

Abstract
Although cancer genomes are replete with noncoding mutations, the effects of these mutations remain poorly characterized. Here we perform an integrative analysis of 930 tumor whole genomes and matched transcriptomes, identifying a network of 193 noncoding loci in which mutations disrupt target gene expression. These “somatic eQTLs” (expression Quantitative Trait Loci) are frequently mutated in specific cancer tissues, and the majority can be validated in an independent cohort of 3,382 tumors. Among these, we find that the effects of noncoding mutations on DAAM1, MTG2 and HYI transcription are recapitulated in multiple cancer cell lines, and that increasing DAAM1 expression leads to invasive cell migration. Collectively the noncoding loci converge on a set of core pathways, permitting a classification of tumors into pathway-based subtypes. The somatic eQTL network is disrupted in 88% of tumors, suggesting widespread impact of noncoding mutations in cancer.
Human cancers are fundamentally heterogeneous, with many distinct subtypes associated with differences in molecular, cellular and clinical characteristics. To gain insight into this complexity, projects such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) have used massively parallel DNA sequencing to construct large catalogs of somatic mutations in many types of tumors1–3. Focusing initially on protein coding regions, several hundred genes were found to be recurrently mutated in cancer, a few of which are targetable therapeutically4.
Since coding regions account for less than 2% of the human genome, attention is now shifting to the greater number of somatic mutations in noncoding regions5. Thus far the clearest role for noncoding mutations in cancer has been in the promoter of the telomerase reverse transcriptase gene (TERT)6–8, with such mutations leading to increases in TERT expression levels in many types of tumors8,9. Although whole-genome sequencing (WGS) of tumor-normal pairs has found recurrent somatic mutations at several other noncoding loci, assessing the function of these mutations, if any, has been challenging6–8. In this respect, the task of functional interpretation is greatly aided by recent efforts of consortia such as ENCODE10,11 and Roadmap12,13, which have published extensive reference maps of non-coding regions and their likely transcriptional regulatory connections to genes. Here, we show that such networks provide critical information for identifying noncoding mutations with functional impacts among the many others that may be spurious6.
RESULTS
Genome-wide identification of somatic eQTLs in cancer
To identify noncoding mutations associated with functional effects, we performed a systematic analysis of 930 tumors integrating whole-genome sequences, matched mRNA expression profiles and reference transcriptional interaction maps. Using WGS of paired normal and tumor tissues in 930 patients across 22 types of cancer from TCGA1 (Fig. 1a), we identified 3.5 × 107 sites with somatic single nucleotide variations (SNVs). We called these SNVs uniformly across all genomes using the MuTect suite14 according to GATK Best Practice recommendations15,16 and those of Melton et al.6 (Fig. 1b). Clusters of noncoding SNVs located within 50 base pairs of one another were grouped, defining recurrently mutated loci (Fig. 1c, Methods, Supplementary Fig. 1).
Figure 1. Mutation calling and somatic eQTL analysis.
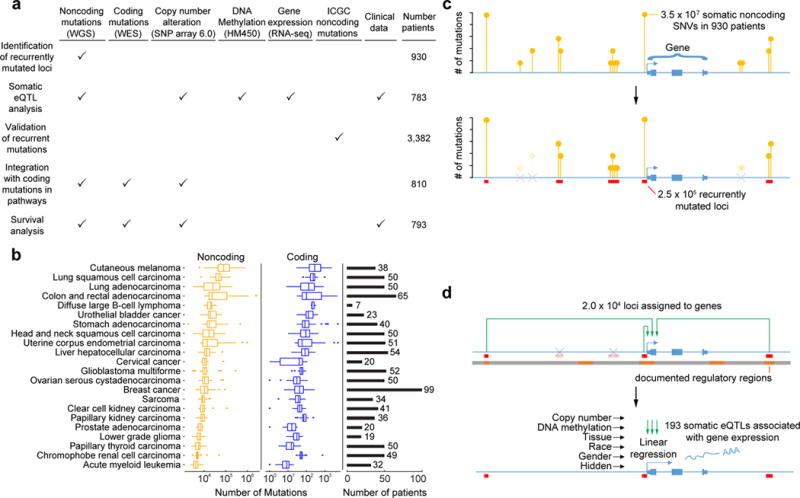
(a) Types of data and numbers of tumors used in this study. (b) Number of mutations called per tumor. Boxplots show the distribution of this number within tumors of each tissue type (center line, median; upper and lower hinges, first and third quartiles; whiskers, highest and lowest values within 1.5 times the interquartile range outside hinges; dots, outliers beyond 1.5 times interquartile range). The number of tumors of each type (sample size) is shown on the right panel. (c) Clustering of somatic noncoding mutations resulting in identification of recurrently mutated loci. (d) Workflow of somatic eQTL analysis.
We then tested each locus for its association with changes in mRNA expression of target genes (Fig. 1d). This task made use of two additional datasets. First, enhancer-gene mappings in GeneHancer17 were used along with promoter-proximal regions, defined as sequences within 1 kb of each transcription start site (TSS), to link recurrently mutated loci to putative target genes considered to be under direct transcriptional control (Methods). Second, for the vast majority of patients with tumor genome sequences, tumor mRNA expression profiles were also available (Fig. 1a). From these data we developed a multivariate linear regression model of the expression change of each target gene, as a function of the mutation status of its linked loci and co-variates including the presence of copy number alterations (CNAs), DNA methylation status, tissue, race, and gender (Fig. 1d, Methods). Conceptually this procedure is similar to identifying expression Quantitative Trait Loci (eQTL), in which inherited nucleotide variants are mapped to downstream functional changes18,19. Here, however, the variants are somatically acquired rather than inherited. Such “somatic eQTL analysis” simplifies the complexity and scope of eQTL mapping to a relatively small number of unlinked genetic variants: on average, 2.6 loci were tested per gene, with a standard deviation of 3.1 and a maximum number of 53 (Supplementary Fig. 1e).
Altogether, this approach identified a cancer transcriptional network of 206 regulatory interactions between 193 somatic eQTLs and 196 gene expression level changes, at a false discovery rate (FDR) of 20% (Figs. 2a, b, Supplementary Fig. 2, somatic eQTLs at different FDR thresholds are provided in Supplementary Table 1 and Supplementary Note). At least one locus in this network was somatically mutated in 88% of cases studied (820 out of 930), suggesting transcriptional dysregulation through noncoding mutations is a general property of most tumors. Somatic eQTLs linked noncoding mutations to the expression levels of thirteen known tumor suppressor or oncogenes4,20,21 (Supplementary Table 1) although, interestingly, known cancer genes were not significantly enriched overall (Fisher’s exact test p-value = 0.3). We also found that 43% of somatic eQTLs disrupt or create a transcription factor binding motif (83 out of 193, Supplementary Table 2, Supplementary Fig. 3), although this percentage was very similar for recurrently mutated loci not detected as somatic eQTLs (40%, 2,409 out of 8,607).
Figure 2. Effect size and recurrence of somatic eQTLs.
