

Abstract
Background
Non-human primates have long been identified to harbour different species of Plasmodium. Long-tailed macaques (Macaca fascicularis), in particular, are reservoirs for P. knowlesi, P. inui, P. cynomolgi, P. coatneyi and P. fieldi. A previous study conducted in Sarawak, Malaysian Borneo, however revealed that long-tailed macaques could potentially harbour novel species of Plasmodium based on sequences of small subunit ribosomal RNA and circumsporozoite genes. To further validate this finding, the mitochondrial genome and the apicoplast caseinolytic protease M genes of Plasmodium spp. were sequenced from 43 long-tailed macaque blood samples.
Results
Apart from several named species of malaria parasites, long-tailed macaques were found to be potentially infected with novel species of Plasmodium, namely one we refer to as “P. inui-like.” This group of parasites bifurcated into two monophyletic clades indicating the presence of two distinct sub-populations. Further analyses, which relied on the assumption of strict co-phylogeny between hosts and parasites, estimated a population expansion event of between 150,000 to 250,000 years before present of one of these sub-populations that preceded that of the expansion of P. knowlesi. Furthermore, both sub-populations were found to have diverged from a common ancestor of P. inui approximately 1.5 million years ago. In addition, the phylogenetic analyses also demonstrated that long-tailed macaques are new hosts for P. simiovale.
Conclusions
Malaria infections of long-tailed macaques of Sarawak, Malaysian Borneo are complex and include a novel species of Plasmodium that is phylogenetically distinct from P. inui. These macaques are new natural hosts of P. simiovale, a species previously described only in toque monkeys (Macaca sinica) in Sri Lanka. The results suggest that ecological factors could affect the evolution of malaria parasites.
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1170-9) contains supplementary material, which is available to authorized users.
Keywords: Long-tailed macaque, Macaca fascicularis, Plasmodium, Population expansion
Background
Species in the genus Plasmodium (Apicomplexa: Haemosporida) are vector-borne blood parasites that infect a wide range of hosts, some of which cause the disease malaria [1] in humans. There are approximately 250 species of Plasmodium identified in mammals, birds and reptiles [2]. The number of Plasmodium species infecting non-human primates (apes, gibbons, New World Monkeys and Old World Monkeys) is estimated to be more than 30 [2] with non-human primates in Asia harbouring approximately 13 of these species [3]. Of these, six species (P. knowlesi, P. inui, P. cynomolgi, P. fieldi, P. coatneyi and P. fragile) infect two or more species of macaques (M. fascicularis, M. nemestrina, M. mulatta, M. arctoides, M. cyclopsis, M. sinica, M. radiate and M. assamensis) and silvered leaf monkeys (Trachypithecus cristatus) in nature [4]. Plasmodium simiovale is restricted to toque macaques (M. sinica) of Sri Lanka, while P. fragile has been identified in macaques (M. mulatta and M. radiata) in both India and Sri Lanka [4]. Of the other six, P. hylobati, P. eylesi, P. jefferyi and P. youngi are found in gibbons (Hylobates lar, H. moloch and H. leusciscus) while P. pitheci and P. silvaticum are found in orangutans (Pongo pygmaeus) of Borneo [5].
The interest in primate malarias escalated with the discovery of the simian malaria P. knowlesi infecting humans in Southeast Asia [6, 7] and the discovery of novel Plasmodium species in non-human primates [8–13]. This was made possible using molecular approaches in the field of malariology [14–18]. In the absense of morphological evidence, species which are phylogenetically well-defined and distinct from extant species can be distinguished using suitable DNA markers. Through phylogenetic analysis of mitochondrial genomes (mtDNA), as well as nuclear and apicoplast genes, unique species of Plasmodium in African apes were discovered (P. billbrayi, P. billcollinsi) and lineages closely related to P. vivax, P. reichnowi and P. ovale in chimpanzees [9, 10, 13] parasites closely related to P. malariae and P. falciparum in bonobos [10] and lineages closely related to P. falciparum and P. vivax in gorillas [8, 11, 12]). Transfers of Plasmodium between different hosts is aided by opportunistic vectors. These vectors are attracted to various vertebrate hosts for blood meals and are not host-specific [19, 20]. Hence, the role of these vectors in the transmission of Plasmodium across host ranges incriminates them as bridge vectors. As bridge vectors, these mosquitoes may lead to the emergence of new zoonotic infections, should they also feed on humans.
