

Abstract
Many eukaryotic transcription factors recognize the epigenetic marker 5-methylcytosine (mC) at CpG sites in DNA. Despite their structural diversity, methyl-CpG-binding proteins (MBPs) share a common mode of recognition of mC methyl groups that involves hydrophobic pockets and weak hydrogen bonds of the CH⋯O type. The zinc finger protein Kaiso possesses a remarkably high specificity for methylated over unmethylated CpG sites. A key contribution to this specificity is provided by glutamate 535 (E535), which is optimally positioned to form multiple interactions with mCpG, including direct CH⋯O hydrogen bonds. To examine the role of E535 and CH⋯O hydrogen bonding in the preferential recognition of mCpG sites, we determined the structures of wild type Kaiso (WT) and E535 mutants and characterized their interactions with methylated DNA by nuclear magnetic resonance spectroscopy (NMR), X-ray crystallography, and in vitro protein–DNA binding assays. Our data show that Kaiso favors an mCpG over a CpG site by 2 orders of magnitude in affinity and that an important component of this effect is the presence of hydrophobic and CH⋯O contacts involving E535. Moreover, we present the first direct evidence for formation of a CH⋯O hydrogen bond between an MBP and 5-methylcytosine by using experimental (NMR) and quantum mechanical chemical shift analysis of the mC methyl protons. Together, our findings uncover a critical function of methyl-specific interactions, including CH⋯O hydrogen bonds, that optimize the specificity and affinity of MBPs for methylated DNA and contribute to the precise control of gene expression.
Graphical abstract
Epigenetic marking of the mammalian genome encodes tissue-specific gene expression, leading to normal development and differentiation of distinct cell lineages.1–3 In mammalian DNA, the principal epigenetic modification is 5-methylcytosine (mC), which is formed by the enzymatic addition of a methyl group to the C5 position of cytosine.4 This epigenetic tag is most commonly found in CpG dinucleotides,5 though methylation at non-CpG sequences occurs more rarely and is important in neurons and embryonic stem cells.6–8 CpG methylation can profoundly alter the structure and transcriptional state of chromatin by blocking or enhancing the binding of regulatory proteins or by directly modifying nucleosome stability and DNA accessibility.9,10 Methylation plays a central role in developmental processes such as X-chromosome inactivation,11 genomic imprinting,12 and transposon suppression13 as well as in carcinogenesis.14 Aberrant methylation can deregulate genes and foster cancer progression: hyper-methylation at regulatory CG islands can silence tumor suppressor genes, while hypomethylation can activate tumor proliferation genes and promote genome instability through DNA rearrangements.15,16
Methylation at CpG sites (i.e., methylation on the cytosines of both DNA strands, forming [mC]pG:Gp[mC]) is read by a set of transcription factors (TFs) known as methyl CpG-binding proteins (MBPs), which translate the signal into a transcriptional response via interaction with chromatin modifiers or help maintain the epigenetic mark by recruitment of DNA methyltransferases.9,17 Until recently, only three classes of mammalian TFs—methyl-CpG-binding domain (MBD) proteins (MeCP2, MBD1, MBD2, and MBD4), C2H2 zinc finger (ZF) proteins [Kaiso (also known as ZBTB33), ZBTB4, ZBTB38, ZFP57, etc.], and SET and RING finger-associated (SRA) domain proteins (UHRF1 and UHRF2)—were known to recognize mCpG sites in DNA.9,17 However, the use of genomewide technologies and high-throughput screens for mCpG “readers” has led to the discovery of a large number of TFs with distinct DNA-binding domains (DBDs) that bind specifically to mCpG sites, both in vitro and in vivo.18 These findings suggest that mCpG-dependent activity may be widespread among mammalian TFs, pointing to potential new mechanisms for epigenetic regulation.
Despite their structurally distinct DNA-binding domains, the MBD and ZF class of MBPs share a common mode of recognition of mCpG sites.17,19 At one (ZF) or both (MBD) DNA strands of the mCpG site, a spatially conserved arginine stacks against the mC methyl group in the major groove and forms a pair of hydrogen bonds with the 3′ guanine (Figure 1 and Figure S1). In addition, the methyl groups of the two symmetric mCs form (i) hydrophobic contacts with nearby aliphatic protein side chains and (ii) noncanonical CH⋯O type hydrogen bonds (direct or water-mediated) with a side chain carboxyl (glutamate or aspartate) or hydroxyl (tyrosine) group or a backbone carbonyl group (Figure 1 and Figure S1). Several studies have recognized the functional importance of CH⋯O hydrogen bonds in protein structure and folding, protein–protein recognition, ligand binding, and enzyme catalysis.20,21 CH⋯O interactions have also been implicated in nucleic acid structure and protein–nucleic acid complexes, including the recognition of 5-methylcytosine in DNA by all three classes of MBPs,22–24 yet direct evidence for their formation at the protein–DNA interface and their contribution to binding energy is currently lacking.
Figure 1.
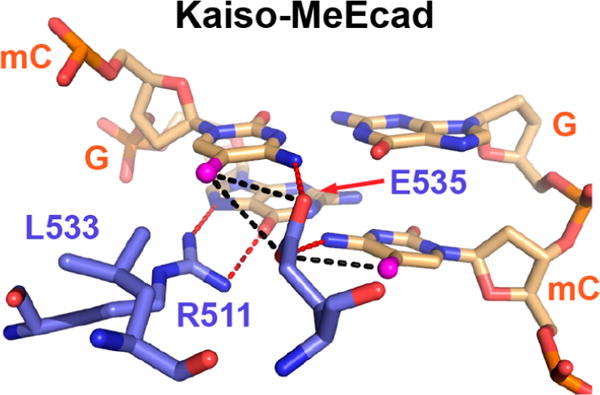
Mode of mCpG recognition in the methylated DNA (MeEcad) complex of Kaiso. Specific major groove interactions are shown between protein residues (blue) and a DNA mCpG site (wheat) in the crystal structure of Kaiso (Protein Data Bank entry 4F6N) bound to methylated DNA.23 mC methyl groups are colored magenta. Proposed canonical (red) and CH⋯O (black) hydrogen bonds are shown as dashed lines.
It is likely that the extent to which MBPs can discriminate methylated from unmethylated CpG sites plays a large part in their selectivity toward target sites and thus in their ability to modulate gene expression. Published studies reveal that preferences of MBPs for methylated over unmethylated CpGs are highly variable, ranging from 2-fold to nearly 2 orders of magnitude.25–28 The ZF proteins Kaiso and Zfp57 have shown exceptionally high selectivity toward mCpG and tend to have more direct or stronger methyl-specific contacts relative to those of other well-characterized ZF MBPs.23,24 What sets Kaiso apart is the optimal orientation of a glutamate (E535), which extends deep into the major groove between the two symmetric mCs and establishes van der Waals contacts and potentially two CH⋯O hydrogen bonds with the mC methyl groups (Figure 1).23 In Zfp57, a corresponding glutamate (E182) adopts two alternative, less optimal conformations and makes a potential CH⋯O hydrogen bond with only one mC methyl group (Figure S1).24 In other ZF proteins, the glutamate is replaced by an aspartate, which cannot protrude as deeply into the major groove and consequently forms weaker or water-mediated CH⋯O hydrogen bonds with the mC methyl groups (Figure S1).29 Importantly, mutation of E535 to alanine in Kaiso and the related protein ZBTB4 abolishes binding to methylated DNA in vitro and in vivo,30 while mutation of the respective glutamate or aspartate has an only modest effect on the mCpG binding affinity of other MBPs.27,31
Kaiso presents a unique opportunity to study the role of methyl-specific contacts, especially CH⋯O hydrogen bonds, in the preferential binding of MBPs to CpG-methylated DNA. Kaiso is a BTB/POZ transcription factor with both a sequence-specific and methylation-dependent DNA binding activity in the cell.32,33 It typically acts by binding to target DNA in gene-regulatory regions via its three adjoining C-terminal C2H2-ZF motifs and recruiting chromatin remodeling factors to gene promoters via its N-terminal POZ domain34,35 to inhibit (and in some cases stimulate36) transcription. Kaiso is known to regulate a large number of genes (for example, those encoding MMP7, MTA2, Cyclin D1, WNT11, and Hif1α) and some miRNAs, which perform diverse functions instrumental to normal development and cancer, including cell cycle regulation, genomic imprinting, hypoxic response, and behavior.37–42 Interactions between the ZF domain and other proteins also play an essential role in Kaiso function; for instance, association with the signaling cofactor p120 catenin in cancer interferes with Kaiso–DNA binding and relieves transcriptional repression.32 Here, to elucidate the role of E535 and CH⋯O hydrogen bonds in the specific recognition of mCpG sites, we examined the methylated DNA binding and structural properties of the zinc finger DNA-binding domain (residues 471–604) of wild type (WT) Kaiso and E535 mutants using X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and biolayer interferometry. Our findings point to a critical role of Kaiso E535 in maintaining mC-specific contacts, including CH⋯O hydrogen bonds that we can visualize directly by NMR, in the preferential recognition of methylated DNA.
