BglA reported here and the native β-glucosidase (3) had almost identical substrate specificity. Interestingly, such substrate specificity is very similar to that of a β-glucosidase purified from the anaerobic rumen bacterium Ruminococcus albus (49).
To assess the ability of BglA to convert cellooligosaccharides to glucose, we added BglA to the saccharification reaction of Avicel by T. reesei cellulase since it is well known that T. reesei secretes insufficient levels of β-glucosidase activity. Glucose, cellobiose, and cellotriose generated by the cellulase mixtures were measured (Fig. 6). In the presence of BglA under the test conditions, cellobiose and cellotriose disappeared almost completely and glucose increased more than sixfold. The amount of glucose in the presence of BglA was more than the total amount of glucose, cellobiose, and cellotriose without the addition of BglA, indicating that the conversion of the oligosaccharides to glucose removed inhibition of the sugars on both endoglucanases and CBHs of T. reesei.
Fig. 6.
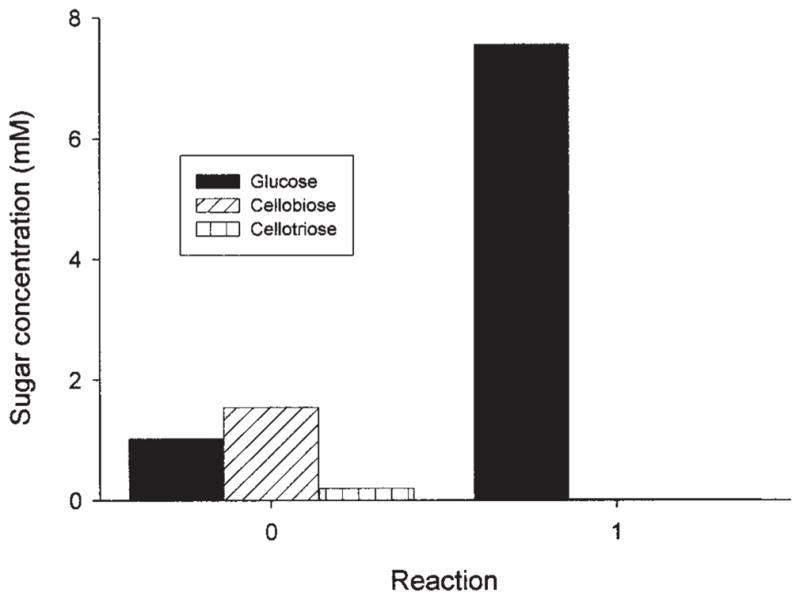
Saccharification of Avicel cellulose by T. reesei cellulase without (panel 0) and with (panel 1) Orpinomyces BglA. Concentrations of sugars were measured using an HPLC system (see Materials and Methods) after 16 h of reaction.
Table 3.
Substrate Specificity of Orpinomyces BglA Purified from S. cerevisiaea
| Substrate (1 mM) | Specific activity (μmol/[min·mg])b |
|---|---|
| p-Nitrophenyl-β-glucopyranoside | 2.10 |
| Cellobiose (β-1,4) | 1.87 |
| o-Nitrophenyl-β-glucopyranoside | 2.99 |
| Sophorose (β-1,2) | 4.53 |
| Laminaribiose (β-1,3) | 6.10 |
Conditions were 40°C and pH 6.0.
Activity on gentibiose (β-1,6-glucoside), methyl-β-glucoside, p-nitrophenyl-β-xyloside, salicin, maltose, sucrose, lactose, xylan (1.0% [w/v]), Avicel (1.0% [w/v]), or carboxymethylcellulose (1.0% [w/v]) was <1.0% of that on pNPG.
