

Abstract
NOD-like receptors (NLRs) play a large role in regulation of host innate immunity, yet their role in periodontitis remains to be defined. NLRX1, a member of the NLR family that localizes to mitochondria, enhances mitochondrial ROS (mROS) generation. mROS can activate the NLRP3 inflammasome, yet the role of NLRX1 in NLRP3 inflammasome activation has not been examined. In this study, we revealed the mechanism by which NLRX1 positively regulates ATP-induced NLRP3 inflammasome activation through mROS in gingival epithelial cells (GECs). We found that depletion of NLRX1 by shRNA attenuated ATP-induced mROS generation and redistribution of the NLRP3 inflammasome adaptor protein, ASC. Furthermore, depletion of NLRX1 inhibited Fusobacterium nucleatum infection-activated caspase-1, suggesting that it also inhibits the NLRP3 inflammasome. Conversely, NLRX1 also acted as a negative regulator of NF-κB signaling and IL-8 expression. Thus, NLRX1 stimulates detection of the pathogen F. nucleatum via the inflammasome, while dampening cytokine production. We expect that commensals should not activate the inflammasome, and NLRX1 should decrease their ability to stimulate expression of pro-inflammatory cytokines such as IL-8. Therefore, NLRX1 may act as a potential switch with regards to anti-microbial responses in healthy or diseased states in the oral cavity.
Keywords: Oral, periodontal, innate immunity, inflammation, Fusobacterium nucleatum
1. Introduction
F. nucleatum, a Gram-negative anaerobe, is one of the most abundant microorganisms present in the oral cavity during periodontal disease [7]. Its ability to co-aggregate with diverse microorganisms makes it a bridge that connects early commensals and late pathogens, which results in dental plaque formation [52]. In addition, F. nucleatum exacerbates periodontitis by inducing apoptosis of immune cells and bone loss [23, 28, 10]. However, the molecular mechanisms by which F. nucleatum promotes dysregulated inflammatory responses in periodontitis remain mostly unknown.
Gingival epithelial cells (GECs) represent the first protective barriers in the oral cavity that are exposed to invading microorganisms. During F. nucleatum infection, GECs secrete chemokines such as IL-8 to recruit neutrophils [55, 11, 26, 42]. In addition, we recently reported that GECs detect stress related to F. nucleatum infection by activating an inflammasome [9], which is relevant for inflammation and other physiological functions of the epithelium [36, 50], and that F. nucleatum infection activates the transcription factor NF-κB and stimulates chemokine expression in GECs. However, how cells coordinate the two types of innate immune responses to F. nucleatum infection has not been investigated.
The innate immune system relies on germline-encoded pattern-recognition receptors (PRRs) to detect conserved pathogen-derived components that are known as pathogen-associated molecular patterns (PAMPs) or host-released factors by damaged or dying cells called damage-associated molecular patterns (DAMPs) [24, 47, 30, 48]. Based on domain homology, PRRs can be classified into 5 families. They are the Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), RIG-I like receptors (RLRs), and AIM2-like receptors (ALRs) [8]. TLRs and CLRs are membrane-spanning PRRs detecting microorganisms in the extracellular milieu or in endosomes, whereas NLRs, RLRs, and ALRs are expressed in the cytosol where they sense intracellular microorganisms [8, 31].
NLRs are typically composed of a central conserved NOD domain, a C-terminal leucine-rich repeat (LRR), and a N-terminal effector domain [45]. According to the N-terminal effector domain, NLRs can be further classified into different groups, e.g. NLRP, NLRC, and NLRX [5]. Each NLR plays a non-redundant role in recognizing specific PAMPs or DAMPs, but the responses they trigger can be categorized as inflammasome-dependent or -independent. For example, NLRP1, NLRP3 and NLRC4 can form inflammasomes to activate caspase-1 upon sensing PAMPs and DAMPs. On the other hand, NOD1, NOD2 and NLRX1 induce pro-inflammatory responses through NF-κB or MAPK signaling during microbial infection [5, 8]. NLRX1 (also known as NOD5 or NOD9) has an atypical N-terminal X domain without obvious homology to other NLRs, but has a mitochondria-addressing sequence that sorts NLRX1 to mitochondria [41, 56, 4].
Due to the localization of NLRX1 in mitochondria, its role in inflammation has been linked to mitochondria, which is involved in multiple immune responses, such as autophagy and inflammasome activation. Proteomic analysis has identified the association between NLRX1 and mitochondrial protein TUFM (Tu translation elongation factor), which interacts with autophagy-related proteins Atg5–Atg12. The indirect association between NLRX1 and Atg5–Atg12 is necessary to induce autophagy during viral infection in mice [33, 32]. In addition, mitochondrial ROS (mROS) has been proposed to be a secondary signal for various inflammatory responses. However, its generation and the mechanism by which it damages pathogens remain elusive. NLRX1 can modulate mROS during infection possibly through interaction with UQCRC2, a subunit in the mitochondrial respiratory chain complex III [4]. Overexpression of NLRX1 enhances ROS generation in response to TNF-α, poly(I:C), Shigella and Chlamydia infection, whereas knockdown of NLRX1 abrogates pathogen-induced mROS generation [56, 2, 59]. Recently it has been shown that mROS can activate the NLRP3 inflammasome [60, 22, 19, 53]. Therefore, the first goal of this study is to examine whether NLRX1 affects NLRP3 inflammasome activation via mROS.