EXPERIMENTAL PROCEDURES
Sample Preparation
A uniformly 15N-labeled Kaiso(471–604) construct containing the optimal DNA-binding domain was expressed and purified as previously described.26 Kaiso E535 mutants were generated using standard methods for site-directed mutagenesis. The lyophilized protein was dissolved in NMR buffer [10 mM Tris (pH 7.0) and 1 mM TCEP] and quantified by the absorbance at 280 nm (NanoDrop, Thermo Scientific). The protein was refolded by gradual addition to NMR buffer supplemented with 3.3 molar equivalents of Zn2+ (~20 μM final protein concentration) and stored at −4 °C for up to 2 weeks. All DNA oligonucleotides, including those with 5-methylcytosine and 5′-biotin modifications, were purchased in purified form from IDT (Coralville, IA) and dissolved in NMR buffer (0.1–0.5 mM). Single-stranded DNA was quantified on a Nanodrop (Thermo Scientific) using the absorbance at 260 nm and extinction coefficients provided by the supplier. Duplex DNA was prepared by mixing equal amounts of complementary oligonucleotides, heating to 95 °C, and annealing by slow cooling to room temperature. DNA constructs are listed in Table S1. Kaiso–DNA (1:1.1) complexes were formed by adding the protein to the DNA, desalted, and buffer-exchanged using an Amicon centrifugal filter device (3 kDa) (EMD Millipore), and further concentrated to 0.1–0.2 mM. NMR samples were supplemented with 5% D2O. Kaiso(E535A)–DNA complexes were prepared with a 2-fold molar excess of DNA due to significant protein precipitation at the 1:1.1 ratio. Complex formation for NMR and crystallographic studies was verified by recording one-dimensional imino proton spectra for the DNA and two-dimensional 1H–15N HSQC spectra for the protein.
Biolayer Interferometry
Kaiso–DNA binding kinetic studies were performed on an Octet RED96 system using streptavidin (SA) biosensors (Pall ForteBio). DNA was labeled with biotin at the 5′ end of the top strand (Table S1). The reactions were performed at 298 K in a 96-well plate using the following steps: (1) sensor check (60 s), (2) immobilization of 5′-biotin-labeled DNA (800 nM) onto the biosensor (80 s), (3) blocking and buffer equilibration (300 s), (4) protein association (5–30 min), (5) protein dissociation (5–360 min), (6) sensor regeneration in 2 mM KCl (5 min) followed by (7) buffer equilibration, and repeat steps 6 and 7 twice. Steps 1, 2, and 7 were performed in buffer 1 [10 mM Tris (pH 7.0), 150 mM KCl, and 1 mM TCEP], and steps 3–5 were performed in buffer 2 (buffer 1 with 0.05% Tween 20 and 1 mg/mL dried milk powder). For step 4, four different protein concentrations ranging from 12.5 to 200 nM were typically used, and each data set was repeated at least twice. Assay conditions were extensively optimized because of strong nonspecific Kaiso interactions (i.e., with the biosensor surface, standard blocking reagents such as BSA, DNA, or itself) as well as the large range of protein dissociation times observed. The binding profiles were typically fitted with a two-state binding model using the Octet software, which allowed us to extract the association and dissociation rate constants (kon and koff, respectively) and to obtain dissociation constants (Kd = koff/kon) (Table 1). kon values were alternatively extracted from the slope of a linear fit of the observed association rate constant (kobs) as a function of protein concentration (reported in Table 1). Significant nonspecific binding effects at protein concentrations above 200 nM precluded us from probing higher concentrations that were necessary for accurate determination of kon in the case of weak protein–DNA binding. In the presence of residual nonspecific binding that was sufficiently faster than the protein–DNA koff, the nonspecific portion was removed from the analysis or a three-state binding model was used instead.
Table 1.
Kaiso–DNA Binding Kinetics and Affinities
| DNA construct | koff(s−1) | kon(M−1 s−1) | Kd (nM) | ΔG (kcal/mol) |
|---|---|---|---|---|
| WT | ||||
| MeKBS | (1.3 ± 0.2) × 10−5a | (6.4 ± 0.5) × 104a | 0.20 ± 0.04 | −13.2 |
| MeCG2 | (2.3 ± 0.2) × 10−5 | (5.3 ± 0.6) × 104 | 0.43 ± 0.06 | −12.7 |
| MeCG2semi | (3.7 ± 0.3) × 10−5 | (4.6 ± 0.8) × 104 | 0.8 ± 0.2 | −12.4 |
| MeCG2hemi | (5.8 ± 0.2) × 10−5 | (4.0 ± 0.5) × 104 | 1.4 ± 0.2 | −12.0 |
| CG2 | (400 ± 26) × 10−5 | b | (88 ± 10) | (−9.6) |
| E535Q | ||||
| MeKBS | (39 ± 4) × 10−5 | (5.4 ± 0.4) × 104 | 7.2 ± 0.9 | −11.1 |
| MeCG2 | (84 ± 4) × 10−5 | (4.2 ± 0.2) × 104 | 20 ± 1 | −10.5 |
| MeCG2semi | (190 ± 6) × 10−5 | (4.2 ± 0.5) × 104 | 45 ± 5 | −10.0 |
| MeCG2hemi | (80 ± 12) × 10−5 | (4.4 ± 0.7) × 104 | 18 ± 4 | −10.5 |
| CG2 | (990 ± 50) × 10−5 | b | (220 ± 30) | (−9.0) |
| E535A | ||||
| MeKBS | (160 ± 16) × 10−5 | b | (35 ± 5) | (−10.1) |
| MeCG2 | (340 ± 22) × 10−5 | b | (76 ± 9) | (−9.7) |
| MeCG2semi | (400 ± 27) × 10−5 | b | (89 ± 11) | (−9.6) |
| MeCG2hemi | (200 ± 14) × 10−5 | b | (44 ± 5) | (−10.0) |
| CG2 | (1400 ± 210) × 10−5 | b | (310 ± 60) | (−8.8) |
koff values represent the mean and standard deviation of four or more individual kinetic measurements. kon values represent the mean and standard deviation of two or more linear fits of the observed association rate constant as a function of protein concentration (also see Experimental Procedures).
kon could not be reliably determined for values in parentheses (see Experimental Procedures for details). Approximate Kd values (shown in parentheses) were estimated using the measured koff and the average value of kon [(4.5 ± 0.5) × 104 M−1 s−1] determined for binding of Kaiso to other DNA sequences.
X-ray Crystallography
Crystallization trials for Kaiso–MeCG2 and Kaiso–KBS complexes were performed using the automated Rigaku Crystalmation system of the Joint Center for Structural Genomics (JCSG) at The Scripps Research Institute (TSRI). A subset of these conditions was replicated manually on a larger scale using sitting drop vapor diffusion, where protein and mother liquor were mixed in a 1:1 ratio in a 2 μL drop. The final crystallization conditions for the Kaiso–MeCG2 complex were 0.1 M Tris (pH 8.5), 0.2 M ammonium acetate, and 30% 2-propanol (4 °C). The final conditions for Kaiso–MeKBS, Kaiso–MeKBShemi, and Kaiso(E535Q)– MeKBS complexes were 0.2 M disodium tartrate (pH ~7.2) and 20% PEG3350 (20 °C). Suitable crystals could not be obtained for Kaiso(E535A)–MeCG2 and –MeKBS complexes, likely because of the low stability and/or conformational exchange. Crystallographic data were collected from cryo-cooled crystals at Stanford Synchrotron Radiation Lightsource (SSRL) beamline 9-2, 11-1, or 12-2 (see Table S2). The data were processed using HKL-2000.43 All structures were determined by molecular replacement with Phaser44 using the published structure of the Kaiso–KBS complex [Protein Data Bank (PDB) 4F6M]23 as a search model. Multiple cycles of manual model building (Coot45) and refinement (Phenix46) were completed. Distance and angle restraints were applied for Zn2+–Cys2His2 coordination. Hydrogen bonding distance restraints were imposed on the three terminal DNA base pairs due to poor electron density. Residues at the N-terminus (471–480) and C-terminus (597–604 in the Kaiso–MeCG2 complex and 602–604 in others) could not be observed. Crystallographic and refinement statistics are summarized in Table S2. Structural figures were rendered using PyMOL.47
NMR Spectroscopy
All NMR spectra of Kaiso–DNA complexes were recorded at 298 K on Bruker 800 and 900 MHz spectrometers equipped with a triple-resonance gradient cryo-probe and room-temperature probe, respectively. Spectra were processed and analyzed using NMRPipe48 and Sparky.49 Standard 1H–15N HSQC spectra of Kaiso–DNA samples (0.1–0.2 mM) in NMR buffer (5% D2O) were acquired. Backbone assignments of Kaiso–MeCG2 and Kaiso–MeKBS complexes were deduced by comparison with published assignments of Kaiso–MeEcad and Kaiso–KBS complexes.23 Weighted average chemical shift perturbations (CSPs) for Kaiso E535Q/A versus WT complexes were calculated from the difference in proton (δH) and nitrogen (δN) chemical shifts in 1H–15N HSQC spectra as follows: CSP = (0.1ΔδN2+ΔδH2H)1/2. Normalized cross peak intensities were obtained by normalizing the peak heights measured in 1H–15N HSQC spectra to a helical residue in the ZF1 helix (value set to 1). DNA methyl resonances were observed at 13C natural abundance using 1H–13C HMQC spectra acquired with a non-uniform sampling (NUS) scheme (30% sampling of 512 complex points) and reconstructed with MDDNMR, as previously described.50 Resonance assignments for 5-methyl-cytosine and certain thymine methyl groups in MeKBS bound to Kaiso were obtained by spectral overlay with complexes containing partially methylated (MeKBSsemi and MeKBShe-mi) and unmethylated (KBSCG2) DNA. DNA methyl assignments for the Kaiso–MeCG2 complex were deduced from those mentioned above.