The current study focuses on the malaria parasites of the long-tailed macaques (Macaca fascicularis) in the state of Sarawak, Malaysian Borneo. These long-tailed macaques have a vast distribution across the Southeast Asia region including Borneo [21] and were identified in the Kapit division of Sarawak, Malaysian Borneo as the natural hosts for P. knowlesi, P. inui, P. cynomolgi, P. coatneyi and P. fieldi using PCR detection assays [18]. Phylogenetic analysis of the small subunit ribosomal RNA (SSU rRNA) and circumsporozoite (csp) genes of Plasmodium parasites derived from these macaques suggested the presence of at least two potentially novel species of Plasmodium [22]. However, these genes are not suitable to discriminate closely related species of Plasmodium [23–25]. The distinct rRNA loci in Plasmodium that are expressed at different stages of the life cycle present potential problems with paralogy [23–25]. On the other hand, the csp gene (a surface protein gene) is under selective pressure of the host immune system [24, 25] and as such is not a neutral marker.
In this study, we have characterised the mitochondrial (mtDNA) and apicoplast caseinolytic protease M (ClpM) genes, which are suitable phylogenetic markers [25–33] to ascertain the existence of novel species of Plasmodium. Based on the mtDNA genomes, the demographic history of these Plasmodium species was also investigated. A previous study conducted in Kapit, Malaysian Borneo estimated a population expansion for the P. knowlesi at 30,000 to 40,000 years before present [18]. Therefore, the demographic histories of the other Plasmodium species infecting these macaques were estimated to investigate whether a similar expansion was observed among the other parasites. Our phylogenetic analyses provide a robust support for the presence of complex Plasmodium infections among the long-tailed macaques of Malaysian Borneo and the existence of a novel species of Plasmodium.
Methods
Detection of Plasmodium spp. in macaque blood samples
A total of 43 long-tailed macaques were captured from different locations in Sarawak [18]. Blood was obtained from anaesthetised animals prior to their release. The blood smears were stained with Giemsa and examined under the microscope. DNA was extracted from the macaque blood samples using the QIAampDNA Mini kit (QIAGEN, Germany) according to the manufacturer’s protocol. All but one (LT7) of 43 long-tailed macaques was found to be infected with multiple species of Plasmodium by species-specific nested PCR assays for P. inui, P. knowlesi, P. cynomolgi, P. fieldi and P. coatneyi [6, 18].
Sequencing of mtDNA and ClpM
The complete Plasmodium mitochondrial genome was amplified by PCR with Plasmodium-specific primers: Pkmt F1 (5’-GGACTTCCTGACGTTTAATAACGAT-3′) and Pkmt R1 (5’-TGGACGTTGAATCCAATAGCGTA-3′) [18]. PCR amplification for each sample was performed in a 20 μL reaction mixture containing 10 mM dNTP mix, 5× buffer A, 5× buffer B, 0.25 μmol/L of each primer (Pkmt F1 and Pkmt R1), 0.8 μL Elongase enzyme (Invitrogen, USA) and 4 to 8 μL of purified genomic DNA under the following conditions: 94 °C for 30 s for first denaturation, 40 cycles at 94 °C for 30 s, 55 °C for 30 s and 68 °C for 5 mins, followed by a final extension for 10 mins at 68 °C. The PCR amplified mtDNA fragment was gel purified using S.N.A.P UV Gel Purification kit (Invitrogen, Life Technologies, USA). Purified mtDNA fragments were cloned into pCR-XL-TOPO vector (Invitrogen, USA) and transformed into One Shot electrocompetent Escherichia coli cells by electroporation and then plated onto LB agar containing (50 μg/mL) kanamycin. Recombinant plasmids containing the mtDNA fragment were purified using the SNAP miniprep kit (Invitrogen, USA). Finally, the mtDNA genome was sequenced using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems) under the following cycling conditions: 96 °C for 1 min for first denaturation, 30 cycles at 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 mins. Sequencing was carried out on a ABI377 sequencer (Applied Biosystems, USA). At least 2 clones were sequenced from each isolate using M13 primers and 14 internal primers (Additional file 1), with both DNA strands sequenced for each clone.