In addition to its role in mitochondria, NLRX1 has been reported to affect NF-κB signaling [43, 57, 56]. Depletion of NLRX1 augments expression of LPS-induced NF-κB-responsive genes, such as IL-1β and IL-6, in mice [61, 56, 3]. Immunoprecipitation experiments showed that NLRX1 binds constitutively to the TLR downstream signaling molecule, TRAF6, under unstimulated conditions. However, upon LPS stimulation, NLRX1 dissociates from TRAF6, leading to NF-κB activation [61]. In addition, TNF treatment enhances I-κB degradation in intestinal epithelial cells in conditional NLRX1 deficient mice [57]. Taken together, these results show that NLRX1 may act as a negative regulator in TLR signaling. Therefore, the second goal of this study is to examine the role of NLRX1 in NF-κB signaling during Fusobacterium nucleatum infection.
2. Materials and methods
2.1. Media, enzymes, oligonucleotides, and antibodies
Defined Keratinocytes-serum free medium (SFM), Bovine Pituitary Extract (BPE), fetal bovine serum (FBS), TRIzol reagent, MitoSOX™ Red Mitochondrial Superoxide Indicator (M36008), JC-1 dye (T3168) and all oligonucleotides were from Thermo Fisher Scientific. ATP, puromycin, polybrene, hemin, and menadione, Triton-X, Tween-20, and paraformaldehyde were from Sigma-Aldrich. Antibodies against NLRX1 (8583), NF-κB RelA (8242) were from Cell Signaling. Anti-ASC (TMS1, ab155970 for Immunoblotting; ab64808 for microscopy) and cleaved caspase-1 p10 (ab108326) were purchased from Abcam. Anti-actin (MAB1501) and cleaved caspase-1 p20 (AB1871) were obtained from Millipore. Anti-TOPO-I (sc-5342) was purchased from Santa Cruz. Gel electrophoresis reagents were from Bio-Rad.
2.2. Cells and bacterial Culture
The human immortalized gingival epithelial cells (GECs) were obtained as previously described [22]. Cells were routinely cultured in defined keratinocyte-SFM with 5 μg/ml plasmocin and grown at 37 °C in a humidified incubator containing 5% CO2. Cells were seeded in antibiotic-free keratinocyte-SFM medium two days before infection and during infection. An extra well was seeded for cell counting on the day of infection
F. nucleatum (ATCC 25586) was cultured anaerobically for 24 h at 37°C in brain-heart infusion broth supplemented with yeast extract (5 mg/ml), hemin (5 μg/ml), and menadione (1 μg/ml). For infection, F. nucleatum was cultured for 16–24 h before harvest, followed by washing and collection in PBS. F. nucleatum was then added into GEC-seeded wells at an M.O.I. of 50 or 100 and incubated at 37 °C with 5% CO2 as indicated for each assay.
2.3. Lentivirus infection and selection of shRNA-expressing Cells
GECs stably expressing shRNA against NLRX1 were generated by transducing the cells with lentiviral particles purchased from Sigma-Aldrich. The vectors encoding the 5 shRNA sequences separately were:
Transduction was carried out following the manufacturer’s instructions in the presence of 8 μg/ml polybrene. Nontarget shRNA control cells were also generated using an irrelevant sequence (SHC002V, Sigma). Briefly, GEC were plated at 70% confluency 24 h prior to transduction, and the corresponding lentiviral transduction particles were added at M.O.I. of 3 overnight. Fresh media was added the next day, and stably infected cells were selected by addition of media containing 0.6 μg/ml puromycin for 1 week.
2.4. Measurement of mitochondrial membrane potential and ROS
Cells were stained with the cationic carbocyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide) or MitoSOX mitochondrial superoxide dye as described previously [22, 21]. In brief, cells were loaded with 7.7 μM JC-1 or 5 μM MitoSOX in HBSS for 10 min at 37°C, washed with HBSS, and treated with 5 mM ATP for 20 min at 37°C followed by 3 washes with HBSS. Finally, the cells were observed under wide-field fluorescence microscope (Leica, Deerfield, IL). Quantitative analysis was performed using Image J software from NIH.
2.5. RNA extraction, reverse transcription, PCR and Quantitative PCR
Total RNA was isolated from GECs after indicated treatments using TRIzol reagent per the manufacture’s protocol. Total RNA was reversed transcribed to cDNA using Taqman Reverse Transcription Reagents kit (Applied Biosystem) followed by PCR or qPCR as previously described [22]. The following primers were used: 5′-GAAACCGCAGATCTCACCAT-3′ and 5′-TTGTAGTGACCCGGAGGAAC-3′ for NLRX1; 5′-AATCTGGCAACCCTAGTCTGCTA-3′ and 5′-AGAAACCAAGGCACAGTGGAA-3′ for IL-8; 5′-TTAAAAGCAGCCCTGGTGAC-3′ and 5′-CTCTGCTCCTCCTGTTCGAC-3′ for GAPDH; 5′-CCGCGGTAATACGTATGTCACG-3′ and 5′-TCCGCTTACCTCTCCAGTACTC-3′ for 16S rRNA of F. nucleatum.
2.6. Immunofluorescence staining
For NF-κB staining, cells infected with F. nucleatum were washed with HBSS, fixed with 4% paraformaldehyde and permeabilized followed by 0.1% Triton-X for 40 min. Following one wash with PBST (PBS containing 0.05% Tween-20) and two washes with PBS, cells were blocked with 2% BSA in PBS for 30 min before hybridization with anti-NF-κB p65 antibody overnight. Cells were then washed with PBST once and PBS twice and incubated with goat anti-rabbit IgG antibody conjugated with red fluorescent Alexa Fluor 546 dye (Invitrogen) for 40 min. DAPI was then used to stain nuclei for another 10 min followed by washing with PBST once and PBS twice, and the fluorescence was visualized under wide-field fluorescence microscope (Leica) or laser scanning confocal microscope (Eclipse Ti C1, Nikon).