Density Functional Theory (DFT) Chemical Shift Calculations
The model fragments included mC8 or mC28 nucleotides truncated at the deoxyribose ring with E535 or Q535 side chains truncated at Cα. Hydrogen atoms were added with Phenix, and their geometry was optimized with Gaussian 0951 using basis sets b3lyp/3–21g* (all hydrogens) and subsequently b3lyp/6–311+g(2d,p) (mC methyl and imino hydrogens) with implicit water solvation while other atoms were held fixed. The 3-fold symmetric methyl group was manually rotated with Chimera52 in 10° increments for 120° from a random starting position to sample all unique orientations. NMR chemical shifts for different methyl rotamers were calculated with Gaussian 09 using the GIAO method and the b3lyp/6-311+g(2d,p) basis set with implicit water solvation and corrected using scaling factors obtained at the same level of theory in water solvent, reported elsewhere.53 Differences between the observed (NMR) and calculated (DFT) absolute methyl proton chemical shift values could be due to unknown C–H distances, improper scaling factors, incomplete structural models, or other deficiencies in the method, which should not affect the chemical shift difference (ΔδH) values being compared here.
RESULTS
DNA Constructs
Kaiso binds to a specific DNA sequence termed the Kaiso-binding sequence (KBS) as well as to methylated cytosines in double-stranded mCpG sites.33 The KBS and methylated DNA motifs derived from the E-cadherin promoter (MeEcad) and used in previous structural studies of Kaiso–DNA complexes23 are shown in Figure 2 and Table S1. For the studies presented here, we chose to use variations on a palindromic sequence containing two CpG sites [CG2 (Figure 2)], which was identified as a Kaiso consensus binding site in vivo and is among the most abundant and conserved human promoter elements.35 This sequence was prepared unmethy-lated (CG2), fully methylated (MeCG2), semimethylated (MeCG2semi), and hemimethylated (MeCG2hemi) (Figure 2). A second series of experiments were performed using a variant of the KBS sequence containing two CpG sites. The sequences of the unmethylated (KBSCG2), fully methylated (MeKBS), semimethylated (MeKBSsemi), and hemimethylated (MeKBShemi) forms are shown in Figure 2. We refer to C8pG9 as the 5′ CpG site and C10pG11 as the 3′ CpG site. For most NMR and X-ray studies, we chose to use constructs based on MeKBS rather than MeCG2, because complexes of Kaiso with MeKBS oligonucleotides were better behaved in solution, produced diffraction-quality crystals with greater efficiency, and avoided complications associated with preparation of homogeneous partially methylated samples with the palindromic MeCG2 sequence.
Figure 2.

Sequences of DNA constructs used in this study and in the published studies for KBS and MeEcad.26 Shown are the central 10 bp around the core Kaiso-binding site (boxed). Residues are numbered as indicated for MeEcad, and methylated cytosines are colored red.
Biolayer Interferometry Studies of Kaiso–DNA Binding
Earlier in vitro studies comparing the binding affinity of Kaiso for DNA in the presence and absence of methylation were qualitative in nature or performed in the absence of salt.23,26,33 To examine the effect of cytosine methylation and the role of E535 in Kaiso binding at salt concentrations that more closely approach physiological levels, we employed biolayer interferometry (Octet) to measure the kinetics of association and dissociation of Kaiso using biotin-tagged DNA immobilized onto a streptavidin-coated biosensor. The kinetic measurements were fitted to the appropriate binding model to extract the association and dissociation rate constants (kon and koff, respectively) and to obtain dissociation constants (Kd) for protein–DNA binding.
Biolayer interferometry results are summarized in Table 1, and representative binding and dissociation curves are shown in Figure S2. We found that Kaiso binds to doubly methylated MeCG2 with subnanomolar binding affinity (Kd = 0.43 nM) and exhibits very slow dissociation kinetics (koff = 2.3 × 10−5 s−1) (Table 1). Symmetric methylation at a single CpG site (MeCG2semi) or double methylation on a single strand (MeCG2hemi) resulted in a small decrease in DNA binding affinity, 2- and 3-fold, respectively, mainly due to faster dissociation. By contrast, the complete absence of methylation (CG2) dramatically reduced DNA binding affinity (Kd ~ 88 nM), due to a roughly 200-fold increase in koff, as compared to that of doubly methylated DNA. These trends agree well with a previous gel shift study of Kaiso binding to the MeEcad motif performed under low-salt conditions.26
Kaiso also binds to the doubly methylated MeKBS motif33 with subnanomolar binding affinity (Kd = 0.20 nM), ~2-fold tighter than the affinity for the consensus MeCG2 motif due to even slower dissociation (Table 1). This implies that Kaiso can recognize double mCpG sites with comparable binding affinity in different sequence contexts.
A previously published structure of Kaiso with MeEcad23 showed that a key contribution to the high selectivity for mCpG comes from residue E535, which protrudes deep into the major groove, stacks against both mC methyl groups, and maintains direct NH⋯O and CH⋯O hydrogen bonds with the two methylated cytosines (Figure 1). The binding data for two Kaiso mutants, E535Q and E535A, with the same methylated DNAs (Table 1) showed that mutation of E535 to alanine substantially reduces the affinity of Kaiso for fully and partially methylated DNA, primarily as a consequence of a dramatic increase in koff relative to that of the WT protein: 150-fold for MeCG2, 110-fold for MeCG2semi, and 35-fold for MeCG2he-mi (Table 1). The koff values are comparable and do not favor doubly methylated MeCG2, suggesting that Kaiso E535A does not discriminate between different methylation states. The more conservative mutation of E535 to glutamine (E535Q), which replaces the negatively charged carboxyl with a carboxamide group, leads to smaller but sizable effects on Kaiso–DNA dissociation and binding affinity. For Kaiso E535Q, koff increased by 37-fold for MeCG2, 50-fold for MeCG2semi, 14-fold for MeCG2hemi, and 30-fold for MeKBS as compared to that of WT Kaiso (Table 1). By contrast, for the unmethylated CG2 motif, koff increased only 2.5- and 3.5-fold for the E535A and E535Q mutants, respectively.
NMR Spectroscopy of Kaiso–DNA Complexes
The backbone amide 1H–15N HSQC NMR spectrum provides an excellent diagnostic for the formation of Kaiso–DNA complexes26 and can be used to pinpoint small structural differences between them. An overlay of the HSQC spectra of Kaiso(471–604) bound to MeKBS, MeKBShemi, and MeKBSsemi is shown in Figure 3A; the chemical shift changes between MeKBS and the two partially methylated complexes are plotted as a function of residue number in Figure 3B. The chemical shift signatures for all complexes are very similar, except in the regions of the protein closest to the substituted mC sites in the major and minor groove. The largest chemical shift changes between the WT and MeKBSsemi complexes are localized to the regions of ZF1 and of the C-terminal tail, which make major and minor groove contacts, respectively, with the 3′ CpG site. Similarly, the largest shift differences between the WT and MeKBShemi complexes reflect subtle changes in DNA contacts associated with the absence of the methyl groups at C26 and C28 (Figure 2). By contrast, the HSQC spectra of E535A and E535Q mutant proteins show significant differences in both chemical shift and cross peak intensity compared to those of the WT protein in complex with MeKBS (Figure 3C,D) and MeCG2 (Figure S3). In particular, the cross peaks of the E535A mutant are severely attenuated for most of the protein when it is bound to MeCG2, but not in the complex with MeKBS (Figure S3C,D), likely due to conformational exchange. Therefore, we used MeKBS rather than MeCG2 for most subsequent NMR studies.