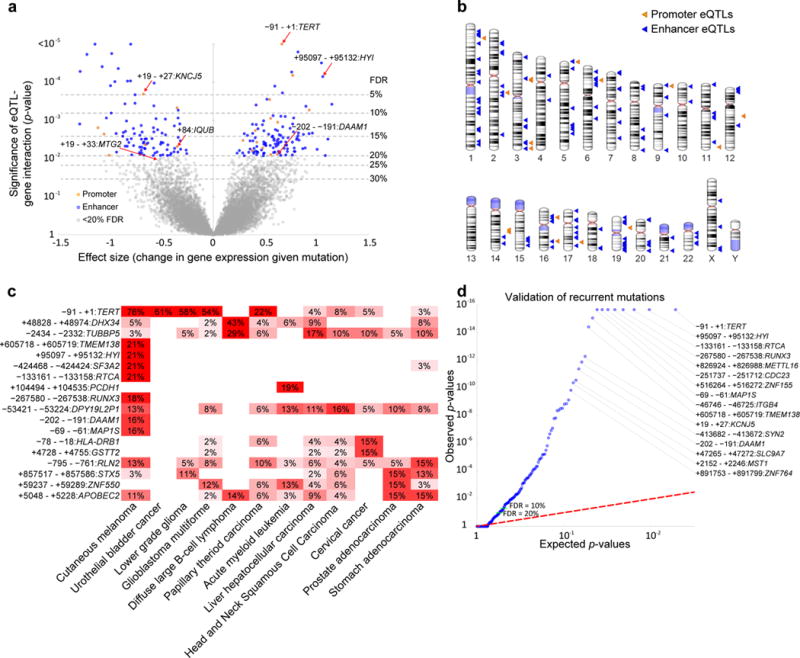
(a) Volcano plot of associations between somatic eQTLs and the expression levels changes of their target genes, evaluated by significance (y-axis, F-test p-value, n = 783 tumors) versus effect size (x-axis). One unit on the x-axis represents one standard deviation of change in gene expression. FDR is calculated using the Storey approach50. Selected somatic eQTLS are labeled by coordinates in base pairs relative to the TSS of the target gene. (b) Ideogram of the 193 significant somaitc eQTLs at FDR < 20%. (c) Heatmap showing the percentage of patients in various cancer tissues with alterations in each somatic eQTL. Somatic eQTLs and cancer tissues with ≥ 15% mutation rates are shown. (d) Validation of somatic eQTL recurrence in a pan-cancer cohort from ICGC. The quantile–quantile plot shows the observed empirical p-values of mutation recurrence (n = 3,382 tumors) compared to the random expectation for the 193 somatic eQTLs. FDR is calculated using the Benjamini-Hochberg approach.
Many of the identified somatic eQTLs were frequently mutated in specific cancer tissues (Fig. 2c, Supplementary Table 3). Beyond the promoter of TERT, which is highly mutated in several tissues as previously noted6–8 (Supplementary Figs. 3a, b), we found recurrently mutated loci associated with expression of DHX34 (mutated in 43% of diffuse large B-cell lymphoma), TUBBP5 (29% of lymphoma and 17% liver cancer), HYI (21% of melanoma), and PCDH1 (19% of acute myeloid leukemia), among others. While most of the somatic eQTLs were mutated in multiple tissues, 12 of the somatic eQTLs were mutated almost exclusively in melanoma (80% or more of the mutations occurred in melanoma). Such enrichment for a single tissue was not seen for any other tissue type.
Somatic eQTLs are recurrently mutated in a second cohort
To systematically validate this network, we examined an independent pan-cancer cohort from ICGC consisting of genome-wide somatic mutation calls for 3,382 patients2. Notably, we found that the majority of the somatic eQTLs identified in the original TCGA discovery set were recurrently mutated in the ICGC validation cohort (107 of the 193 at FDR < 20%, Fig. 2d). These included 10 out of the 12 melanoma eQTLs, which again were frequently and almost exclusively mutated in the melanoma samples in ICGC (Fisher’s exact test p-value = 4.1 × 10−12, Supplementary Table 4). For example, a somatic eQTL associated with increased HYI mRNA expression level was mutated in 21% of US melanomas (TCGA) and 18% of Australian melanomas (ICGC).
Increasing DAAM1 expression leads to cell invasion
We next sought to examine in more detail the somatic eQTL located −191 bp upstream of DAAM1 (Fig. 2a, Methods), which is recurrently mutated in melanoma patients with metastatic disease in both cohorts (Figs. 2c, d). The DAAM1 protein forms a complex with Dishevelled and RhoA to recruit the actin cytoskeleton, which is thought to increase the motility and invasiveness of cancer cells in response to Wnt signaling22–24. Mutations at this somatic eQTL are associated with increased DAAM1 mRNA expression levels due potentially to the loss of an E2F motif and the gain of an Ets motif (Fig. 3a, NC_000014.8:g.59655190G>A). To confirm a causal relationship between the somatic eQTL and gene expression level changes, wild-type and mutant DAAM1 regulatory elements were inserted upstream of Green Fluorescent Protein (GFP) (Fig. 3b). Analysis by flow cytometry showed that the mutated regulatory element leads to a significantly higher percentage of cells expressing GFP in melanoma, sarcoma and breast cancer cell lines (Figs. 3c, d, Supplementary Fig. 4). Furthermore, the GFP-expressing cells had significantly higher levels of GFP expression with the mutant rather than the wild type DAAM1 element in all four cell lines tested (Fig. 3d, Supplementary Figs. 4c, e, g).
Figure 3. Functional validation of the mutated DAAM1 regulatory element.
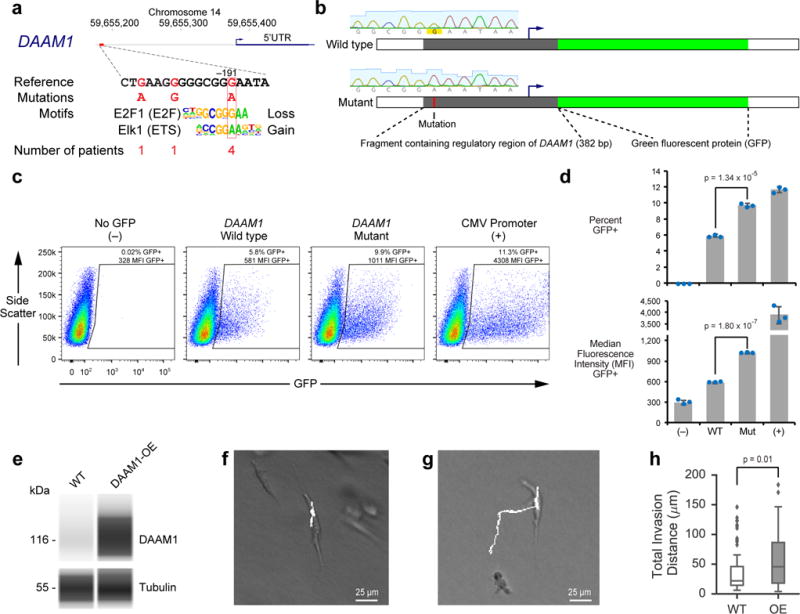
(a) A somatic eQTL in the DAAM1 promoter region is associated with increased mRNA expression levels. (b) Schematic of wild type and mutant GFP reporter constructs along with the Sanger sequencing traces confirming the sequence of the key nucleotide. (c) Flow cytometry analysis of A375 human melanoma cells 48 hours after transient transfection. The polygon delineated by black lines shows the gated region used to define GFP+ cells. (d) Bar graphs (average ± standard deviation across 3 cell culture replicates; p-values from two-tailed t-tests) showing the percentage of GFP+ cells and the median fluorescence intensity of the GFP+ cells. Individual data points are in Supplementary Table 5. (e) Protein electropherogram analysis of wild type and DAAM1 overexpressing MDA-MB-231 cells using the antibodies against DAAM1 and tubulin. The complete electropherogram is in Supplementary Fig. 6e. The image is representative of two independent cell culture experiments. (f, g) Sample trajectories of (f) wild type and (g) DAAM1-overexpressing cells embedded in 2.5 mg/mL 3D collagen hydrogels. (h) Total invasion distance travelled by individual cells (p-value from two-tailed Mann–Whitney U test; 95% confidence intervals of mean are (32.3 μm, 48.2 μm) and (47.6 μm, 67.0 μm) for wild type and DAAM1-overexpressing cells, respectively). Imaging and quantitation was performed on 74 and 83 cells in the wild type and DAAM1-overexpression groups, respectively. Box-plot elements are defined as Fig. 1b.