Nuclear NF-κB p65 intensity was quantified by CellProfiler. Cells with nuclear p65 mean intensity over 0.1 were considered as positive, and at least 350 cells were analyzed under each condition.
For ASC staining, the procedure is like for NF-κB staining except the cells were permeabilized for 20 min and anti-ASC and goat anti-rabbit IgG (H+L) secondary antibody conjugated with Alexa Fluor 488 were used. Fluorescence intensity was analyzed using ImageJ. Mean fluorescence intensity of ASC was normalized to total fluorescence intensity of nuclei. At least 150 cells were analyzed under each condition.
2.7. Nuclear/cytosol fractionation and Western blots
To determine NF-κB protein levels in nuclear and cytosol, these extracts were prepared using Nuclear/Cytosol Fractionation kit from BioVision. Briefly, 2 × 106 uninfected or infected cells were collected and lysed with Cytosol Extraction Buffer-A (CEB-A) containing DTT and protease inhibitors. Following vigorously vortex and cold incubation on ice, CEB-B were added and mixed before centrifugation at 16,000 g at 4°C. The supernatant (cytoplasmic extract) was collected for further analysis, and the pellet was resuspended using Nuclear Extraction Buffer (NEB). The mixture was vortexed for 15s every 10 min for a total 40 min and finally centrifuged at 16,000 g for 10 min at 4°C. The supernatant (nuclear extract) was collected for future use.
Preparation of supernatants for caspase-1 detection and Western blot were performed as previously described [22, 9].
2.8. MTS assay
MTS assay was conducted as previously described using CellTiter 9 AQueous One Solution Cell Proliferation Assay kit from Promega [21].
2.9. Statistical analysis
Immunoblot and fluorescence microscopy data were quantitatively analyzed using ImageJ software or CellProfiler software. Statistics were carried out using two-way ANOVA with Bonferroni’s post hoc test. For experiments examining knockdown efficiency, one-way ANOVA was used. A p value less than 0.05 was considered significant.
3. Results
3.1. Attenuated ATP-induced mROS in NLRX1-deficient GECs
We have previously reported that a DAMP, extracellular ATP, induces mROS generation to activate the NLRP3 inflammasome in GECs [22]. However, the mechanism by which mROS were generated remained to be investigated. NLRX1, the only NLR localized in mitochondria, has been shown to mediate mROS production during microbial infection [56, 2, 59]. Therefore, we depleted GECs of NLRX1 using shRNA specific against NLRX1 or tGFP as a control. qRT-PCR and Western blot confirmed that NLRX1 was efficiently depleted in GECs transduced with shRNA sequence 325 and 446, compared with the tGFP control (Fig. 1, A and B).
Fig. 1. Depletion of NLRX1 reduced ATP-induced mROS in GECs.
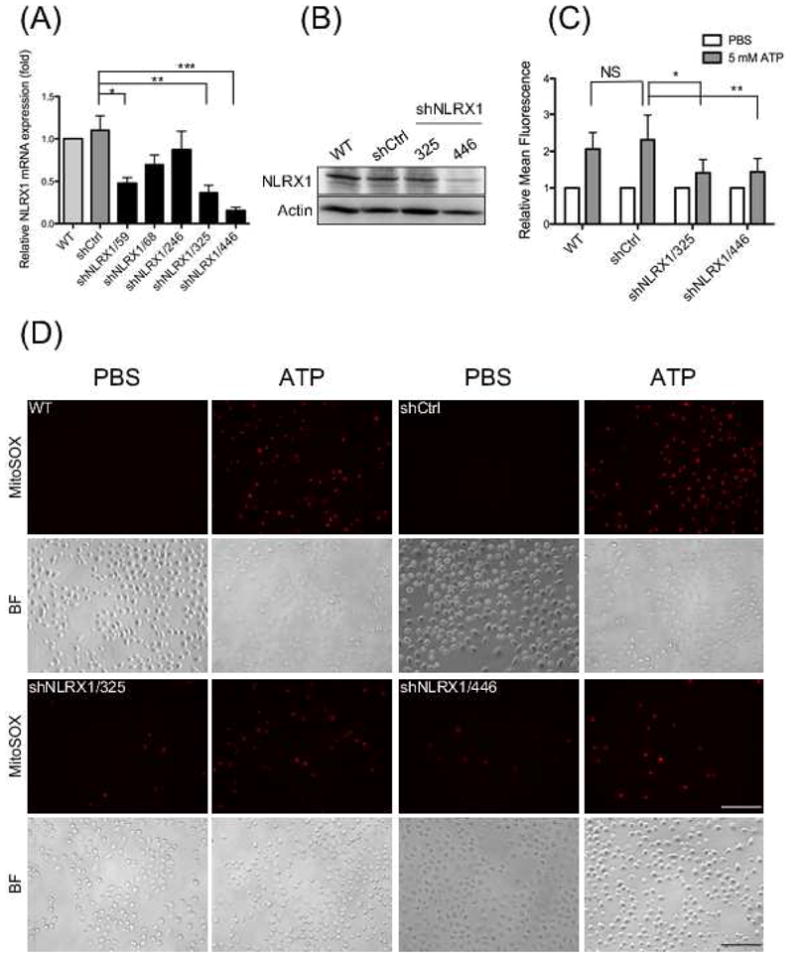
GECs were transduced with lentivirus carrying specific shRNA targeting NLRX1 (A) NLRX1 mRNA levels were measured by qRT-PCR in GECs expressing different shRNA. (B) Whole cell lysates were collected to measure protein expression levels of NLRX1. Actin was used as an internal control. (C and D) mROS generation was detected by fluorescence microscopy using MitoSOX after 5 mM ATP stimulation for 20 min. Quantification of mROS production were analyzed by ImageJ as in (C). BF represents bright field. Mean fluorescence intensity was obtained by dividing total fluorescence intensity against total number of cells in each image. Relative mean fluorescence is determined by normalizing with control group in each cell lines. Scale bars represents 100 μm. Statistical significance determined using one-way ANOVA for (A) and two-way ANOVA for (C). *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001. Data were from a representative of at least three independent experiments performed in duplicates or triplicates that gave similar results.