Figure 3.
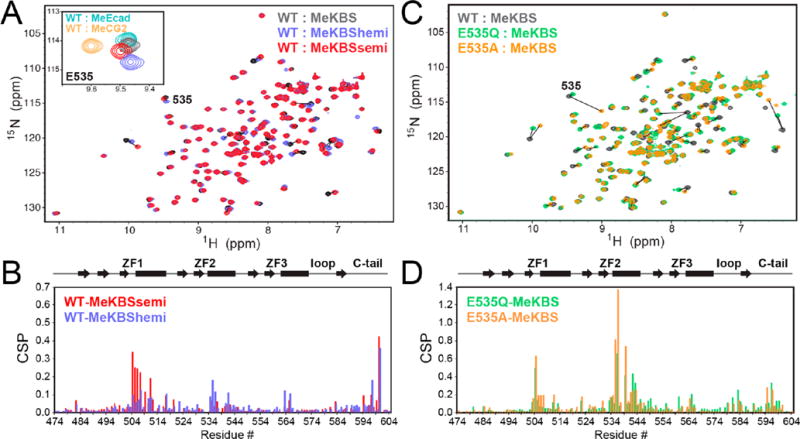
NMR spectra of complexes of Kaiso with methylated DNA. (A) Overlay of 1H–15N HSQC spectra of WT Kaiso bound to MeKBS (gray), MeKBShemi (blue), and MeKBSsemi (red). The inset shows a close-up of the E535 cross peak in these complexes, together with Kaiso–MeEcad (cyan) and Kaiso–MeCG2 (yellow) complexes. (B) Weighted average 1H–15N chemical shift differences [CSP = (0.1ΔδN2 + ΔδH2)1/2] between the backbone amide resonances of Kaiso in complex with MeKBS and MeKBShemi (blue) and in complex with MeKBS and MeKBSsemi (red), plotted as a function of residue number. (C) Overlay of 1H–15N HSQC spectra of Kaiso–MeKBS complexes of WT Kaiso (gray), E535Q (green), and E535A (orange). (D) CSPs between the backbone amide resonances of the MeKBS complexes of WT Kaiso and E535Q (green) and E535A (orange) as a function of residue number. Note that the y-axis scales in panels B and D differ by a factor of 2.
Crystal Structures of Kaiso–DNA Complexes
Crystal structures were determined for complexes of Kaiso bound to MeCG2 (2.4 Å), MeKBS (2.4 Å, two structures), and MeKBShemi (2.0 and 2.1 Å, two structures) and for Kaiso E535Q bound to MeKBS (2.3 Å) (statistics reported in Table S2). Unfortunately, complexes of Kaiso E535A with methylated DNA failed to crystallize and could be examined by only NMR. The independently determined structures of the MeKBS complexes [MeKBS(1) and MeKBS(2)] are almost identical, as are the two structures of the MeKBShemi complex (Figure S4). The structures of the MeCG2 and MeKBS complexes in the vicinity of the 5′ mCpG-binding site are shown in panels A and B of Figure 4 and are superimposed in Figure 4C to illustrate the small structural differences. Omit maps are shown in Figure S5. In each complex, Kaiso recognizes the 5′ mCpG site in the same way as in the Kaiso–MeEcad complex23 (Figure 4D). However, the higher resolution of the structures reported here reveals new details, such as a conserved water molecule that forms a hydrogen bonded network linking R511 NηH, E535 Oε1, and the N7 atom of G27 (Figure S6C). The R511 guanidinium group forms a direct NH⋯N hydrogen bond to the N7 atom of G9 and a bifurcated hydrogen bond to the O6 atoms of G9 and G27 (Figure S6C), likely the structural basis for its important role in recognition of the guanine bases in the two 5′ CpG sites. The carboxyl oxygen atoms of E535 form hydrogen bonds across the major groove to the N4 amino nitrogens of mC8 and mC28. The distances between the mC8 methyl group and the Oε1 and Oε2 atoms of E535 (Table S3) are well within the van der Waals distance cutoff of 3.7 Å, consistent with CH⋯O hydrogen bond formation in each of the complexes.20,54 Oε1 is also in close contact with the mC28 methyl [mC28(C5A)–E535(Oε1) distance of 3.4–3.5 Å]. Similar backbone amide chemical shifts are observed for E535 in 1H–15N HSQC spectra of each complex (Figure 3A, inset), consistent with the conserved DNA contacts seen in the X-ray structures.
Figure 4.
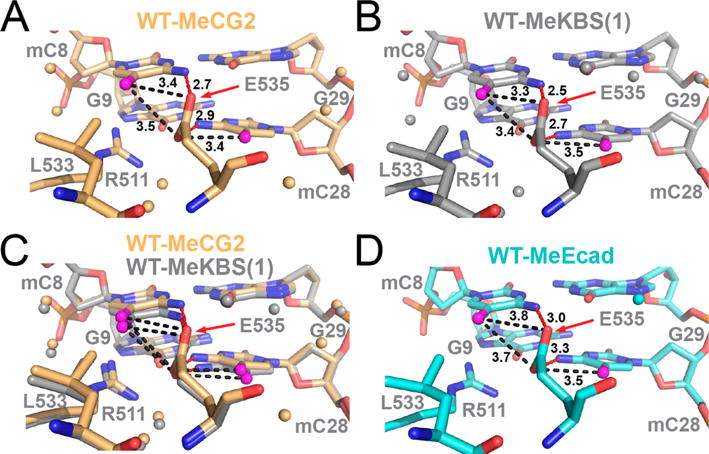
Structural comparison of the 5′ mCpG E535-binding interface in complexes of WT Kaiso with methylated DNA. (A) Complex of Kaiso with MeCG2 (yellow). (B) Complex of Kaiso with MeKBS (gray); MeKBS(1) refers to structure (1) reported in Table S2. (C) Overlay of the complexes of WT Kaiso with MeKBS and MeCG2. (D) Complex of Kaiso with MeEcad (cyan, PDB entry 4F6N). In each panel, the methyl carbon (C5A) of mC8 and mC28 is colored magenta and water molecules are shown as small spheres. Dashed lines show NH⋯O hydrogen bonds (red) between mC8(N4) and E535(Oε2), and between mC28(N4) and E535(Oε1), and potential CH⋯O hydrogen bonds (black) between mC8(C5A) and E535(Oε2) and between mC28(C5A) and E535(Oε1) (alternate CH⋯O hydrogen bonds with the E535 acceptor swapped are also possible but not shown for the sake of clarity). Donor–acceptor bond lengths are labeled in angstroms.
The crystal structures of Kaiso bound to MeKBShemi indicate a similar mode of DNA binding, where the 5′ hemi-mCpG sites are recognized the same way as symmetrically methylated mCpG sites (Figure 5A,B). The absence of the methyl group at C28 in MeKBShemi is accompanied by a slight shift of the E535 side chain away from the mC8 methyl, relative to its position in the MeCG2 and MeKBS complexes (Figure 5B); as a result, the potential CH⋯O hydrogen bonding distances are increased by 0.1–0.3 Å (Table S3). This subtle conformational change is reflected in a noticeable change in the 15N chemical shift for the backbone amide of E535 (Figure 3A, inset).
Figure 5.
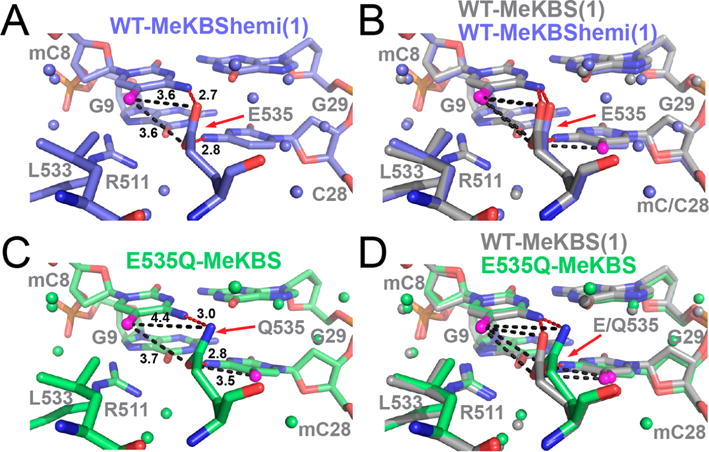
Effect of hemimethylation and E535Q mutation on the 5′ mCpG E535-binding interface in complexes of Kaiso with methylated DNA. (A) Complex of WT Kaiso with MeKBShemi (blue) (structure (1) reported in Table S2). (B) Overlay of the complexes of WT Kaiso with MeKBS and MeKBShemi. (C) Complex of Kaiso E535Q with MeKBS (green). (D) Overlay of the complexes of Kaiso WT and E535Q with MeKBS. In each panel, the methyl carbon (C5A) of mC8 and mC28 is colored magenta, water molecules are shown as small spheres, and donor–acceptor bond lengths are labeled in angstroms (see Figure 4 for a detailed description).