We also explored the functional relationship between increased DAAM1 expression and cell motility, using an established 3D collagen hydrogel matrix model25. Genome-wide mRNA sequencing was performed on cells grown within low or high density collagen, mimicking the stiffness of normal or tumor tissues and eliciting less or more invasive phenotypes, respectively26,27 (Methods). In these experiments DAAM1 was one of the most up-regulated transcripts under invasive conditions28 (Supplementary Fig. 5). To test whether invasion was functionally dependent on DAAM1, we quantified cell migration behavior after DAAM1 expression was increased artificially by exogenous overexpression (OE, Fig. 3e, Supplementary Fig. 6e, Methods). When cells overexpressing DAAM1 were embedded in the 3D collagen hydrogel, they migrated with significantly greater persistence than did wild type cells (p = 0.008, two-sided Mann–Whitney U test; Supplementary Fig. 6a). Cells overexpressing DAAM1 also invaded for longer distances than wild type cells (p = 0.01, two-sided Mann–Whitney U test; Figs. 3f-h), while retaining the same velocities as wild type cells (Supplementary Fig. 6b, c). This invasive phenotype was observed in the absence or presence of additional Wnt5a signaling (Supplementary Fig. 6d). These results suggest that increased DAAM1 expression levels allows cells to more efficiently invade the local microenvironment, thereby linking this noncoding mutation to DAAM1 overexpression and cell invasion.
Noncoding mutations dysregulating MTG2 and HYI
Beyond DAAM1, we examined two additional somatic eQTLs, one in the promoter of MTG2 (+19 – +33) and another in the enhancer of HYI (+95097 – +95132) (Methods). The first eQTL was associated with decreased MTG2 mRNA expression levels, likely due to the disruption of a HIF1b binding motif by the G-to-A mutation at +19 bp downstream of the TSS (Fig. 4a, NC_000020.10:g.60758100G>A). This somatic eQTL was present in several types of cancer including lung adenocarcinoma and sarcoma. Using another GFP-based reporter assay of promoter activity, we found that this G-to-A mutation greatly decreased reporter gene expression in both A549 lung epithelial carcinoma cells and U2OS bone osteosarcoma cells (Fig. 4b). The second eQTL was present in 21% of melanomas (Fig. 2c) and was associated with increased HYI mRNA expression levels, likely due to G-to-A or GG-to-AA substitutions altering an Ets family binding motif (Fig. 4c, NC_000001.10:g.43824528G>A, NC_000001.10:g.43824529G>A, or NC_000001.10:g.43824528_43824529GG>AA). As this somatic eQTL was present in an enhancer region, we used a luciferase-based reporter assay where regulatory elements were cloned upstream of a mini-promoter and luciferase. We found that two out of the three HYI enhancer variants led to increased expression levels relative to wild type in both A375 melanoma cells and MDA-MB-231 breast cancer cells (Fig. 4d).
Figure 4. Additional case studies.
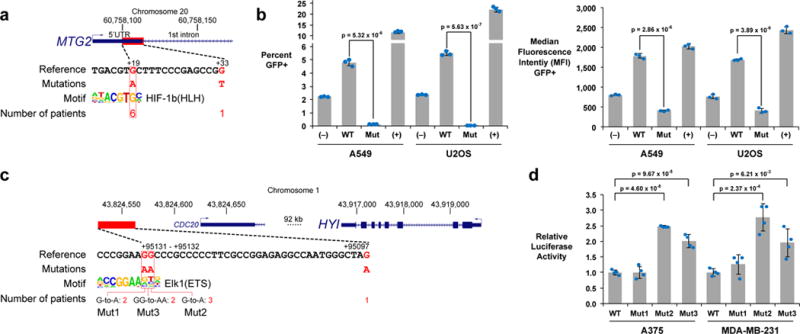
(a) The somatic eQTL associated with downregulation of MTG2 is located in its 5′ UTR and frequently alters a potential HIF-1b binding motif. (b) Flow cytometry analysis of A549 lung epithelial carcinoma cells and U2OS bone osteosarcoma cells 48 hours after transient transfection with MTG2 GFP reporter constructs. Bar graphs (mean ± standard deviation across three cell culture replicates; p-values from two-tailed t-tests) showing the percentage of GFP+ cells and the median fluorescence intensity of GFP+ events. (c) The somatic eQTL associated with upregulation of HYI is located 95 kb downstream of the TSS and frequently alters a potential Ets family binding motif. (d) Luciferase assay results (mean ± standard deviation across four cell culture replicates; p-values from two-tailed t-tests) for the HYI somatic eQTLs 48 hours after transient transfection in A375 melanoma cells and MDA-MB-231 breast cancer cells. Individual data points are available in Supplementary Tables 5 and 6.
Noncoding and coding mutations converge on pathways
Next, we investigated the relationship between the 196 genes transcriptionally regulated by somatic eQTLs and the 138 genes previously documented to have recurrent coding mutations in cancer21. This combined set of genes was analyzed by Network-Based Stratification29,30 (NBS; Fig. 5a), which uses a reference molecular network to implicate network regions associated with the genetic alterations in a tumor and groups tumors into subtypes based on similarity of these implicated regions. As a reference molecular network we used ReactomeFI31, documenting 229,300 interactions among 12,177 human gene products pertaining to previously reported protein-protein, transcriptional and metabolic interactions.
Figure 5. Identification of molecular networks and associated tumor subtypes incorporating noncoding mutations.