We next measured ATP-induced mROS generation in cells stained with MitoSOX, which specifically detects mitochondrial superoxide. Fluorescence microscopy showed that ATP stimulated a large increase in mROS production in wild-type GECs and GECs transduced with shRNA control, but lower levels of mROS were observed in the NLRX1-depleted cells. Quantification analysis of the fluorescence images showed that ATP-promoted ROS production decreased ~25% in NLRX1-depleted cells compared with wild type cells (Fig. 1C and D). These results suggest that NLRX1 enhances ATP-triggered mROS generation.
3.2. Depletion of NLRX1 reduced ATP-induced mitochondrial damage
Recent studies revealed a critical role of mitochondria in NLRP3 inflammasome activation [53, 64]. Thus, NLRP3 agonists such as ATP disrupt the mitochondrial membrane potential, which results in mROS generation and subsequent release of oxidized mitochondrial DNA to the cytosol, where it activates the NLRP3 inflammasome [53]. We therefore examined the effect of NLRX1 on mitochondrial membrane potential (Δψm) in GECs with the mitochondrial membrane potential probe, JC-1. The dye JC-1 shits profoundly from red to green in both WT and shCtrl GECs upon ATP stimulation, suggesting that ATP triggered a loss in Δψm. However, depletion of NLRX1 reduced the ATP-elicited Δψm decrease (Fig. 2A). Quantitative analysis of the fluorescence micrographs confirmed that depletion of NLRX1 abrogates the ATP-induced Δψm decrease (Fig. 2B).
Fig. 2. Suppression of ATP-elicited mitochondrial dysfunction in NLRX1-depleted GECs.

(A) GECs depleted of NLRX1 were stained with 5 μg/ml JC-1 followed by 5 mM ATP treatment for 20 min. The treated cells were subject to fluorescence microscopy for the analysis of the loss of mitochondrial membrane potential (ΔΨm). Red indicates JC-1 aggregates in healthy cells with high membrane potential. The red aggregates shits to green monomer in cytosol when mitochondrial potential is depolarized. Scale bars: 100 μm. (B) Quantification of mitochondrial membrane depolarization was obtained by dividing total fluorescence intensity of red J-aggregate over green monomer followed by normalization to the PBS control group. Data shown are representative of two or more independent experiments performed in duplicates (means ± SD). Statistical significance was determined using two-way ANOVA. ***P-value < 0.001. (C) Cytosolic fractions were isolated from GECs treated with or without 5 mM ATP for 1 h followed by Phenol-Chloroform-Isoamyl Alcohol precipitation. The cytosolic DNA were subject to PCR to detect mitochondrial gene CytB and genomic GAPDH. Data shown are representative of two or more independent experiments (means ± SD).
ATP-induced mROS also induces the cytosolic translocation of mitochondrial DNA (mtDNA), which is recognized by the NLRP3 inflammasome [40, 53]. We therefore examined whether NLRX1 had an effect on ATP-triggered mtDNA release to cytosol in GECs. Cytosol were separated and collected from ATP-treated or untreated cells to measure mitochondrial DNA encoding Cyt b by PCR. Incubation with ATP significantly increased the level of cytosolic mtDNA in GECs, whereas NLRX1 depletion abrogated the enhancement (Fig. 2C and 2C). Genomic DNA encoding Gapdh was not detected in the cytosolic samples, excluding the possibility of nonspecific contamination from organelles. Taken together, these results suggest that NLRX1 protects against ATP-caused mitochondrial damage.
3.3. Diminished NLRP3 activation in NLRX- depleted GECs
Mitochondrial dysfunction-caused cytosolic translocation of mitochondrial DNA is sensed by NLRP3, which triggers NLRP3 inflammasome assembly [53]. Assembly of the NLRP3 inflammasome requires recruitment of the adaptor protein ASC (apoptosis-associated speck-like protein containing CARD) at perinuclear mitochondria-associated membranes (MAMs) of the endoplasmic reticulum (ER) [64, 38, 35]. To determine whether NLRX1 is involved in the redistribution of ASC, the cells were treated with ATP for 10 min followed by immunofluorescence assays. Under untreated conditions, ASC was barely detectable due to its general distribution throughout the cytosol. However, in ATP-stimulated GECs with or without shRNA against tGFP, ASC became predominantly perinuclear, and NLRX1-depletion blocked the ATP-induced ASC redistribution (Fig. 3A, 3B, and Fig. S1). In addition, the protein abundance of ASC was not altered by the treatment (Fig. 3C). We also examined whether NLRX1 contributes to F. nucleatum infection-triggered ASC redistribution. As Fig. S2 shows, F. nucleatum infection leads to ASC to become concentrated to in the perinuclear region, consistent with our previous observation that NLRP3 inflammasome is activated by infection [9]. NLRX1 depletion also suppressed the perinuclear relocalization of ASC by infection (Fig. S2). Taken together, these data suggest that NLRX1 abolishes ATP- and F. nucleatum- triggered ASC redistribution rather than affecting ASC expression.