The structure of the MeKBS complex of the Kaiso E535Q mutant in the vicinity of the 5′ mCpG site is shown in Figure 5C and compared with that of the WT complex in Figure 5D. The E535Q mutation, which weakens binding to MeKBS, MeCG2, and MeCG2semi DNA by up to 50-fold (Table 1), disrupts a canonical NH⋯O hydrogen bond to mC8(N4) and a potential CH⋯O hydrogen bond to mC8(C5A) by removing the acceptor atom [E535(Oε2)]. A small rotation of the Q535 side chain moves the Cδ and Nε2 atoms away from the mC8 methyl by >0.5 Å relative to the WT complex (Figure 5D and Table S3), weakening van der Waals interactions. The rotation seems to prevent a steric clash between the Q535 and mC8 amino groups but keeps the two nitrogens within 3.0 Å of each other, which may allow a weaker Nε2(Hε2) ⋯N4 hydrogen bond to form (bond angle of ~130°). This conformational adjustment has only a small impact on the polar and hydrophobic contacts between Q535 and the symmetrically positioned mC28.
The X-ray structures determined in this work provide new insights into the protein–DNA interactions in the 3′ mCpG site. In contrast to the 5′ mCpG site, which is recognized by way of direct contacts between protein side chains and the mC8 and mC28 bases, interactions between Kaiso and the 3′ mC bases (mC10 and mC26) are mostly water-mediated (Figure 6 and Figure S6). In the structures of the MeCG2, MeKBS, and MeKBShemi complexes, a conserved water molecule is bound close to the edge of mC10, where it acts as a bifurcated acceptor of hydrogen bonds from the C5A methyl [water O–mC10(C5A) distance of 3.2–3.4 Å] and the N4 amino group [water O–mC10(N4) distance of 2.8–3.0 Å] (Figure 6, Figure S5, and Figure S6B). The bound water is also within hydrogen bonding distance of the N4 amino group of mC26 and the side chain hydroxyl oxygen atoms of T507 and S508.
Figure 6.
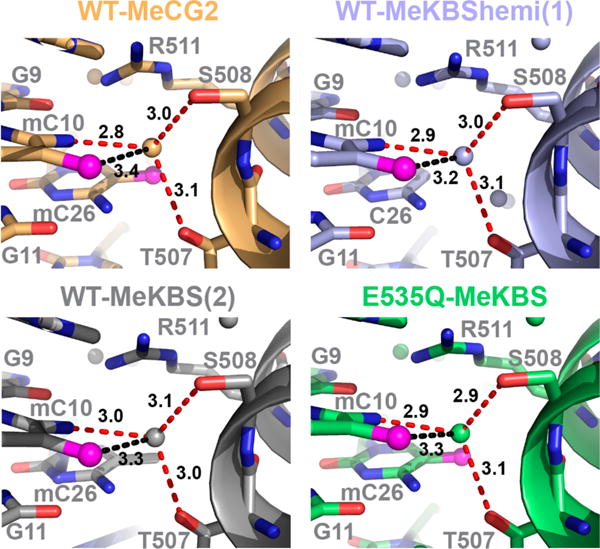
Conservation of the structure of the 3′ mCpG site in the complexes of MeCG2 (yellow), MeKBS (gray), and MeKBShemi (blue) with WT Kaiso and MeKBS with E535Q Kaiso (green). Potential water-mediated NH⋯O (red) and CH⋯O (black) hydrogen bonds between Kaiso T507/S508 and mC10 are shown by dashed lines, and distances are labeled in angstroms.
Direct Evidence for CH···O Hydrogen Bonds from NMR
Because the methyl hydrogen atoms cannot be directly visualized in our current crystal structures, the evidence for CH⋯O hydrogen bonds to the 5-methylcytosines remains indirect and is based on the observation of a C–O distance shorter than the van der Waals contact distance. We therefore adopted a combined approach using NMR and DFT calculation to seek direct evidence for the methyl–oxygen hydrogen bonds. This approach was inspired by a study that showed that methyl protons in S-adenosylmethionine bound to a lysine methyltransferase experience downfield (toward higher parts per million) chemical shift changes (~0.5 ppm) when involved in CH⋯O hydrogen bonds.55
Unlike the Kaiso proteins, the DNA was not isotopically labeled because of the prohibitively large cost of incorporating 13C-labeled methylcytosines site-specifically into DNA. We therefore used a sensitive 1H–13C HMQC experiment with a time-saving non-uniform sampling (NUS) scheme to detect DNA methyl peaks at 13C natural abundance and low sample concentrations (~0.1–0.2 mM) (Figure 7). Unambiguous mC methyl resonance assignments for the Kaiso–MeKBS complex were made by comparison with the spectra of the corresponding complexes with semi-, hemi-, and unmethylated DNA (Figure 7A). The methyl HMQC cross peaks of mC8, and also mC28, exhibit similar chemical shifts in the WT Kaiso complexes, regardless of the DNA sequence or methylation state (Figure 7A). The methyl proton resonance of mC8, but not that of mC28, is shifted upfield in spectra of MeKBS (ΔδH ~ 0.17 ppm) or MeCG2 (ΔδH ~ 0.14 ppm) bound to the E535Q mutant (Figure 7B,C), indicating a change in the local environment of the mC8 methyl group. The chemical shift changes are consistent with disruption of a CH⋯O hydrogen bond between the E535 carboxyl and the mC8 methyl group upon substitution with glutamine, which eliminates the E535(Oε2) acceptor closest to the mC8 methyl carbon and weakens or disrupts the interaction of E535(Oε1) with the mC8 methyl (Table S3).
Figure 7.
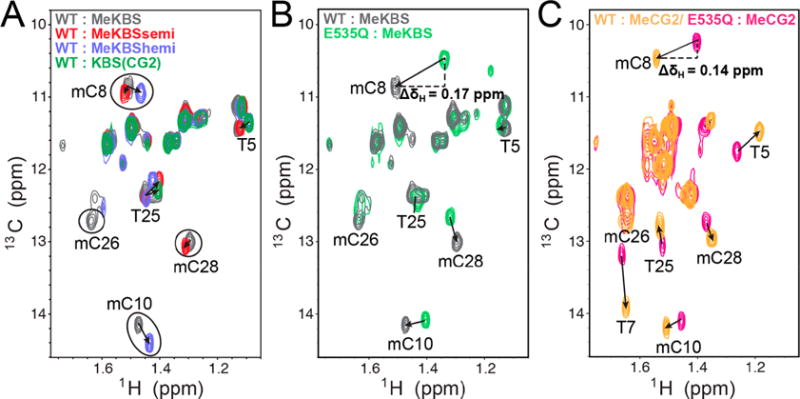
Direct NMR evidence for a CH⋯O hydrogen bond between Kaiso E535 and mCpG methyl groups in DNA. (A) Methyl region of the natural-abundance 1H–13C HMQC spectrum of DNA [MeKBS, gray; MeKBSsemi, red; MeKBShemi, blue; unmethylated KBS(CG2), green] in complex with WT Kaiso. (B) Overlay of the methyl region of the natural-abundance 1H–13C HMQC spectrum of MeKBS with Kaiso WT (gray) and E535Q (green). (C) Overlay of the methyl region of the natural-abundance 1H–13C HMQC spectrum of MeCG2 with Kaiso WT (yellow) and E535Q (magenta).
Given its stronger effect on the affinity of Kaiso for methylated DNA, the effect of the E535A mutation on the DNA methyl spectrum is expected to be even larger than for E535Q. Unfortunately, extensive line broadening and/or overlap of DNA methyl resonances in E535A complexes precluded measurement of the corresponding mC8 methyl proton shifts (Figure S7).