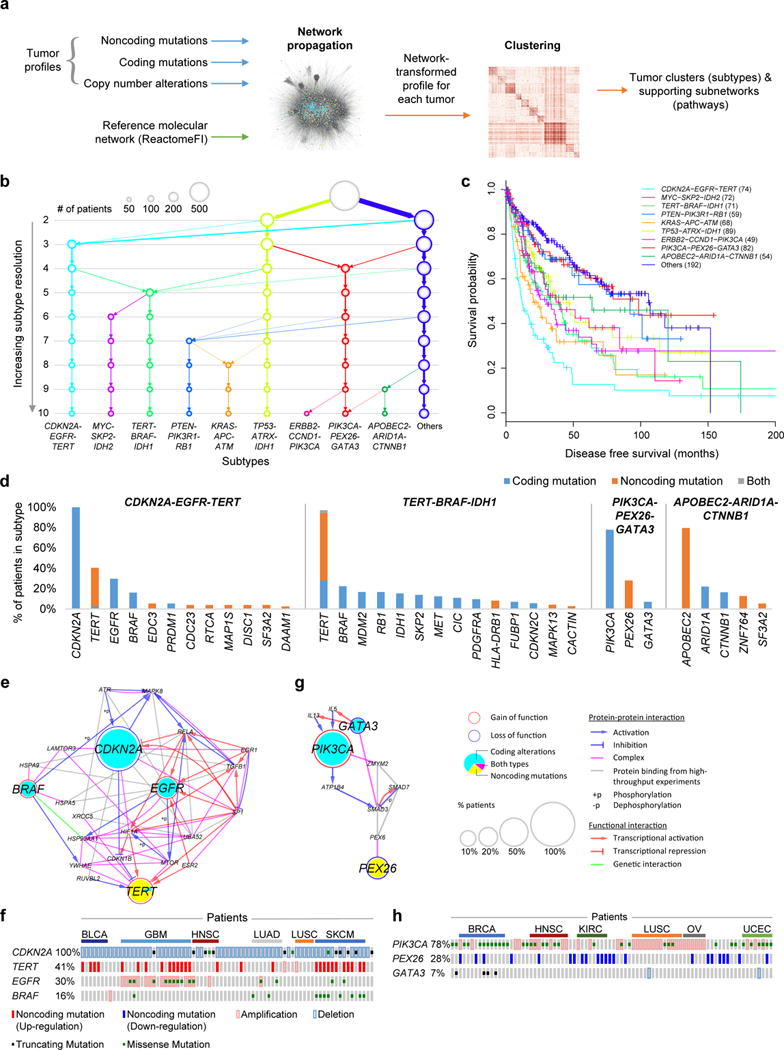
(a) Workflow of Network-Based Stratification. (b) Resulting hierarchy of subtypes, at increasing resolution from 2-10 subtypes. (c) Disease-free survival probabilities (y-axis) are plotted against time after diagnosis in months (x-axis) for each of the identified cancer subtypes (colors). Patients with censored survival data are indicated by a “+” at the censoring time (last follow-up). (d) Signature genes are shown for each subtype with a large proportion of patients with noncoding mutations (x-axis), ordered by the percent of patients with alterations (y-axis). (e, g) Pathways characterizing (e) CDKN2A-EGFR-TERT or (g) PIK3CA-PEX26-GATA3 subtypes, defined as subnetwork regions extracted from ReactomeFI by Network-Based Stratification. (f, h) Mutation matrix of the (f) CDKN2A-EGFR-TERT or (h) PIK3CA-PEX26-GATA3 pathway subtypes showing individual tumors (columns, ordered by cancer tissues) with indicated types of mutations on signature genes for that subtype (rows).
This approach identified a collection of network regions (henceforth called “pathways” for simplicity) that stratified tumors into a hierarchy of increasingly specific subtypes (Fig. 5b). At a resolution of ten subtypes, each subtype was enriched in 2-5 tumor tissues and tumors of each tissue could be subdivided into 1-3 subtypes (Supplementary Fig. 7). Nonetheless, these subtypes differed significantly in their implications for disease-free survival, beyond the baseline survival for each tissue (p = 3.3 × 10−6, log likelihood ratio test controlling for the tissue types as covariates; Fig. 5c, Supplementary Fig. 8).
Subtypes aggregating noncoding and coding mutations
Among the ten subtypes, four were of particular interest as they contained a large proportion of patients with noncoding mutations (Fig. 5d). The “CDKN2A-EGFR-TERT subtype” (Figs. 5e, f) was defined by disruption of the CDKN2A coding sequence, sometimes in combination with noncoding mutations to the TERT promoter, EGFR activation, or BRAF activation. CDKN2A encodes p14ARF, which can form a complex with Hif-1ɑ and inhibit HIF-1-mediated transcription of TERT32,33. These loss-of-function mutations in CDKN2A may release a key brake on the activity of hTERT. Separately, gain-of-function mutations in EGFR may lead to increased levels of mTOR phosphorylation and activation34, which can up-regulate telomerase activity by forming a complex with hTERT35. The synergy between BRAF and TERT mutations has been previously noted and attributed to modulation of TERT transcription through BRAF-RAS-ERK signaling36. This pathway was also linked to DAAM1 promoter mutations (Fig. 5d), validated previously, as Daam1 forms a complex with Dishevelled (Dvl3)22,23, which indirectly regulates transcription of CDKN2A and EGFR through inhibition of Notch137. This subtype was the most aggressive, with median disease free survival time at 13 months (Fig. 5c).
A second subtype of interest, the “TERT-BRAF-IDH1 subtype” (Supplementary Fig. 9) was characterized by tumors with TERT noncoding mutations or amplifications, combined in some patients with coding alterations to functionally related genes such as BRAF and SKP2. Beyond the synergy between BRAF and TERT mutations as described above, Skp2 is essential for ubiquitination and degradation of p27Kip1 (encoded by CDKN1B)38, which inhibits the activity of hTERT39. Amplification of SKP2 in this pathway may thus increase the activity of hTERT.
A third subtype, “PIK3CA-PEX26-GATA3” (Figs. 5g, h), integrated coding alterations activating PIK3CA and inactivating GATA3 with noncoding alterations downregulating PEX26. In this pathway, members of peroxisomal biogenesis factor (Pex26 and Pex6) appear to indirectly interact with PIK3CA and GATA3 through the binding of SMAD family members (Smad3 and Smad7)40.
Finally, the fourth subtype, “APOBEC2-ARID1A-CTNNB1”, was characterized by the co-occurrence of noncoding mutations within an enhancer of APOBEC2 and coding alterations in ARID1A and CTNNB1. APOBEC2 encodes a nucleic-acid editing enzyme with well-known mutagenic effects in cancer41. Although ARID1A and CTNNB1 are also known cancer drivers, the connections to APOBEC are unanticipated and create a compelling opportunity for further study.
DISCUSSION
Relative to coding changes, interpretation of noncoding mutations poses particular challenges due to the very large number of events and a limited understanding of their functional consequences. Dealing with these challenges requires strategies to boost signal-to-noise, which we have pursued here by integrating mutations with key structural and functional data on transcriptional networks. Structurally, maps of enhancer- and promoter-gene interactions amplify signal by selecting noncoding mutations within defined regulatory regions of specific target genes. These mutations are then characterized functionally as somatic eQTLs by requiring their presence to be significantly associated with expression changes in tumors. The result is a global network of transcriptional regulatory interactions in cancer supported by multiple lines of evidence. Given that most tumors we analyzed had noncoding mutations affecting some part of this network, such mutations appear to represent a widespread feature of cancer biology.
Of the approximately two hundred noncoding mutations that have been previously identified as recurrent in cancer6–8, one third were also identified here as recurrently mutated loci (Fig. 1c), including well known mutations in the promoters of PLEKHS1 and DPH3. Notably though, with the exception of TERT, these mutations did not associate significantly with mRNA expression level changes. This suggests that the effects of these mutations are through mechanisms outside of transcriptional regulation, or that the effects on mRNA expression are weaker than could be detected given our statistical power (Supplementary Fig. 2c). On the other hand, hundreds of somatic eQTLs were identified, all of which were unanticipated other than those in the promoter of TERT. Many of the affected genes are not yet widely appreciated as cancer drivers, motivating further studies on the mechanistic basis of noncoding mutations in cancer.
Given an association between gene expression changes and a somatic mutation, it is important to consider whether this association reflects a causal relationship. Although it is tempting to assume that the occurrence of a mutation drives gene expression changes, the opposite could be true, where the change in gene expression levels drives the appearance of the mutation (e.g., by increased opening and exposure of chromatin). It is also possible that both effects could be due to a third causal factor. However, the three examples we tested experimentally do support a causal link from mutation to expression changes. These results include transcriptional alterations of DAAM1, impacting cell migration (Fig. 3, Supplementary Fig. 4); MTG2, which encodes a GTPase that regulates mitochondrial ribosomes42 (Figs. 4a, b); and HYI, which encodes a putative hydroxypyruvate isomerase and may be involved in carbohydrate transport and metabolism43 (Figs. 4c, d).