Fig. 3. NLRX1-depletion led to lower ATP-mediated NLRP3 inflammasome activation.

(A) GECs were treated or untreated with 5 mM ATP for 10 min before fixation. The cells were then immunostained for ASC (green) antibody and subjected to confocal microscopy. Nuclei were stained by DAPI (blue). Scale bar, 20 μm. (B) Fluorescence microscopy images were quantified by dividing mean ASC fluorescence intensity over total DAPI fluorescence intensity of each cell. Statistical significance was determined using two-way ANOVA. **P-value < 0.01; ***P-value = 0.001. (C) Western blot analysis of ASC protein levels of whole cell lysates from GECs treated as in (A). Actin was chosen as loading control. (D) Caspase-1 protein levels were examined by Western blot after GECs were left untreated or treated with 5 mM ATP for 1 h. Actins was used as internal control. Data shown are representative of two or more independent experiments (means ± SD).
NLRP3 inflammasome assembly with ASC leads to autoprocessing and activation of caspase-1, which in turn cleaves pro-IL-1β to IL-1β [1]. To determine whether the compromised redistribution of ASC affected NLRP3 inflammasome activation, we measured the protein level of activated caspase-1 by Western blots. As expected, ATP stimulation significantly enhanced caspase-1 activation in wt and shCtrl GECs. The level of caspase-1 was also enhanced in NLRX1-depleted cells after ATP stimulation, but to a lesser extent than in wild-type cells. Caspase-1 activation was impaired more severely in cells transduced with shNLRX1 sequence 446, which may be due to the better knockdown efficiency of NLRX1 with this sequence (Fig. 3D). Taken together, these data suggest that NLRX1 regulates positively NLRP3 inflammasome activation.
3.4. NLRX1 enhances caspase-1 activation during F. nucleatum infection
Previous studies have shown that F. nucleatum infection activates the NLRP3 inflammasome [58, 9]. Since NLRX1 modulates ATP-induced NLRP3 inflammasome activation, we hypothesized that NLRX1 is also involved in F. nucleatum mediated NLRP3 inflammasome activation. We therefore examined caspase-1 activation during infection of GECs by F. nucleatum, and observed that infection-induced caspase-1 activation decreased in NLRX1-deficient cells, compared with controls. Cell viability was not affected by the depletion of NLRX1 and was not significantly affected by infection (Fig. 4 and Fig. S3) [9] [25, 15]. Taken together, these results suggest that NLRX1 positively modulates F. nucleatum infection-elicited NLRP3 inflammasome activation.
Fig. 4. NLRX1 depletion decreased F. nucleatum-triggered caspase-1 activation.
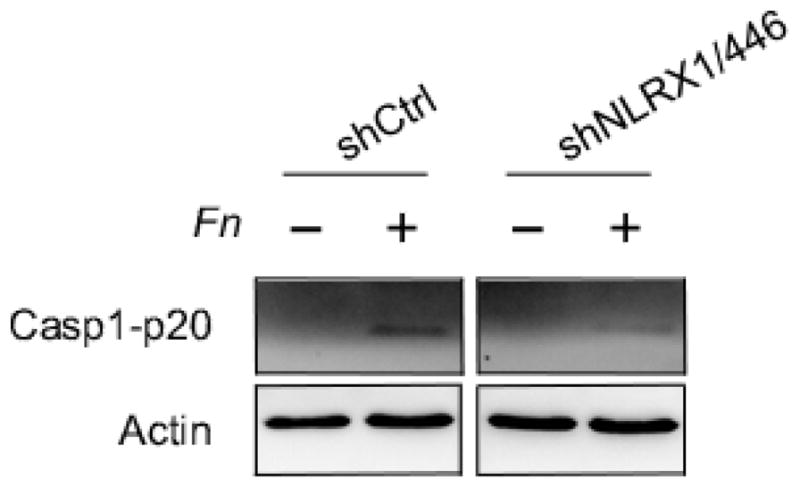
GECs were infected with or without F. nucleatum (Fn) at an MOI of 100 for 24 h. Supernatants were collected and subjected to TCA precipitation. Cells were lysed in RIPA buffer. Western blot was assayed to determine the protein levels of caspase-1 in supernatants (extracellular) and whole cell lysates (intracellular). Actin served as loading control. Data are representative of three independent experiments.
3.5. Enhanced IL-8 gene expression in NLRX1-deficient cells during F. nucleatum infection
IL-8 is expressed via NF-κB signaling to recruit neutrophils in response to F. nucleatum infection [11, 63]. However, the mechanism whereby F. nucleatum increases IL-8 expression remains elusive. NLRX1 has been reported to negatively regulate NF-κB signaling by targeting the IKK complex [61]. Therefore, we examined whether NLRX1 participates in F. nucleatum-induced IL-8 expression. Cells were infected with or without F. nucleatum at a multiplicity of infection (M.O.I.) of 50 and 100, and IL-8 expression was then measured by qPCR. As shown in Fig. 5A, IL-8 gene expression increased as a function of F. nucleatum dose in shCtrl and wildtype cells, but infection in NLRX1-deficient cells led to significantly higher IL-8 expression. 16S rRNA gene was examined by RT-PCR to confirm infection (Fig. 5B). These data suggest that NLRX1 negatively regulates F. nucleatum infection-induced IL-8 expression.