DFT Calculation of mC Methyl Proton Chemical Shifts
To corroborate our NMR studies, we performed DFT calculations for WT Kaiso and the E535Q mutant bound to MeKBS (Figure 8). Chemical shifts were predicted using crystal structure-based fragments that included the mC8 or mC28 nucleotides and the intact E535 and Q535 side chains; control calculations were performed with the side chain truncated at the Cα atom. Because the position of the methyl protons is unknown, the 3-fold symmetric methyl group was rotated through 120° around its C–C axis to sample all unique rotameric states (Figure 8A,B). The difference in the mC8 methyl proton chemical shift relative to the “no side chain” control is shown as a function of rotation angle in Figure 8C. A subset of the methyl rotamers position a methyl proton within hydrogen bonding distance of one of the E535 carboxyl oxygen atoms. In principle, there are two alternate CH⋯O hydrogen bonding schemes in which a cytosine methyl proton interacts with the Oε1 or Oε2 atom of the E535 carboxyl group (Figure 8A,B). For mC8 in WT MeKBS [structure (1) in Table S2], the optimal rotamer for formation of a linear CH⋯Oε1 hydrogen bond (bond angle of ~170°) yielded the most downfield methyl proton chemical shift and largest difference relative to the same rotamer in the absence of the E535 side chain (ΔδH ~ 0.26 ppm) (Figure 8C, magenta bar). A smaller downfield shift (ΔδH ~ 0.18 ppm) is predicted for the methyl rotamer that leads to optimal CH⋯Oε2 hydrogen bonding (bond angle of ~166°) (Figure 8C, orange bar). The decrease in ΔδH correlates with disruption of the optimal CH⋯O hydrogen bonding schemes. Given that CH⋯O interactions are weak,20 there is unlikely to be a strong preference for hydrogen bonding to either Oε1 or Oε2. Population-weighted averaging of the chemical shift over both configurations, and potentially other configurations that form suboptimal hydrogen bonds to both carboxyl oxygens (Figure 8C, gray bar; ΔδH ~ 0.13 ppm), is expected to reduce ΔδH from the maximal predicted value. Smaller changes in ΔδH with the methyl rotamer are calculated for WT–MeKBShemi complexes (~0.07–0.11 ppm), where the C⋯Oε1/Oε2 distances are longer (~3.6–3.7 Å) than for WT–MeKBS complexes (~3.3–3.5 Å).
Figure 8.
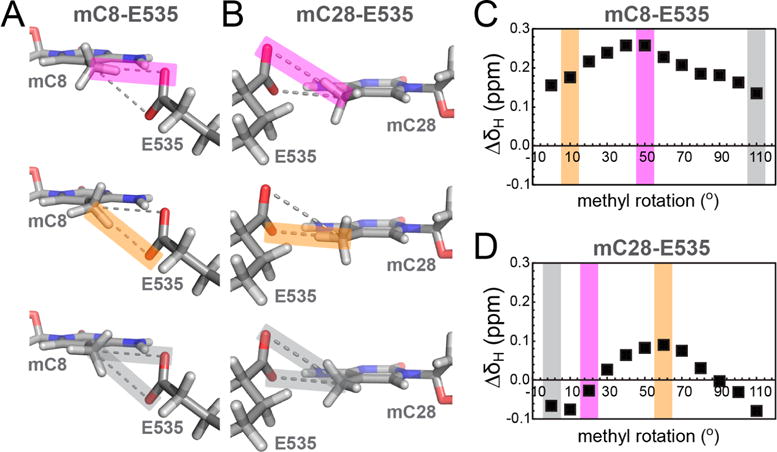
DFT chemical shift calculations for DNA mC methyl protons in Kaiso–MeKBS complexes. (A and B) Fragments from the crystal structure of the WT Kaiso–MeKBS complex showing DNA methyl group orientations that represent a nearly linear hydrogen bond alignment between mC8(C5A) (A) or mC28(C5A) (B) and E535(Oε2) (top, magenta), E535(Oε1) (middle, orange), or where on average the minimal effect on the proton chemical shift is observed, respectively (bottom, gray). (C and D) Representative plots of the difference in predicted methyl proton chemical shifts (ΔδH, ■) for mC8 and mC28, respectively, relative to the “no side chain” control. The chemical shift differences were calculated using DFT and are plotted as a function of counterclockwise methyl group rotation. The colored bands correspond to the methyl rotamers depicted in panel A or B.
A similar trend with methyl group orientation was observed for the mC28–E535 pair (Figure 8B). DFT calculations show that the most downfield methyl proton chemical shift relative to the “no side chain” control (ΔδH ~ 0.09 ppm) occurs for the methyl rotamer with optimal geometry for hydrogen bonding to the E535(Oε1) acceptor (~3.5 Å) (Figure 8D, orange bar). In contrast to the mC8–E535 pair, the E535(Oε2) acceptor is too distant (>4.0 Å) from the mC28 methyl for CH⋯O hydrogen bonding,20,56 and accordingly, the calculated ΔδH is very small (–0.03 ppm) for rotamers that place a methyl proton closest to Oε2 (Figure 8D, magenta bar).
To provide a direct comparison with the NMR studies, we used DFT to estimate the mC8 and mC28 methyl proton chemical shift differences between the complexes of the WT and E535Q with MeKBS. The crystal structures show that, while the mC28(C5A)–Q535(Oε1) distance is the same in the WT and E535Q complexes (~3.5 Å), the mC8(C5A)–Q535(Oε1) distance increases from ~3.4 Å (WT) to ~3.7 Å (E535Q), leading to a weaker CH⋯O interaction in the mutant. In addition, the E535Q mutation replaces the Oε2 atom with nitrogen, thus eliminating the methyl–E535(Oε2) hydrogen bonding interactions observed in the WT complex. Thus, on the basis of the crystal structures, we expect the level of mC8 CH⋯O hydrogen bonding to be substantially reduced in the E535Q complex, which should manifest in an upfield shift of the mC8 methyl proton resonance. This is confirmed by the DFT calculations, which predict an upfield mC8 methyl proton shift (ΔΔδH = 0.25 ppm, averaged over all methyl rotamers) in the E535Q–MeKBS complex. This is in good agreement with the experimental chemical shift difference [0.17 ppm (Figure 7)], given that we observe an ~0.05 ppm overall standard deviation in ΔδH for the same methyl rotamer in different crystal structures of the WT–MeKBS complex.55 For mC28, DFT predicts an average upfield shift of 0.06 ppm for the methyl proton resonance in the E535Q complex, compared to the small downfield shift (0.02 ppm) observed experimentally.
DISCUSSION
Exceptional Specificity of Kaiso for Binding Methylated over Unmethylated CpG Sites
The binding data show that Kaiso has a remarkable ability to discriminate between methylated and unmethylated CpG sites. Methylation of the cytosines on both strands at the first CpG site within the C8GC10G core binding site (Figure 2) enhances Kaiso binding by 2 orders of magnitude [Kd(CG2) ~ 88 nM vs Kd(MeCG2semi) ~ 0.8 nM (Table 1)] and is necessary and sufficient for subnanomolar binding. In contrast, methylation at the second CpG site contributes only a little to further stabilize the Kaiso–DNA complex [Kd(MeCG2) ~ 0.4 nM (Table 1)]. One other member of the zinc finger class of MBPs, Zfp57,24 has also been reported to have a strong preference for CpG methylation and, like Kaiso, recognizes one of the mC methyl groups through hydrophobic contacts and a direct CH⋯O hydrogen bond (Figure S1). All other well-characterized zinc finger MBPs show only a marginal preference for CpG methylation (2–3-fold), which correlates with seemingly weaker hydrophobic and methyl CH⋯O hydrogen bonding interactions.29,31,57
Our findings suggest that the major determinants of this remarkable specificity for methylated DNA are interactions with a single 5-methylcytosine in the 5′ mC8pG9 site. Kaiso recognizes this site with the side chains of R511, L533, and E535, which form a hydrophobic pocket around the mC8 methyl group (Figure 4 and Figure S6).22,29 R511 and L533 are in van der Waals contact with the mC8 methyl group, while E535 forms van der Waals contacts and CH⋯O hydrogen bonds with the cytosine methyl groups on both strands (mC8 and mC28) (Figure 4 and Figure S6). E535 and R511 also participate in an extensive network of direct and water-mediated NH⋯O and NH⋯N hydrogen bonds to mC8, mC28, G9, and G27 (Figure S6). The importance of the interactions with the 5′ mCpG site is emphasized by the large increase in koff and in the free energy of binding (ΔG increases by 2.8 kcal/ mol) to unmethylated (CG2) compared to semimethylated (MeCG2semi) DNA (Table 1). In contrast, the small increase of 0.3 kcal/mol in ΔG for binding to doubly methylated compared to semimethylated MeCG2 DNA suggests that methylation at the 3′ mCpG site contributes very little to DNA binding affinity. This is consistent with the observation of fewer protein–base hydrogen bonding interactions, most of which are water-mediated, in the crystal structures of Kaiso–DNA complexes. Apart from a bifurcated hydrogen bond from the methyl and N4 amino group of mC10 to water and van der Waals contacts between R511 and the mC26 methyl group (Figure 6 and Figure S6), none of these interactions could discriminate between cytosine and 5-methylcytosine. Finally, the relatively small increase in ΔG (0.7 kcal/mol) for hemimethylated MeCG2, where C8 and C10 are methylated, versus doubly methylated MeCG2 signifies that interactions with mC8, rather than mC28, largely account for the exceptional specificity of Kaiso for methylated DNA. This is supported by the X-ray structures, which show that a majority of methyl-specific interactions with the 5′ mCpG site are focused on mC8 as well as by mutational studies (Kaiso E535Q), where disruption of interactions with mC8, but not with mC28, leads to a large loss (~50-fold) of binding affinity.