Acknowledgments
We wish to thank Patrick Kane for excellent technical input in the kinetic analysis. This work was supported by grants from the US Department of Energy (DE-FG02-93ER20127), the Consortium for Plant Biotechnology Research (OR220 72-64), Aureozyme, and Georgia Research Alliance (TDP98.001-UGA). L. G. L. received a distinguished professorship from Georgia Power and E. A. X. received a scholarship from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
References
- 1.Ohmiya K, Sakka K, Karita S, Kimura T. Gen Eng Rev. 1997;14:365–414. doi: 10.1080/02648725.1997.10647949. [DOI] [PubMed] [Google Scholar]
- 2.Filho EXF. Can J Microbiol. 1996;42:1–5. doi: 10.1139/m96-001. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Li XL, Ljungdahl LG. Appl Environ Microbiol. 1994;60:64–70. doi: 10.1128/aem.60.1.64-70.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borneman WS, Akin DE, Ljungdahl LG. Appl Environ Microbiol. 1989;55:1066–1073. doi: 10.1128/aem.55.5.1066-1073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li XL, Chen H, Ljungdahl LG. Appl Environ Microbiol. 1997;63:628–635. doi: 10.1128/aem.63.2.628-635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li XL, Chen H, Ljungdahl LG. Appl Environ Microbiol. 1997;63:4721–4728. doi: 10.1128/aem.63.12.4721-4728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Li XL, Blum DL, Ljungdahl LG. FEMS Microbiol Lett. 1998;159:63–68. doi: 10.1111/j.1574-6968.1998.tb12842.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Li X-L, Blum DL, Ximenes EA, Ljungdahl LG. Appl Biochem Biotechnol. 2003;105–108:775–785. doi: 10.1385/abab:108:1-3:775. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Li XL, Ljungdahl LG. J Bacteriol. 1997;179:6028–6034. doi: 10.1128/jb.179.19.6028-6034.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blum DL, Li XL, Chen H, Ljungdahl LG. Appl Environ Microbiol. 1999;65:3990–3995. doi: 10.1128/aem.65.9.3990-3995.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Li XL, Ljungdahl LG. Proc Natl Acad Sci USA. 1995;9:2587–2591. doi: 10.1073/pnas.92.7.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herr D, Baumer F, Dellweg H. Appl Microbiol Biotechnol. 1978;5:29–36. [Google Scholar]
- 13.Chen H, Ximenes EA, Li X-L, Ljungdahl LG. In: Cellulose Degradation. Ohmiya K, Hayashi K, Sakka K, Kobayashi Y, Karita S, Kimura T, editors. Uni Publishers; Tokyo, Japan: 1999. pp. 173–181. [Google Scholar]
- 14.Laemmli UK. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 15.Fairbanks G, Steak TS, Wallach DFH. Biochemistry. 1971;10:2606–2616. doi: 10.1021/bi00789a030. [DOI] [PubMed] [Google Scholar]
- 16.Rutenburg AM, Goldbarg JA, Rutenburg SH, Lang RT. J Histochem Cytochem. 1960;8:268–272. doi: 10.1177/8.4.268. [DOI] [PubMed] [Google Scholar]
- 17.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Anal Chem. 1956;28:350–356. [Google Scholar]
- 18.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–273. [PubMed] [Google Scholar]
- 19.Miller GL. Anal Chem. 1959;31:426–428. [Google Scholar]
- 20.Li X-L, Chen H, He Y, Blum DL, Ljungdahl LG. Abstracts of 97th General Meeting of the American Society for Microbiology. American Society for Microbiology; Washington, DC: 1997. p. 424. [Google Scholar]
- 21.Steenbakkers PJM, Harhangi HR, Bosscher MW, van der Hooft MMC, Keltjens JT, van der Drift C, Vogels GD, Op den Camp HJM. Biochem J. 2003;370:963–970. doi: 10.1042/BJ20021767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steenbakkers PJM, Li XL, Ximenes EA, Arts JG, Chen H, Ljungdahl GL, Op den Camp HJM. J Bacteriol. 2001;183:5325–5333. doi: 10.1128/JB.183.18.5325-5333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Béguin P, Lemaire M. Crit Rev Biochem Mol Biol. 1996;31:201–236. doi: 10.3109/10409239609106584. [DOI] [PubMed] [Google Scholar]