A portion of the Plasmodium apicoplast ClpM gene (of the AAA+ motif region) was amplified by PCR with primers ACLP-F1 (5′-GGTAGTTGGATTTTATGTGG-3′) and ACLP-R1 (5′- CGWGCTCCATATAAAGGAT-3′) with the latter modified from a previous study [26]. PCR amplification of the ClpM gene was performed in a 20 μL reaction mixture containing 2 μL of purified genomic DNA, 200 μM each deoxyribonucleotide triphosphate (dNTP) (Promega,Madison WI, USA), Phusion HF buffer (1.5 mM MgCl2), 0.02 U/ μL of Phusion DNA polymerase (ThermoScientific, USA) and 0.5 μM of each primer (ACLP-F1 and ACLP-R1) under the following conditions: 98 °C for 30s for first denaturation, 35 cycles at 98 °C for 7 s, 60 °C for 20s and 72 °C for 30s, followed by a final extension for 10mins at 72 °C. PCR products were separated by electrophoresis in a 1% agarose gel, stained with SYBR® DNA gel stain (Invitrogen, USA) and visualised under a UV transilluminator (GBOX from Syngene). Those samples positive for Plasmodium were each purified using Gel/PCR DNAFragment Extraction kit (Geneaid, Taiwan), cloned into the Zero Blunt® vector (ThermoFisher Scientific), transformed into One Shot® TOPO10 Chemically Competent E. coli using heat-shock, and then plated onto LB agar containing (50 μg/mL) kanamycin. To determine whether the transformed E. coli harbouring recombinant plasmid with ClpM insert, the colonies were examined by PCR using the ClpM-specific primers ACLP-F1 and ACLP-R1. Recombinant plasmids containing the ClpM gene fragment were purified using PureLink® Quick Plasmid DNA Miniprep Kits (Invitrogen, USA). The ClpM gene was sequenced using the BigDye® Terminator Cycle Sequencing kit (Applied Biosystems, USA) as described for the mtDNA genome. The products were then sequenced on a AB1377 sequencer (Applied Biosystems). At least 2 clones from each PCR set of each sample were sequenced using M13 primers. In addition to the Plasmodium sp. from macaque blood samples, ClpM gene sequences were also derived from 2 human infections in the Kapit division of Sarawak, Malaysian Borneo.
Sequence analyses of mtDNA of Plasmodium spp.
The mtDNA genome sequences were aligned using the Lasergene package (DNASTAR). Measures of polymorphism and genetic variation were performed using DnaSP v5.10.00 [34]. Pairwise genetic distances of the mtDNA sequences were estimated using PAUP version 4.0b10 [35] based on Bradley & Baker’s [36] genetic species concept [37–39]. A minimum-spanning network connecting the mtDNA haplotypes of each Plasmodium species, based on the statistical parsimony method, was constructed using Network5000 [40].
The demographic expansion of each species of Plasmodium was tested based on pairwise mismatch distribution using Arlequin v3.1 software [41]. The observed mismatch distribution was compared with the estimated mismatch distributions under the sudden demographic expansion model using a generalised least-square approach [42]. Deviations from the population expansion model were tested using the Harpending’s raggedness index [43] with a parametric bootstrap of 1000 replicates.