Critical Role of Kaiso E535 in Methylated CpG Site Recognition
A key contribution to this specificity for methylated DNA is the interaction of glutamate 535 with the mCpG site. Mutation of E535 to alanine causes a dramatic loss of the free energy of binding [2.8–3.1 kcal/mol (Table 1)] for both double and semimethylated DNA relative to the WT protein. This corroborates previous gel shift binding studies30 and is consistent with disruption of multiple E535–mCpG interactions, including methyl CH⋯O hydrogen bonds and van der Waals contacts that are specific to the methyl groups of the two symmetric 5-methylcytosines (Figure 4 and Figure S6). In fact, the E535A mutation is so deleterious to methylated DNA binding that crystals of sufficient quality for X-ray diffraction could not be obtained. Observation of the complex by NMR under low-salt conditions shows that the E535A mutation perturbs the local structure and/or dynamics in many sites across the entire protein, particularly in ZF2, ZF1, and the C-terminal tail (Figure 3C,D and Figure S3).
While the E535Q mutation is conservative compared to the E535A mutation, it nevertheless results in chemical shift perturbations at similar sites (Figure 3C,D and Figure S3), consistent with similar but more subtle rearrangement of the protein–DNA interface. Although binding to methylated DNA is greatly impaired (Kd is increased ≤50-fold, and ΔG is increased by ~2 kcal/mol), the E535Q mutant retains the ability to discriminate, albeit less effectively, between methylated and unmethylated DNA (Table 1). This is not the case for E535A, which strongly impairs binding and largely abrogates selectivity for methylated DNA. The profound effect of E535 mutants on Kaiso DNA binding affinity, structure, and dynamics underscores the critical role of E535 in recognition of the 5′ mCpG step. E535 acts as an anchor that protrudes deep into the major groove to establish optimal contacts with the methylated cytosines. Most other known zinc finger MBPs contain a shorter side chain (Figure S1) that is incapable of making direct methyl-specific interactions simultaneously with both symmetric 5-methylcytosines. Thus, it is conceivable that engineering a glutamate side chain at that position in other MBPs might improve their specificity for methylated DNA.
CH···O Hydrogen Bonds Mediate Recognition of Both Methylated CpG Sites
CH⋯O hydrogen bonds are weak, are largely electrostatic in nature, and have donor–acceptor (C–O) distances (~3.0–3.7 Å) that are longer than those of canonical hydrogen bonds and angles that tend to deviate from linearity (110–180°).20,54 In all of the crystal structures of WT Kaiso with methylated DNA, one E535 carboxyl oxygen atom or both (Oε1 and Oε2) are within CH⋯O hydrogen bonding distance (≤3.7 Å) of the mC8 and mC28 methyl carbon (C5A) (Figure 4 and Table S3). In fact, the proximity and orientation of E535, which has two hydrogen bond acceptors, allow for two alternate linear CH⋯O hydrogen bond configurations (bond angles of ~165–175°) for both mC8 and mC28 and additional less linear configurations in which methyl CH⋯O hydrogen bonds are formed simultaneously to both carboxyl oxygen atoms (Figure 8A).
The improved resolution of the current Kaiso–MeKBS/ MeCG2 X-ray structures (2.0–2.4 Å) as compared to that of the published Kaiso–MeEcad structure (2.8 Å)23 provides new insights into the mechanism of recognition of the 3′ mCpG site. A conserved water molecule mediates NH⋯O and CH⋯O hydrogen bonds between the mC10 amino and methyl groups and T507/S508 hydroxyl groups (Figure 6). This water-mediated CH⋯O hydrogen bond may contribute to the 0.3 kcal/mol difference in the binding free energy for the complexes with semimethylated versus doubly methylated MeCG2 (Table 1). The frequent occurrence of direct or water-mediated methyl CH⋯O hydrogen bonds in the complexes between methylated DNA and methyl CpG-binding proteins makes a strong case that these interactions are important for selective 5-methylcytosine recognition.23,27,29,58,59
Direct NMR Evidence for CH···O Hydrogen Bonds between Kaiso and Methylated CpG
On the basis of the crystal structures, the E535Q mutation impairs the NH⋯O and CH⋯O hydrogen bonds between E535 and mC8, leaving neighboring interactions between Q535 and MeKBS largely intact. This presents an ideal opportunity to study the formation of a CH⋯O bond between the glutamate carboxyl group and the mC methyl group. Natural-abundance NMR of various methylated DNA constructs in complex with WT Kaiso and E535 mutants, together with DFT chemical shift calculations, has enabled us to directly detect the presence of these methyl CH⋯O hydrogen bonds.
Comparison of the natural-abundance 1H–13C HMQC methyl spectra of complexes of WT Kaiso and E535Q with MeKBS and MeCG2 oligonucleotides reveals a downfield chemical shift (ΔδH = 0.17 and 0.14 ppm, respectively) of the mC8 methyl proton resonance in the WT complex compared to the E535Q mutant complex (Figure 7B). For the mC28 methyl protons, a much smaller upfield shift (ΔδH = 0.02 ppm) is observed (Figure 7B). These chemical shift changes are consistent with a model in which the E535Q mutation disrupts CH⋯O hydrogen bonds between the E535 carboxyl and the mC8 methyl group, while preserving a CH⋯O hydrogen bond between E535 and the mC28 methyl group. This model is also in line with the crystallographic data for WT Kaiso– and E535Q–MeKBS complexes shown in Figures 4 and 5. Further agreement between this model and the crystal structures is shown by the small downfield shift (ΔδH = 0.05 ppm) in the mC8 methyl proton resonance in the MeKBS complex relative to the MeKBShemi complex with WT Kaiso (Figure 7A), consistent with the slightly shorter CH⋯O distances observed in the corresponding crystal structures (Table S3). Interestingly, the mC10 methyl proton resonance in the 3′ mCpG site also shows a small downfield shift (ΔδH ~ 0.07 ppm) in the WT versus E535Q complex (Figure 7B), which may indicate weakening of the water-mediated methyl CH⋯O hydrogen bond (Figure 6) that is coupled to the distant E535Q mutation. A coupling between the E535Q mutation and the 3′ mCpG site is also suggested by the protein backbone chemical shift perturbations observed by NMR (Figure 3B, D).
Finally, the structure-based DFT methyl proton chemical shift calculations are consistent with the NMR data and provide further support for CH⋯O hydrogen bonding between the E353 carboxyl and the mC8 methyl group. The DFT calculations show that, depending on the methyl rotamer, hydrogen bonding to E535 Oε1, Oε2, or both simultaneously leads to a marked downfield shift (0.18–0.26 ppm) of the mC8 methyl proton resonance. The DFT calculations also recapitulate the difference in mC8 chemical shift between the WT and E535Q complexes. To the best of our knowledge, this is the first direct evidence for the presence of CH⋯O hydrogen bonding between a methyl-CpG DNA-binding protein and 5-methylcytosine. Importantly, the calculations predict much larger downfield shifts for the mC8 methyl proton resonance than for that of mC28, in full agreement with the experimental data and indicating stronger methyl CH⋯O hydrogen bonding interactions with mC8. In turn, this provides a molecular-level explanation for the critical role played by E535 in recognition of 5-methylcytosine and for the extraordinary ability of Kaiso to discriminate, by 2 orders of magnitude in binding affinity, between methylated and unmethylated CpG sites in DNA.
Supplementary Material
Acknowledgments
The authors thank Gerard Kroon for help with the setup and analysis of NMR spectra, Maria Martinez-Yamout for valuable discussions, and Euvel Manlapaz for technical assistance. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
Funding
This work was supported by Grant GM36643 from the National Institutes of Health (P.E.W.) and by the Skaggs Institute for Chemical Biology. E.N.N. was supported by a Robert Black fellowship from the Damon Runyon Cancer Research Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.8b00065.