Finally, the somatic eQTL analysis introduced here contrasts with germline eQTL studies in several key aspects. First, in GWAS and germline eQTL studies, testing of multiple SNPs is complicated by the strong codependencies among neighboring SNPs at a genomic locus – so-called linkage disequilibrium44,45. In contrast, somatic mutations near to one another in the genome are not in linkage disequilibrium since these alterations, by definition, arise independently in each tumor. Second, population stratification caused by racial diversity has been a major confounder in analysis of germline variants44,45. It is less of a concern for somatic variants, since these are derived from comparisons between tumor and normal genomes from the same individual, eliminating many, if not all, effects due to ancestry. Nonetheless, we controlled for racial diversity and found the impact on somatic eQTL discovery was minimal. Given these aspects, somatic eQTL analysis may have future interest alongside classical eQTLs as a general mode of mapping transcriptional regulatory architecture.
METHODS
Calling and clustering of somatic noncoding mutations
Somatic noncoding mutations of 930 tumors were called as described in the main text. Clusters of noncoding mutations within d = 50 bp from each other were merged using BEDTools46 until no such locus was located within d bp from any other. Loci with mutations in k < 5 tumors were removed from further analyses. The above parameters d and k were chosen to aggregate mutations within short distance with a modest requirement of recurrence. We achieved very similar results when d was within the range of 20 to 60 (inclusive). Whenever a subset of 930 tumors was used in subsequent analyses (Fig. 1a), this set was again filtered to remove those altered in fewer than k tumors within the subset. We also calculated a “concentration score” to penalize loci where mutations were spread over a large region rather than concentrated at a single base pair, as might be expected for sites affecting gene transcription. Within each locus, we selected the mutated position present in the largest number of patients. The proportion of patients affected at that position (out of all patients affected by mutations at that locus) was defined as the concentration score. Loci scoring < 35% were removed from further study. It is worth noting that the threshold of concentration score is somewhat arbitrary and could lead to missing certain loci with multiple closely located somatic mutations. It should also be noted that by clustering noncoding mutations into loci, we assume that all SNVs in a locus act in a similar way. This assumption is consistent with the previously identified SNVs in TERT promoter. Our analysis does not attempt to detect loci in which different SNVs alter gene expression in opposite directions.
RNA-seq, CNA and DNA methylation data processing
RNA-seq, CNA (SNP 6.0) and DNA methylation (Illumina HM450) data for TCGA tumors were downloaded from Firehose (see URLs). The data were processed as follows. First, for RNA-seq the RSEM count for a gene (RNA-Seq by Expectation Maximization)47 was normalized by dividing by the 75th percentile of RSEM values within the tumor sample and multiplying by 1000, as per TCGA practice (see URLs). Genes were retained if the normalized RSEM > 1 in > 50% of tumors, resulting in 16,413 expressed genes. Normalized RSEM values were log2 transformed and z-score standardized for subsequent analyses. Second, for copy number alterations (CNAs) we used the output of GISTIC2, which indicates gene-level CNAs for all samples. The CNAs are in units of (copy number – 2), so that normal copy number (no amplification or deletion) has a value of 0, whereas genes with amplifications have positive values and genes with deletions have negative values. A gene is assigned the highest amplification or the lowest deletion value among the markers it covers. Among the 783 patients with both mRNA expression and genome sequence, 761 also had copy number data available. The remaining patients were assigned 0 for all CNAs. Third, methylation probes were mapped to the promoter regions of genes (± 1 kb from TSS), and each gene was assigned the mean methylation (beta) values of these probes. Among the 783 TCGA patients with both mRNA expression and genome sequences, 605 had methylation data available. Methylation data for the remaining patients were imputed using mean values for the DNA methylation of each gene.
Linking recurrently mutated loci to transcriptional target genes
Our recurrently mutated loci were extended by 100 bp on each side when mapping to the promoters or enhancers. Transcriptional regulatory interactions from recurrently mutated loci to target genes were defined whenever a locus had 50% of its sequence overlap with either the promoter region of a gene (± 1 kb from its TSS) or a gene enhancer region defined by GeneHancer17. In case an enhancer is shorter than a locus, the mapping was performed when 50% of the enhancer sequence overlaps with the locus.
Somatic eQTL analysis using multivariate linear regression
For each gene target linked to recurrently mutated loci, we fit a regression model of the normalized gene expression level e as a function of l, the alteration status of its recurrently mutated loci (1 = mutated; 0 = wt), controlling for the impact of CNA status c (0 = wt; positive values for amplifications; negative values for deletions), DNA methylation m (mean beta-values), 21 tumor tissues t (binary variables), 3 races r (binary variables: Asian; Black or African American; White), gender g (1 = Female; 0 = male) and 20 hidden factors h (real values) as covariates:
| (Eq. 1) |
The hidden factors h were identified using probabilistic estimation of expression residuals (PEER)48,49, while accounting for the effect of known covariates t, r and g. The number of hidden factors was determined by the posterior variance of the factor weights, as previously recommended49. The parameters β were estimated from data from 783 tumors with matched RNA-seq and WGS data. Somatic eQTLs were identified as follows. First, for each gene, we selected features by adding an L1-norm to the objective function based on least squared error between true and predicted gene expression level.
| (Eq. 2) |
The sparsity parameter λ was optimized by cross validation. For genes in which the L1-norm resulted in β1 = 0 for all loci, we decreased λ to include at least one locus. Second, to assess whether mutation status of any locus contributed significantly to gene expression, the accuracy of the complete model was compared to that of a simple model under null hypothesis of no genetic associations (i.e., β1 = 0 for all loci). The F-test p-value between the two nested models was used as the test statistic. Third, having derived an F-test p-value for each gene, q-values were calculated using the Storey approach50 with a threshold of FDR ≤ 20%. And finally, for each gene that passed the selection, this threshold was mapped back to the equivalent F-test p-value of each locus. Loci with F-test p-value below or equal to this threshold were included in the final list and defined as somatic eQTLs.
We elected to perform one test per gene for three reasons. First, in GWAS and typical (germline) eQTL studies, linkage disequilibrium complicates the simultaneous testing of multiple SNPs in a single model, because these SNPs are usually codependent. Unlike inherited SNPs, somatic mutations observed in a tumor population are not in linkage disequilibrium no matter how closely they are located. Therefore, a simple F-test, which assumes independent influences of multiple factors, is sufficient to simultaneously test whether any loci are associated with gene expression. Second, for each gene, all eQTLs share the same set of covariates along with the associated phenotype of mRNA expression level. If multiple eQTLs are associated with gene expression levels, they can be covariates of one another. It is then convenient to fit them all in a single model and enjoy the benefit of gene-based approaches such as feature selection by L1 regularization. Third, there is precedent in the literature to fit gene-level models in eQTL studies51,52.