- 24.Fannuti G, Ponyi T, Black GW, Hazlewood GP, Gilbert HJ. J Biol Chem. 1995;270:29,314–29,322. doi: 10.1074/jbc.270.49.29314. [DOI] [PubMed] [Google Scholar]
- 25.von Heijne G. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harhangi HR, Steenbakkers PJM, Akmanova A, Jetten MSM, van der Drift C, Op den Camp HJM. Biochem Biophys Acta. 2002;1574:293–303. doi: 10.1016/s0167-4781(01)00380-3. [DOI] [PubMed] [Google Scholar]
- 27.Hays WS, Jenison SA, Yamada T, Pastuszyn A, Glew RH. Biochem J. 1996;319:829–837. doi: 10.1042/bj3190829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoue M, Shibuya M, Yamamoto K, Ebizuka Y. FEBS Lett. 1996;389:273–277. doi: 10.1016/0014-5793(96)00601-1. [DOI] [PubMed] [Google Scholar]
- 29.Gräbnitz F, Seiss M, Rücknagel KP, Staudenbauer WL. Eur J Biochem. 1991;200:301–309. doi: 10.1111/j.1432-1033.1991.tb16186.x. [DOI] [PubMed] [Google Scholar]
- 30.Paavilainen S, Hellman J, Korpela T. Appl Environ Microbiol. 1993;59:927–932. doi: 10.1128/aem.59.3.927-932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breves R, Bronnenmeier K, Wild N, Lottspeich F, Staudenbauer WL, Hofemeister J. Appl Environ Microbiol. 1997;63:3902–3910. doi: 10.1128/aem.63.10.3902-3910.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liebl W, Gabelsberger J, Schleifer KH. Mol Gen Genet. 1994;242:111–115. doi: 10.1007/BF00277355. [DOI] [PubMed] [Google Scholar]
- 33.Henrissat B, Bairoch A. Biochem J. 1993;293:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanz-Aparicio J, Hermoso JA, Martinez-Ripoll M, Lequerica JL, Polaina J. J Mol Biol. 1998;275:491–502. doi: 10.1006/jmbi.1997.1467. [DOI] [PubMed] [Google Scholar]
- 35.Penttilä ME, André L, Saloheimo M, Lehtovaara P, Knowles JKC. Yeast. 1987;3:175–185. doi: 10.1002/yea.320030305. [DOI] [PubMed] [Google Scholar]
- 36.Penttilä ME, André L, Lehtovaara P, Bailey M, Teeri TT, Knowles JKC. Gene. 1988;63:103–112. doi: 10.1016/0378-1119(88)90549-5. [DOI] [PubMed] [Google Scholar]
- 37.Cummings C, Fowler T. Curr Genet. 1996;29:227–233. [PubMed] [Google Scholar]
- 38.Li XL, Ljungdahl LG. Appl Environ Microbiol. 1996;62:209–213. doi: 10.1128/aem.62.1.209-213.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothstein SJ, Lanhners KN, Lazarus CM, Baulcombe DC, Gatenby AA. Gene. 1987;55:353–356. doi: 10.1016/0378-1119(87)90296-4. [DOI] [PubMed] [Google Scholar]
- 40.van Rensburg P, Van Zyl WH, Pretorius IS. Yeast. 1998;14:67–76. doi: 10.1002/(SICI)1097-0061(19980115)14:1<67::AID-YEA200>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 41.Ngsee JK, Hansen W, Walter P, Smith M. Mol Cell Biol. 1989;9:3400–3410. doi: 10.1128/mcb.9.8.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orlean P, Kuranda MJ, Albright CF. Methods Enzymol. 1991;194:682–696. doi: 10.1016/0076-6879(91)94050-m. [DOI] [PubMed] [Google Scholar]
- 43.Herbaud M, Fevre M. Appl Environ Microbiol. 1990;56:3164–3169. doi: 10.1128/aem.56.10.3164-3169.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teunissen MJ, Lahaye DHTP, Huis In’t Veld JHJ, Vogels GD. Arch Microbiol. 1992;158:276–281. [Google Scholar]
- 45.Li XL, Calza RE. Enzyme Microb Technol. 1991;13:622–628. [Google Scholar]
- 46.Li XL, Calza RE. Biochem Biophys Acta. 1991;1080:148–154. doi: 10.1016/0167-4838(91)90142-m. [DOI] [PubMed] [Google Scholar]
- 47.Ward M. In: EMBO-ALKO Workshop on Molecular Biology of Filamentous Fungi. Nevalainen H, Pentillä M, editors. Foundation for Biotechnical and Industrial Fermentation Research; Espoo, Finland: 1989. pp. 119–128. [Google Scholar]
- 48.Archer DB, Peberdy JF. Crit Rev Biotechnol. 1997;17:273–306. doi: 10.3109/07388559709146616. [DOI] [PubMed] [Google Scholar]
- 49.Ohmiya K, Shirai M, Kurachi Y, Shimizi S. J Bacteriol. 1985;161:432–434. doi: 10.1128/jb.161.1.432-434.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]