Figures and tables with details about DNA constructs, Kaiso–DNA binding kinetics, crystallographic statistics and omit maps, comparisons between crystal structures, and NMR studies of complexes of Kaiso with DNA (PDF)
Accession Codes
The coordinates for the X-ray crystal structures have been deposited in the RCSB Protein Data Bank (PDB) as entries 5VMV (Kaiso–MeCG2), 5VMU [Kaiso–MeKBS(1)], 5VMW [Kaiso–MeKBS(2)], 5VMX [Kaiso–MeKBShemi(1)], 5VMY [Kaiso–MeKBShemi(2)], and 5VMZ (Kaiso E535Q–MeKBS). The backbone amide chemical shifts for Kaiso–DNA complexes have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) as entries 27364 (Kaiso–MeCG2), 27366 (Kaiso E535Q–MeCG2), 27367 (Kaiso E535A–MeCG2), 27368 (Kaiso–MeKBS), 27369 (Kaiso E535Q–MeKBS), 27370 (Kaiso E535A–MeKBS), 27371 (Kaiso–MeKBSsemi), and 27372 (Kaiso–MeKBShe-mi).
ORCID
H. Jane Dyson: 0000-0001-6855-3398
Peter E. Wright: 0000-0002-1368-0223
Notes
The authors declare no competing financial interest.
References
- 1.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 2.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 3.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2009;66:596–612. doi: 10.1007/s00018-008-8432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–966. doi: 10.1101/gr.083451.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, Wei CL. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, Zhu H, Chang Q, Gao Y, Ming GL, Song H. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fournier A, Sasai N, Nakao M, Defossez PA. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Briefings Funct Genomics. 2012;11:251–264. doi: 10.1093/bfgp/elr040. [DOI] [PubMed] [Google Scholar]
- 10.Dantas Machado AC, Zhou T, Rao S, Goel P, Rastogi C, Lazarovici A, Bussemaker HJ, Rohs R. Evolving insights on how cytosine methylation affects protein-DNA binding. Briefings Funct Genomics. 2015;14:61–73. doi: 10.1093/bfgp/elu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet. 2011;12:429–442. doi: 10.1038/nrg2987. [DOI] [PubMed] [Google Scholar]
- 12.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Cold Spring Harbor Perspect Biol. 2011;3:a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 14.Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- 15.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck-Koehntop BA, Defossez PA. On how mammalian transcription factors recognize methylated DNA. Epigenetics. 2013;8:131–137. doi: 10.4161/epi.23632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565. doi: 10.1038/nrg.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Zhang X, Blumenthal RM, Cheng X. A common mode of recognition for methylated CpG. Trends Biochem Sci. 2013;38:177–183. doi: 10.1016/j.tibs.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz S, Trievel RC. Carbon-oxygen hydrogen bonding in biological structure and function. J Biol Chem. 2012;287:41576–41582. doi: 10.1074/jbc.R112.418574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodea S, Funk MA, Balskus EP, Drennan CL. Molecular Basis of C-N Bond Cleavage by the Glycyl Radical Enzyme Choline Trimethylamine-Lyase. Cell Chem Biol. 2016;23:1206–1216. doi: 10.1016/j.chembiol.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 23.Buck-Koehntop BA, Stanfield RL, Ekiert DC, Martinez-Yamout MA, Dyson HJ, Wilson IA, Wright PE. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci U S A. 2012;109:15229–15234. doi: 10.1073/pnas.1213726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Olanrewaju YO, Zhang X, Cheng X. DNA recognition of 5-carboxylcytosine by a Zfp57 mutant at an atomic resolution of 0.97 A. Biochemistry. 2013;52:9310–9317. doi: 10.1021/bi401360n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buck-Koehntop BA, Martinez-Yamout MA, Dyson HJ, Wright PE. Kaiso uses all three zinc fingers and adjacent sequence motifs for high affinity binding to sequence-specific and methyl-CpG DNA targets. FEBS Lett. 2012;586:734–739. doi: 10.1016/j.febslet.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Toh H, Sasaki H, Zhang X, Cheng X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26:2374–2379. doi: 10.1101/gad.202200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walavalkar NM, Cramer JM, Buchwald WA, Scarsdale JN, Williams DC., Jr Solution structure and intramolecular exchange of methyl-cytosine binding domain protein 4 (MBD4) on DNA suggests a mechanism to scan for mCpG/TpG mismatches. Nucleic Acids Res. 2014;42:11218–11232. doi: 10.1093/nar/gku782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Olanrewaju YO, Zheng Y, Hashimoto H, Blumenthal RM, Zhang X, Cheng X. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859–4867. doi: 10.1093/nar/gku134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasai N, Nakao M, Defossez PA. Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. 2010;38:5015–5022. doi: 10.1093/nar/gkq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto H, Olanrewaju YO, Zheng Y, Wilson GG, Zhang X, Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–2313. doi: 10.1101/gad.250746.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daniel JM, Reynolds AB. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 1999;19:3614–3623. doi: 10.1128/mcb.19.5.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniel JM, Spring CM, Crawford HC, Reynolds AB, Baig A. The p120ctn-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 2002;30:2911–2919. doi: 10.1093/nar/gkf398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso, Mol. Mol Cell. 2003;12:723–734. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Raghav SK, Waszak SM, Krier I, Gubelmann C, Isakova A, Mikkelsen TS, Deplancke B. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPβ and KAISO. Mol Cell. 2012;46:335–350. doi: 10.1016/j.molcel.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 36.Dai SD, Wang Y, Zhang JY, Zhang D, Zhang PX, Jiang GY, Han Y, Zhang S, Cui QZ, Wang EH. Upregulation of δ-catenin is associated with poor prognosis and enhances transcriptional activity through Kaiso in non-small-cell lung cancer. Cancer Sci. 2011;102:95–103. doi: 10.1111/j.1349-7006.2010.01766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park Ji, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, McCrea PD. Kaiso/p120-catenin and TCF/β-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Spring CM, Kelly KF, O’Kelly I, Graham M, Crawford HC, Daniel JM. The catenin p120ctn inhibits Kaiso-mediated transcriptional repression of the β-catenin/TCF target gene matrilysin. Exp Cell Res. 2005;305:253–265. doi: 10.1016/j.yexcr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Ogden SR, Wroblewski LE, Weydig C, Romero-Gallo J, O’Brien DP, Israel DA, Krishna US, Fingleton B, Reynolds AB, Wessler S, Peek RM., Jr p120 and Kaiso regulate Helicobacter pylori-induced expression of matrix metalloproteinase-7. Mol Biol Cell. 2008;19:4110–4121. doi: 10.1091/mbc.E08-03-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donaldson NS, Pierre CC, Anstey MI, Robinson SC, Weerawardane SM, Daniel JM. Kaiso represses the cell cycle gene cyclin D1 via sequence-specific and methyl-CpG-dependent mechanisms. PLoS One. 2012;7:e50398. doi: 10.1371/journal.pone.0050398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pierre CC, Longo J, Bassey-Archibong BI, Hallett RM, Milosavljevic S, Beatty L, Hassell JA, Daniel JM. Methylation-dependent regulation of hypoxia inducible factor-1 alpha gene expression by the transcription factor Kaiso. Biochim Biophys Acta, Gene Regul Mech. 2015;1849:1432–1441. doi: 10.1016/j.bbagrm.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Liu W, Black S, Turner O, Daniel JM, Dean-Colomb W, He QP, Davis M, Yates C. Kaiso, a transcriptional repressor, promotes cell migration and invasion of prostate cancer cells through regulation of miR-31 expression. Oncotarget. 2016;7:5677–5689. doi: 10.18632/oncotarget.6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology Macromolecular Crystallography Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The PyMOL Molecular Graphics System. Schrödinger, LLC; Portland, OR: 2013. [Google Scholar]
- 48.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 49.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2006. [Google Scholar]
- 50.Aoto PC, Fenwick RB, Kroon GJA, Wright PE. Accurate scoring of non-uniform sampling schemes for quantitative NMR. J Magn Reson. 2014;246:31–35. doi: 10.1016/j.jmr.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 52.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 53.Pierens GK. 1H and 13C NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J Comput Chem. 2014;35:1388–1394. doi: 10.1002/jcc.23638. [DOI] [PubMed] [Google Scholar]
- 54.Yesselman JD, Horowitz S, Brooks CL, Trievel RC. Frequent side chain methyl carbon-oxygen hydrogen bonding in proteins revealed by computational and stereochemical analysis of neutron structures. Proteins: Struct, Funct, Genet. 2015;83:403–410. doi: 10.1002/prot.24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horowitz S, Yesselman JD, Al-Hashimi HM, Trievel RC. Direct evidence for methyl group coordination by carbon-oxygen hydrogen bonds in the lysine methyltransferase SET7/9. J Biol Chem. 2011;286:18658–18663. doi: 10.1074/jbc.M111.232876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desiraju GR. The C-h⋯o hydrogen bond: structural implications and supramolecular design. Acc Chem Res. 1996;29:441–449. doi: 10.1021/ar950135n. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG, Cheng X. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol Cell. 2017;66:711–720.e3. doi: 10.1016/j.molcel.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 59.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.