Fig. 5. NLRX1 depletion enhanced IL-8 expression upon F. nucleatum infection.

(A) GECs were left uninfected or infected with F. nucleatum at an M.O.I. of 50 for 24 h. RNA extracted from the cells were reverse transcribed to cDNA followed by qPCR to detect the IL-8 transcripts. Statistical significance was determined using two-way ANOVA. ***P-value < 0.001. (B) PCR were used to detect 16s rRNA of F. nucleatum to confirm the infection in (A). GAPDH was used as an internal control. Data are representative of three independent experiments performed in triplicates (means ± SD).
3.6. NLRX1 modulates IL-8 expression during F. nucleatum infection via the transcription factor NF-κB
NLRX1 has been previously reported to negatively regulate NF-κB signaling [61]. To determine whether NLRX1 is involved in F. nucleatum infection-induced IL-8 expression, the nuclear translocation of RelA (p65) subunit of NF-κB was examined in NLRX1 knockdown GECs by confocal microscopy. As expected, infection by F. nucleatum resulted in nuclear translocation of RelA in 30% of wildtype and shCtrl cells, but RelA translocation increased to 60% in NLRX1-deficient cells (Fig. 6A, 6B and Fig. S4). In addition, the cytosolic and nuclear extracts from infected cells were analyzed for RelA by immunoblotting. NLRX1-deficient cells showed increased RelA levels in the nucleus, but reduced RelA in the cytosol during infection, compared to wildtype and shCtrl cells (Fig. 6C). These results suggest that NLRX1 negatively regulates IL-8 expression through its action on NF-κB.
Fig. 6. NLRX1 regulated F. nucleatum-elicited NF-κB nuclear translocation.


(A) Confocal microscopy of GECs uninfected or infected with F. nucleatum at an M.O.I. of 50 for 1h followed by fixation. Cells were then immunostained with anti-NF-κB p65 (red) and stained nuclei with DAPI (blue). Scale bar, 20 μm. (B) Quantitative analysis of fluorescence microscopy images obtained under condition as in (A). Fluorescence intensity of p65 in nuclei and total cells was measured by CellProfiler. Statistical significance was determined using two-way ANOVA. ***P< 0.001. (C) Nuclear and cytosol fraction of cells were isolated followed by immunoblotting using anti-NF-κB p65 antibody. TOPO I and actin served as loading control for nuclear and cytosol fractions, separately. All data are representative of at least three independent experiments performed in duplicates.
4. Discussion
The role of NLRX1 in innate immunity is still under investigation. Initial studies showed that NLRX1 negatively regulates RIG-I-MAVS-dependent IFN-β expression to prevent excessive inflammation during viral infection [62, 3, 43]. In addition, SIV infection leads to upregulation of NLRX1 and downregulation of interferon-stimulated genes (ISGs) at early stages of infection to promote viral dissemination in rhesus monkeys [6]. More recent studies demonstrated that NLRX1 attenuates STING-dependent ISGs upon HIV-1 or DNA virus infection in human macrophages and dendritic cells [14]. However, the role of NLRX1 in immune responses to bacterial infection remains less clear.
F. nucleatum induces NF-κB-mediated IL-8 expression in GECs, which results in neutrophil recruitment to eradicate infection [20, 63]. However, the pattern recognition receptors (PRRs) responsible for sensing the bacteria remain elusive. IL-8 is expressed following stimulation of extracellular Toll-like receptors (TLRs) during most bacterial infections. Nevertheless, it has been reported that some F. nucleatum strains upregulate IL-8 through TLR-independent mechanisms [63, 34]. Transcriptome analysis and a study using TLR-deficient HEK293T cells have shown that mitogen-activated protein kinase (MAPK) signaling also contribute to IL-8 upregulation upon F. nucleatum infection [17, 46]. The ability of F. nucleatum to invade cells suggests that intracellular or endosomal PRRs may also contribute to IL-8 production [46, 16]. Consistent with this possibility, it has been shown that endosomal TLR9 up-regulates IL-8 in response to F. nucleatum DNA in GECs [29]. Furthermore, the crystal structure of NLRX1 suggests that its leucine-rich repeat (LRR) domain could directly sense PAMPs, such as nucleic acids [18]. Here, we demonstrated that depletion of NLRX1 enhanced RelA nuclear translocation, thus augmenting IL-8 expression. Previous research showing that NLRX1 negatively regulates the NF-κB pathway through suppression of TRAF6 is consistent with our results [3, 43, 61]. However, IL-8 can also be regulated by the MAPK pathway, which is also downstream from TRAF6 [37]. Therefore, further study is needed to validate whether NLRX1 modulates F. nucleatum-triggered IL-8 expression via the NF-κB pathway alone.
We recently reported that F. nucleatum infection activates the NLRP3 inflammasome and facilitates the release of DAMPs such as HMGB1 and ASC to amplify inflammation in GECs [9]. Here, we have demonstrated that NLRX1 behaves as a positive regulator in F. nucleatum infection-elicited NLRP3 inflammasome activation. Consistent with our finding, it has been proposed that NLRX1 modulates NLRP3 inflammasome activation during Chlamydia infection [13, 2].