Power analysis
Statistical power depends on various parameters, including the number of samples, the eQTL effect size, the noise, and the significance threshold. Instead of a simulation based on a model of noise, we evaluated statistical power using the actual data. All locus-gene pairs were plotted in Supplementary Fig. 2c, evaluated by the number of patients with mutations (x-axis) versus the change in gene expression given the mutation (y-axis; defined by ; one unit of W represents one standard deviation of change in residual gene expression). Power was defined as 1 – P(Type II error) at a significance level of P(Type I error) = 0.0085, which is approximately at 20% FDR. We calculated power using the “pwr.f2.test” function in R, where the effect size was calculated based on the proportion of explained variance by two nested models ( ). Our somatic eQTL analysis has 50% power to detect a somatic eQTL with 5 mutations if W > 1.2 or with 10 mutations if W > 0.9.
Independent validation of recurrence
To validate the recurrence of mutations in the identified somatic eQTLs, we downloaded simple somatic mutations (substitutions) called from the WGS of n = 3,382 publicly available non-US donors from the ICGC2. For each eQTL, the number of mutated patients k was used as the test statistic. To determine whether k was greater than expected due to the background mutation rate (BMR), we developed an approach for estimating BMR that was conceptually similar to MutSigCV53. First, a large pool of 20,000 candidate background sequences was created by randomly reassigning (without replacement) the location of the eQTL to the same type of noncoding genomic regions (promoters or putative enhancers17), while retaining the eQTL’s length. Each of these 20,000 sequences was placed in a three-dimensional feature space taking into account nucleotide content, DNA replication timing and gene expression. Nucleotide content was represented as the percentages of all possible mono- (A/T vs. C/G), di- (e.g., AA, AC and AG) and tri-nucleotides (e.g., AAA, AAC and AAG), encoded as a 44-dimensional vector. This information was then compressed into a single feature representing nucleotide content, using the Pearson’s correlation between the vector of the candidate sequence and the vector of the original eQTL. DNA replication timing was obtained from ENCODE via the UCSC Genome Browser54. To create a single replication timing feature, we used the average wavelet-smoothed signal from the following 14 cell lines: BJ, GM06990, GM12801, GM12812, GM12813, GM12878, HeLa-S3, HepG2, HUVEC, IMR-90, K-562, MCF-7, NHEK and SK-N-SH, according to the method of Melton and colleagues6. For gene expression, the median expression value of the nearest gene (log2 transformed RNA-seq data, 783 TCGA patients) was used as a feature. The above three features were z-score standardized. Within this feature space, the top 5% (1,000 out of 20,000) background sequences with the smallest Euclidean distance to the eQTL of interest were selected. For each patient, a patient-specific BMR was estimated as the number of sequences with at least one mutation in that patient out of the 1,000 selected sequences. Finally, we estimated the probability of having observed k or more mutations in n patients in the eQTL of interest using a Poisson binomial model:
| (Eq. 3) |
where Fl is the set of all subsets of k integers that can be selected from {1, 2, …, n}, pi or pj is the probability that patient i or patient j is mutated, A is a set of k integers that can be selected from {1, 2, …, n} and Ac is the complement of A. In practice, we used an approximation for the Poisson binomial in the poibin python package (see URLs).
Transcription factor binding motif analysis
Each reference and somatically altered nucleotide site, along with ± 7 bp flanking sequence, was analyzed using HOMER55 (see URLs). HOMER searches for matches within a library of 319 vertebrate motifs (position weight matrices). Specifically, we ran the “findMotifs.pl” program with default parameters to find motifs from FASTA files. The reference and altered sequences were used as the background for each other to control the nucleotide context. The command line is: findMotifs.pl seqList_mappable_alt.fa fasta log/ -fastaBg seqList_mappable_ref.fa -p 16 -find ~/soft/homer/data/knownTFs/vertebrates/known.motifs.
The list of somatic eQTLs disrupt or create transcription factor binding motifs in four or more patients were reported in Supplementary Table 2.
Prioritizing somatic eQTLs for subsequent functional validation
The three somatic eQTLs selected for functional studies (DAAM1, HYI1 and MTG2) were chosen based on the specific biological interest of the authors and several rules-of-thumb:
The somatic eQTL alters a known transcription factor binding motif in many patients;
The somatic eQTL falls in open chromatin in previously mapped cell lines and conditions (e.g., in regions with markers such as H3K27ac and H3K4me1)11;
The affected target gene has high endogenous mRNA expression levels in cell lines56 that match where the somatic eQTL was detected; and
The somatic eQTL is not present in a region with repetitive DNA.
Note that none of this information was used to filter loci prior to somatic eQTL analysis, as it is not complete, conclusive or cancer-specific.
Generation of reporter plasmids
To examine the effect of the DAAM1 somatic eQTL on gene expression levels, the wild type and mutant regulatory regions, from −233 bp to +148 bp relative to the TSS, including the somatic eQTL at −202 – −191 bp, were synthesized and cloned upstream of GFP (Fig. 3b). For MTG2, the cloned region spanned −200 bp to +200 bp relative to the TSS, including the somatic eQTL at +19 – +33 bp.
For the somatic eQTL located in the HYI enhancer, the region corresponding to +94931 bp to +95332 bp relative to the TSS, including the somatic eQTL at +95097 – +95132 bp, was cloned into the firefly luciferase reporter plasmid pGL4.23 (Promega). The mutations were generated using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs). All inserts for the GFP and luciferase reporter plasmids were confirmed to match the human reference genome hg19 by Sanger sequencing.
Promoter and enhancer activity assays
Cell lines used to evaluate of promoter activity were plated in 6-well dishes at 300,000 cells per well, three replicates per group. The next day, plasmid DNA (1 μg) was transfected using Lipofectamine 3000 (ThermoFisher). Forty-eight hours after transfection, cells were harvested and suspended in ice cold PBS with 1% fetal bovine serum. GFP expression was measured by flow cytometry on a FACSCalibur or FACSCanto (BD Biosciences). Flow cytometry data were analyzed with FlowJo v10 (BD Biosciences). Cells with typical forward (size) and side (granularity) scatter properties were further analyzed for GFP expression. As a negative control, cells were transfected with an empty lentiGuide-Puro plasmid (Addgene) for the DAAM1 experiments (Figs. 3c, d; Supplementary Fig. 4) or a promoter-less GFP plasmid (pRMT-tGFP, Origene) for the MTG2 experiments (Fig. 4b). As a positive control for all GFP experiments, we used a plasmid with the cytomegalovirus promoter upstream of GFP. All flow cytometry experiments were performed at least three times. Early pilot experiments were often performed on single or duplicate samples with then the final triplicate version often performed at least twice.
To evaluate the activity of the enhancer region of HYI, A375 and MDA-MB-231 cells were plated in white, opaque, 96-well plates at 10,000 cells per well, four replicates per group. Cells were transfected 24 hours later using lipofectamine 3000 with 33 ng of total DNA: 27.5 ng of the firefly pGL4.23 constructs and 5.5 ng of control Renilla pGL4.75 (Promega) plasmid. Firefly and Renilla activity were measured 48 hours after transfection using the Dual-Glo Luciferase Assay System (Promega) as per the manufacturer’s instructions. Luciferase values were collected on a BioTek Synergy HT, and data collected via software Gen5 2.01.14. To calculate relative luciferase values, background signal was first subtracted from each channel. Then firefly luminescence was divided by Renilla luminescence. The average value for the wildtype enhancer was set to 1, and the mutated samples were evaluated in comparison to this control. Experiments in both cell lines were performed three times, with each experiment consisting of samples in quadruplicate.