DAMPs released from infected or stressed cells, such as extracellular ATP, could activate the NLRP3 inflammasome through P2X4/P2X7-gated Pannexin-1 channels, which promote mROS generation [22]. Previous reports identified a role for NLRX1 in enhancing ROS production during Shigella infection [56, 61]. Thus, internalized F. nucleatum could destabilize host-cell mitochondria, which results in mROS production, which in turn is regulated by NLRX1 [54, 39, 51, 12, 27]. Interestingly, a recent study revealed up-regulation of NLRP3 expression but down-regulation of NLRX1 in Helicobacter pylori-infected bone-marrow derived macrophages [44, 49].
In this study, we uncovered a surprising role for NLRX1 as a molecular switch between NLRP3 inflammasome activation and NF-κB signaling in GECs during F. nucleatum infection. Depletion of NLRX1 decreases NLRP3 inflammasome activation due to ATP stimulation and F. nucleatum infection. Therefore, we hypothesize that NLRX1 promotes dysregulated inflammatory responses during F. nucleatum infection via the NLRP3 inflammasome in periodontitis. Conversely, commensals or F. nucleatum in healthy individuals may still induce NF-κB-mediated IL-8 expression, but it is repressed by NLRX1. Thus, we propose that NLRX1 should enhance the innate immune response during infection by pathogens but behave as a break to prevent excessive inflammation under normal circumstances (Fig. 7).
Fig. 7.

Model of F. nucleatum infection-triggered inflammatory responses in gingival epithelial cells during healthy (dotted line) or diseased status (solid line). Pathogens and danger signals (DS) such as ATP activate the NLRP3 inflammasome, which is enhanced by NLRX1. Bacteria under non-pathogenic conditions can also stimulate NF-κB activation, which is dampened by NLRX1.
Supplementary Material
Acknowledgments
This work was supported by funds from the University of the Pacific, Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) in Brazil, and the National Institutes of Health grant R01 DE016593.
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi: 10.1016/j.redox.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J Biol Chem. 2010;285:41637–45. doi: 10.1074/jbc.M110.137885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34:854–65. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci. 2009;122:3161–8. doi: 10.1242/jcs.051193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbe F, Douglas T, Saleh M. Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev. 2014;25:681–97. doi: 10.1016/j.cytogfr.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, et al. Rapid Inflammasome Activation following Mucosal SIV Infection of Rhesus Monkeys. Cell. 2016;165:656–67. doi: 10.1016/j.cell.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolstad AI, Jensen HB, Bakken V. Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin Microbiol Rev. 1996;9:55–71. doi: 10.1128/cmr.9.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bui FQ, Johnson L, Roberts J, Hung SC, Lee J, Atanasova KR, et al. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1beta and the danger signals ASC and HMGB1. Cell Microbiol. 2016;18:970–81. doi: 10.1111/cmi.12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaushu S, Wilensky A, Gur C, Shapira L, Elboim M, Halftek G, et al. Direct recognition of Fusobacterium nucleatum by the NK cell natural cytotoxicity receptor NKp46 aggravates periodontal disease. PLoS Pathog. 2012;8:e1002601. doi: 10.1371/journal.ppat.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–5. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182:6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elwell C, Mirrashidi K, Engel J. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol. 2016;14:385–400. doi: 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, Konig R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe. 2016;19:515–28. doi: 10.1016/j.chom.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gursoy UK, Kononen E, Uitto VJ. Intracellular replication of fusobacteria requires new actin filament formation of epithelial cells. APMIS. 2008;116:1063–70. doi: 10.1111/j.1600-0463.2008.00868.x. [DOI] [PubMed] [Google Scholar]
- 16.Han YW, Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H, et al. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–6. doi: 10.1128/iai.68.6.3140-3146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa Y, Mans JJ, Mao S, Lopez MC, Baker HV, Handfield M, et al. Gingival epithelial cell transcriptional responses to commensal and opportunistic oral microbial species. Infect Immun. 2007;75:2540–7. doi: 10.1128/IAI.01957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity. 2012;36:337–47. doi: 10.1016/j.immuni.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35:253–61. doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang GT, Zhang HB, Dang HN, Haake SK. Differential regulation of cytokine genes in gingival epithelial cells challenged by Fusobacterium nucleatum and Porphyromonas gingivalis. Microb Pathog. 2004;37:303–12. doi: 10.1016/j.micpath.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Huang PR, Hung SC, Pao CC, Wang TC. N-(1-pyrenyl) maleimide induces bak oligomerization and mitochondrial dysfunction in Jurkat Cells. Biomed Res Int. 2015;2015:798489. doi: 10.1155/2015/798489. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, et al. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS One. 2013;8:e70210. doi: 10.1371/journal.pone.0070210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huynh T, Kapur RV, Kaplan CW, Cacalano N, Kinder Haake S, Shi W, et al. The role of aggregation in Fusobacterium nucleatum- induced immune cell death. J Endod. 2011;37:1531–5. doi: 10.1016/j.joen.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 24.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 25.Ji S, Shin JE, Kim YC, Choi Y. Intracellular degradation of Fusobacterium nucleatum in human gingival epithelial cells. Mol Cells. 2010;30:519–26. doi: 10.1007/s10059-010-0142-8. [DOI] [PubMed] [Google Scholar]
- 26.Ji S, Shin JE, Kim YS, Oh JE, Min BM, Choi Y. Toll-like receptor 2 and NALP2 mediate induction of human beta-defensins by Fusobacterium nucleatum in gingival epithelial cells. Infect Immun. 2009;77:1044–52. doi: 10.1128/IAI.00449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–59. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan CW, Lux R, Huynh T, Jewett A, Shi W, Haake SK. Fusobacterium nucleatum apoptosis-inducing outer membrane protein. J Dent Res. 2005;84:700–4. doi: 10.1177/154405910508400803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim Y, Jo AR, Jang da H, Cho YJ, Chun J, Min BM, et al. Toll-like receptor 9 mediates oral bacteria-induced IL-8 expression in gingival epithelial cells. Immunol Cell Biol. 2012;90:655–63. doi: 10.1038/icb.2011.85. [DOI] [PubMed] [Google Scholar]
- 30.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–64. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, Ingle H, Prasad DV, Kumar H. Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol. 2013;39:229–46. doi: 10.3109/1040841X.2012.706249. [DOI] [PubMed] [Google Scholar]
- 32.Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy. 2013;9:432–3. doi: 10.4161/auto.23026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36:933–46. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Redline RW, Han YW. Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4-mediated placental inflammatory response. J Immunol. 2007;179:2501–8. doi: 10.4049/jimmunol.179.4.2501. [DOI] [PubMed] [Google Scholar]
- 35.Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111:7403–8. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martel J, Lai HC, Ko YF, Young JD, Ojcius DM. Alternative functions for the multifarious inflammasome. Biomed J. 2016;39:183–7. doi: 10.1016/j.bj.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClure R, Massari P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front Immunol. 2014;5:386. doi: 10.3389/fimmu.2014.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–60. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 39.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109:11282–7. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Neill LA. Innate immunity: squelching anti-viral signalling with NLRX1. Curr Biol. 2008;18:R302–4. doi: 10.1016/j.cub.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 42.Park SR, Kim DJ, Han SH, Kang MJ, Lee JY, Jeong YJ, et al. Diverse Toll-like receptors mediate cytokine production by Fusobacterium nucleatum and Aggregatibacter actinomycetemcomitans in macrophages. Infect Immun. 2014;82:1914–20. doi: 10.1128/IAI.01226-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parvatiyar K, Cheng G. NOD so fast: NLRX1 puts the brake on inflammation. Immunity. 2011;34:821–2. doi: 10.1016/j.immuni.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Philipson CW, Bassaganya-Riera J, Viladomiu M, Kronsteiner B, Abedi V, Hoops S, et al. Modeling the Regulatory Mechanisms by Which NLRX1 Modulates Innate Immune Responses to Helicobacter pylori Infection. PLoS One. 2015;10:e0137839. doi: 10.1371/journal.pone.0137839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One. 2008;3:e2119. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quah SY, Bergenholtz G, Tan KS. Fusobacterium nucleatum induces cytokine production through Toll-like-receptor-independent mechanism. Int Endod J. 2014;47:550–9. doi: 10.1111/iej.12185. [DOI] [PubMed] [Google Scholar]
- 47.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–36. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 48.Said-Sadier N, Ojcius DM. Alarmins, inflammasomes and immunity. Biomed J. 2012;35:437–49. doi: 10.4103/2319-4170.104408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salama NR, Hartung ML, Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–99. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santana PT, Martel J, Lai HC, Perfettini JL, Kanellopoulos JM, Young JD, et al. Is the inflammasome relevant for epithelial cell function? Microbes Infect. 2016;18:93–101. doi: 10.1016/j.micinf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. doi: 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma A, Inagaki S, Sigurdson W, Kuramitsu HK. Synergy between Tannerella forsythia and Fusobacterium nucleatum in biofilm formation. Oral Microbiol Immunol. 2005;20:39–42. doi: 10.1111/j.1399-302X.2004.00175.x. [DOI] [PubMed] [Google Scholar]
- 53.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin JE, Baek KJ, Choi YS, Choi Y. A periodontal pathogen Treponema denticola hijacks the Fusobacterium nucleatum-driven host response. Immunol Cell Biol. 2013;91:503–10. doi: 10.1038/icb.2013.35. [DOI] [PubMed] [Google Scholar]
- 55.Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol. 2011;13:25–36. [PubMed] [Google Scholar]
- 56.Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tattoli I, Killackey SA, Foerster EG, Molinaro R, Maisonneuve C, Rahman MA, et al. NLRX1 Acts as an Epithelial-Intrinsic Tumor Suppressor through the Modulation of TNF-Mediated Proliferation. Cell Rep. 2016;14:2576–86. doi: 10.1016/j.celrep.2016.02.065. [DOI] [PubMed] [Google Scholar]
- 58.Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MT, et al. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem. 2012;287:32791–9. doi: 10.1074/jbc.M112.401737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Unger BL, Ganesan S, Comstock AT, Faris AN, Hershenson MB, Sajjan US. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J Virol. 2014;88:3705–18. doi: 10.1128/JVI.03039-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–17. doi: 10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–53. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao TS, Ting JP. NLRX1 has a tail to tell. Immunity. 2012;36:311–2. doi: 10.1016/j.immuni.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang G, Chen R, Rudney JD. Streptococcus cristatus modulates the Fusobacterium nucleatum-induced epithelial interleukin-8 response through the nuclear factor-kappa B pathway. J Periodontal Res. 2011;46:558–67. doi: 10.1111/j.1600-0765.2011.01373.x. [DOI] [PubMed] [Google Scholar]
- 64.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.