DAAM1 overexpression
Wild type MDA-MB-231 breast cancer cells were transfected with a plasmid encoding the full DAAM1 gene cDNA (Origene catalog number: RC217675). Cells were then selected using G418 (500 μg/mL) for 7 days to ensure stable expression of the DAAM1 construct. DAAM1-overexpression was verified by extracting total protein and quantitating using the Wes electropherogram (Proteinsimple) with anti-DAAM1 antibody (clone WW-3, cat# sc-100942, lot# B1815, Santa Cruz, 1:250 dilution) and anti-tubulin antibody (clone YL1/2, cat# MAB1864, lot# 2886723, Millipore, 1:250 dilution). DAAM1 expression was 5.5 fold greater in cells with DAAM1-overexpression construct relative to wild type (Supplementary Fig. 6e).
3D collagen cell migration assay
Collagen matrices were prepared by mixing cells suspended in culture medium and 10× reconstitution buffer, one-to-one with soluble rat tail type I collagen in acetic acid (Corning)25. Sodium hydroxide was used to normalize pH (pH 7.0, 10 – 20 μL 1M NaOH), and the mixture was placed in 48-well culture plates for polymerization at 37°C. Final gel volumes were approximately 200 μL with final collagen concentration set to 2.5 mg/mL. The polymerized cell-laden hydrogels were incubated for 24 hours under a standard cell culture environment before imaging. Gels were then transferred to a microscope stage-top incubator, and cells were imaged at low magnification (10×) every 2 minutes for 48 hours. Coordinates of the cell location at each time frame were determined using image recognition software (Metamorph/Metavue, Molecular Devices). Tracking data were processed to calculate cell speed using an extension of previously published scripts57. Cell migration assays (Fig. 3f-h) were performed two times and both attempts showed the same trend.
RNA sequencing from cells in 3D culture
In Supplementary Figure 5, cell migration assays were performed using wild type MDA-MB-231 breast cancer cells and HT-1080 fibrosarcoma cells. 3D collagen I gels were seeded in 3 independent experiments and harvested after 24 hours of culture for RNA extraction and directly homogenized in Trizol reagent (Thermofisher). Total RNA was purified using High Pure RNA Isolation Kit (ROCHE) and the integrity of the sample verified using RNA Analysis ScreenTape (Agilent Technologies). Total RNA samples were sequenced using the TruSeq Stranded mRNA Sample Prep Kit (Illumina) and the Illumina MiSeq platform at a depth of > 25 million reads per sample. Paired-end reads were aligned to the hg19 UCSC human genome reference using Bowtie258 and streamed to eXpress59 for transcript abundance quantification.
Tumor genetic profiles integrating noncoding and coding alterations
Integrated genetic alteration profiles were constructed for the 810 tumors with WGS, WES and CNA data (Fig. 1a) as follows. Known oncogenes or tumor suppressors21 were combined with the set of target genes of eQTLs identified by the somatic eQTL analysis (see above); each of these genes was then classified as wild type (0) or altered (1) in each tumor, constituting its tumor genetic profile. In this profile, an alteration was defined as follows: Most oncogenes (e.g., EGFR) were considered altered (activated) if impacted by a missense mutation, in-frame indel, or copy-number amplification. For oncogenes typically altered only by amplification21 (CCND1, MDM2, MDM4, MYC, MYCL, MYCN, NCOA3 and SKP2), only copy number amplifications were considered as alterations and not SNVs or indels. Tumor suppressors (e.g., CDKN2A) were considered altered (inactivated) if there was any type of non-silent mutation or a copy number deletion. For each target gene, we defined a dominant direction of regulation d ∊ {+1, −1} as the sign of the coefficient (β1 in Eq. 1) of its most significantly associated eQTL. Noncoding mutations in eQTLs that lead to a transcriptional change in the dominant direction were considered alterations of such genes. For TERT, copy number amplifications in the coding region was also considered as alterations, since both promoter mutations and gene amplifications have been associated with growth advantage of tumor cells and poor prognosis of patients60,61.
Network-Based Stratification (NBS) to identify tumor subtypes
Network propagation29 was used to compute the pairwise similarities among tumor genetic alteration profiles (see above) within the Reactome functional interaction network (ReactomeFI)31. Each tumor genetic profile was propagated across this network based on a random walk model (equivalent to heat diffusion) with a restart probability of 0.5. After convergence, the score of each gene (temperature) represents its network proximity to genetic alterations. The top 70 principal components of these scores, representing the tumor’s network-transformed profile (Fig. 5a), were analyzed using the sklearn.cluster.SpectralClustering package62 (affinity = k-Nearest-Neighbors, assign-labels = discretize, n_clusters = [2..10]). This method first constructs a similarity graph on all pairs of tumors, where each tumor is connected to the k others with shortest Euclidean distance. We chose k = 170, which ensures the similarity graph to be connected, as previously recommended62. Next this graph is analyzed to partition tumors into subtypes at different resolutions (number of subtypes n = [2..10]). Following spectral clustering, each set of n (parent) subtypes was compared to the n + 1 (child) subtypes to track the similarity of tumor assignments (Fig. 5b). An arrow is drawn from a parent to child subtype if they share ≥ 18 tumors.
Characterizing tumor subtypes with signature genes and subnetworks
For each subtype, we defined a set of “signature genes” as those that had higher network-transformed scores in that subtype than others (t-test, Benjamini Hochberg FDR < 0.1) and, among these, were more frequently altered in that subtype (Fisher Exact Test, FDR < 0.05, Figs. 5b-e). To identify subnetworks, this set was expanded to include “intermediate genes” with relatively high network-transformed score (t-test, FDR < 0.05) that lie on the shortest paths between each pair of signature genes. The union of the signature and intermediate genes was used to induce a subnetwork within ReactomeFI31, referenced in the main text as the corresponding “pathway” impacted in that subtype (Figs. 5d). An additional filter was applied in Fig. 5e and Supplementary Fig. 9a, where we only visualized the signature genes with 10 or more mutations and the shortest paths among them with at most one intermediate gene. All networks were visualized in Cytoscape63.
Survival analysis
We used the “coxph” package in the R statistics software to fit Cox proportional hazard models64. P-values were calculated by log likelihood ratio test. To evaluate whether the subtype classifications provide additional prognostic power beyond the baseline survival expectancy due to cancer tissue, we compared the likelihood for the complete model, including NBS-derived molecular subtypes s and cancer tissues c as covariates, against that of a null model that includes cancer tissues c only:
| (Eq. 4) |
| (Eq. 5) |
where is the baseline hazard function. Then a log-likelihood ratio statistic was defined as:
| (Eq. 6) |
Finally, a chi-squared test p-value was calculated based on D with the number of degrees of freedom equal to the number of NBS-derived molecular subtypes.
Data availability
Somatic mutations of 930 tumors are publicly available (see URLs). RNA-seq data is accessible through the GEO Series accession number GSE101209.
Code availability
Custom code for annotating mutations, somatic eQTL analysis, validation of recurrence, motif analysis and Network-Based Stratificaion are available through GitHub (see URLs).
Supplementary Material
Acknowledgments
The results published here are in whole or part based upon data generated by the TCGA Research Network (see URLs). We would also like to acknowledge the clinical contributors and the data producers from the ICGC who have generated the particular datasets and made them available for public analysis. This work was supported by NIH grants to T.I. (U24CA184427, U54CA209891, P50GM085764, P41GM103504 and R01HG009979) and H.C. (DP5OD017937). G.X. is supported by a UCSD CTRI grant (UL1TR001442). S.I.F. and D.O.V. are supported by a Burroughs Wellcome Fund Career Award at the Scientific Interface (1012027), an NSF CAREER Award (1651855) and UCSD CTRI and FISP pilot grants. We would like to thank members of the Ideker laboratory for valuable comments and critical reading of the manuscript. Finally, we wish to thank the patients and their families for their contributions of valuable data without which this project would not have been possible.
Footnotes
URLs. TCGA Research Network, http://cancergenome.nih.gov/; Firehose, https://confluence.broadinstitute.org/display/GDAC/Home; TCGA RNASeq data description, https://wiki.nci.nih.gov/display/TCGA/RNASeq+Version+2; The poibin python package, https://github.com/tsakim/poibin; HOMER, http://homer.ucsd.edu/homer/index.html; Somatic mutations of 930 tumors, http://ideker.ucsd.edu/papers/wzhang2017/; GitHub site for custom code, https://github.com/wzhang1984/Noncoding-tumor-mutation-paper.
AUTHOR CONTRIBUTIONS
W.Z. and T.I. conceived the study. W.Z. designed and performed most of the analyses. G.X. performed mutation calling of 358 tumors. C.M. and M.S. provided mutation calling of 572 tumors. A.B., K.S.S., J.P.S., K.M.O. and E.K.F. performed the somatic eQTL reporter assays. A.B. and J.F.K. analyzed the flow cytometry and luciferase assay data. A.B., J.P.S. and K.L. performed protein electropherogram analysis. D.O.V., K.C. and S.I.F. performed 3D cell culture assays. M.K.Y. and H.C. helped W.Z. in designing the somatic eQTL analysis. J.K.H. helped W.Z. in network analysis. T.I., J.F.K and W.Z. wrote the paper and formulated all figures.
COMPETING FINANCIAL INTERESTS
Trey Ideker is co-founder of Data4Cure, Inc. and has an equity interest. Trey Ideker has an equity interest in Ideaya BioSciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Cancer Genome Consortium et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hofree M, et al. Challenges in identifying cancer genes by analysis of exome sequencing data. Nat Commun. 2016;7:12096. doi: 10.1038/ncomms12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iorio F, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khurana E, et al. Role of non-coding sequence variants in cancer. Nat Rev Genet. 2016;17:93–108. doi: 10.1038/nrg.2015.17. [DOI] [PubMed] [Google Scholar]
- 6.Melton C, Reuter JA, Spacek DV, Snyder M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet. 2015;47:710–716. doi: 10.1038/ng.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46:1160–1165. doi: 10.1038/ng.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46:1258–1263. doi: 10.1038/ng.3141. [DOI] [PubMed] [Google Scholar]
- 9.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman MM, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roadmap Epigenomics Consortiumet al., editors. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol. 2015;33:364–376. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics. :11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishilevich S, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017;2017 doi: 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogelstein B, et al. Cancer Genome Landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 2001;107:843–854. doi: 10.1016/s0092-8674(01)00614-6. [DOI] [PubMed] [Google Scholar]
- 23.Liu W, et al. Mechanism of activation of the Formin protein Daam1. Proc Natl Acad Sci U S A. 2008;105:210–215. doi: 10.1073/pnas.0707277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, et al. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS One. 2012;7:e37823. doi: 10.1371/journal.pone.0037823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraley SI, et al. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraley SI, et al. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci Rep. 2015;5:14580. doi: 10.1038/srep14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velez DO, et al. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat Commun. 2017;8:1651. doi: 10.1038/s41467-017-01556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofree M, et al. Network-based stratification of tumor mutations. Nat Methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fatyol K, Szalay AA. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (HIF-1alpha) and inhibits HIF-1-mediated transcription. J Biol Chem. 2001;276:28421–28429. doi: 10.1074/jbc.M102847200. [DOI] [PubMed] [Google Scholar]
- 33.Nishi H, et al. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT) Mol Cell Biol. 2004;24:6076–6083. doi: 10.1128/MCB.24.13.6076-6083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan QW, et al. EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci Signal. 2009;2:ra4. doi: 10.1126/scisignal.2000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawauchi K, Ihjima K, Yamada O. IL-2 increases human telomerase reverse transcriptase activity transcriptionally and posttranslationally through phosphatidylinositol 3′-kinase/Akt, heat shock protein 90, and mammalian target of rapamycin in transformed NK cells. J Immunol. 2005;174:5261–5269. doi: 10.4049/jimmunol.174.9.5261. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci U S A. 2016;113:14402–14407. doi: 10.1073/pnas.1611106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper MT, Bray SJ. Frizzled regulation of Notch signalling polarizes cell fate in the Drosophila eye. Nature. 1999;397:526–530. doi: 10.1038/17395. [DOI] [PubMed] [Google Scholar]
- 38.Spruck C, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 39.Lee SH, et al. IFN-gamma/IRF-1-induced p27kip1 down-regulates telomerase activity and human telomerase reverse transcriptase expression in human cervical cancer. FEBS Lett. 2005;579:1027–1033. doi: 10.1016/j.febslet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Warner DR, Roberts EA, Greene RM, Pisano MM. Identification of novel Smad binding proteins. Biochem Biophys Res Commun. 2003;312:1185–1190. doi: 10.1016/j.bbrc.2003.11.049. [DOI] [PubMed] [Google Scholar]
- 41.Okuyama S, et al. Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis. Int J Cancer. 2012;130:1294–1301. doi: 10.1002/ijc.26114. [DOI] [PubMed] [Google Scholar]
- 42.Hirano Y, Ohniwa RL, Wada C, Yoshimura SH, Takeyasu K. Human small G proteins, ObgH1, and ObgH2, participate in the maintenance of mitochondria and nucleolar architectures. Genes Cells. 2006;11:1295–1304. doi: 10.1111/j.1365-2443.2006.01017.x. [DOI] [PubMed] [Google Scholar]
- 43.Ashiuchi M, Misono H. Biochemical evidence that Escherichia coli hyi (orf b0508, gip) gene encodes hydroxypyruvate isomerase. Biochim Biophys Acta. 1999;1435:153–159. doi: 10.1016/s0167-4838(99)00216-2. [DOI] [PubMed] [Google Scholar]
- 44.Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8:e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 46.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol. 2010;6:e1000770. doi: 10.1371/journal.pcbi.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michaelson JJ, Alberts R, Schughart K, Beyer A. Data-driven assessment of eQTL mapping methods. BMC Genomics. 2010;11:502. doi: 10.1186/1471-2164-11-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gamazon ER, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansen RS, et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinz S, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu PH, Giri A, Sun SX, Wirtz D. Three-dimensional cell migration does not follow a random walk. Proc Natl Acad Sci U S A. 2014;111:3949–3954. doi: 10.1073/pnas.1318967111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao Y, Bryan TM, Reddel RR. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci. 2008;99:1092–1099. doi: 10.1111/j.1349-7006.2008.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie H, et al. TERT promoter mutations and gene amplification: promoting TERT expression in Merkel cell carcinoma. Oncotarget. 2014;5:10048–10057. doi: 10.18632/oncotarget.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Von Luxburg U. A tutorial on spectral clustering. Stat Comput. 2007;17:395–416. [Google Scholar]
- 63.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andersen PK, Gill RD. Cox’s Regression Model for Counting Processes: A Large Sample Study. Ann Stat. 1982;10:1100–1120. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Somatic mutations of 930 tumors are publicly available (see URLs). RNA-seq data is accessible through the GEO Series accession number GSE101209.