Tests of neutrality based on Tajima’s D [44], Fu and Li’s D and F [45] and Fay and Wu’s H [46] statistics were calculated using the software DnaSP v5.10.00 [342]. The mtDNA genome sequence of P. coatneyi (GenBank accession no. AB354575) was used as the outgroup to calculate these statistics.
The Bayesian Markov Chain Monte Carlo (MCMC) method implemented in the BEAST package v1.7.5 was used to infer the time to the most recent common ancestor (TMRCA) and the past population dynamics of each species of Plasmodium [47]. The mean substitution rate of mtDNA and TMRCA of each species of Plasmodium was estimated based on a time-calibrated Bayesian phylogenetic analysis of non-human primate malarias (P. gonderi, Plasmodium sp. (Mandrill), P. simiovale, P. fragile, P. cynomolgi, P. knowlesi and P. fieldi) and human malarias (P. falciparum, P. vivax, P. malariae and P. ovale), assuming a strict co-divergence of the parasites with their host lineages, the divergence of the malaria parasites (P. gonderi and P. sp. in Mandrill) found in African Old World Monkeys from those parasites found in macaques in Southeast Asia when Macaca branched from Papio approximately between 6 and 14.2 million years ago [38, 48]. The GenBank accession numbers of all the referral sequences are provided in Additional file 2. The General Time Reversal (GTR) nucleotide substitution model with gamma distribution (G) and a proportion on invariable sites (I), a molecular clock model (uncorrelated relaxed clock) and a coalescent model (Bayesian skyline) were used for this analysis with 250 to 500 million generations of MCMC chains with the first. The best-fit nucleotide substitution model based on the likelihood ratio test and the AIC was selected using Modeltestv3.7 [49]. The convergence of the chain was confirmed by inspecting the MCMC samples using the program Tracer v1.5 with the first 10% sampling of the MCMC chains was discarded as burn-in (default), where the sample size (ESS) is greater than 200 for all continuous parameters [50]. The trees produced by BEAST were then annotated using Tree Annotater and finally the maximum clade credibility tree was visualised using the FigTree v1.3.1 program.
Past populations dynamics (change in effective population size (Ne)) of a single species through time [51] were analysed using the estimated mean substitution rate recorded from the previous analysis with 500 million MCMC chains. All parameters in the analyses were determined to have reached convergence when ESS for all parameter was more than 200. Both the log and tree output files were then used to draw the Bayesian skyline reconstruction plot in Tracer v1.5.
The Plasmodium mtDNA sequences generated in this study were deposited in GenBank under the accession numbers KX645877-KX645965 and KU254034-KU254057 (Additional file 3).
Sequence analyses of partial apicoplast ClpM gene of Plasmodium spp.
The ClpM sequence data were aligned using Lasergene package (DNASTAR). The genetic distance of the partial apicoplast ClpM sequences were estimated using PAUP version 4.0b10 [35] based on Bradley & Baker’s (2001) genetic species concept [37, 39]. For Bayesian phylogenetic inference, the Bayesian Markov Chain Monte Carlo (MCMC) method implemented in the BEAST package v1.7.5 was used [48]. The GenBank accession numbers of all the referral sequences are provided in Additional file 4.
At least 2 independent runs were performed for each genome and the convergence for all parameters were estimated based on the values of ESS more than 200 for all analyses implemented in BEAST.
The partial ClpM sequences of Plasmodium species generated in this study were submitted to GenBank under the accession numbers KX158739-KX158831 (Additional file 5).
Results
Mitochondrial DNA
A total of 113 complete Plasmodium mtDNA genome sequences (5908 – 5938 bp) were generated from the 43 long-tailed macaques sampled. The Bayesian phylogenetic inference of these sequences with several other reference sequences from other studies showed close phylogenetic relationships of 89 sequences to P. inui, four to P. simiovale, 13 to P. cynomolgi, three to P. coatneyi and four to P. knowlesi (Fig. 1).
Fig. 1.
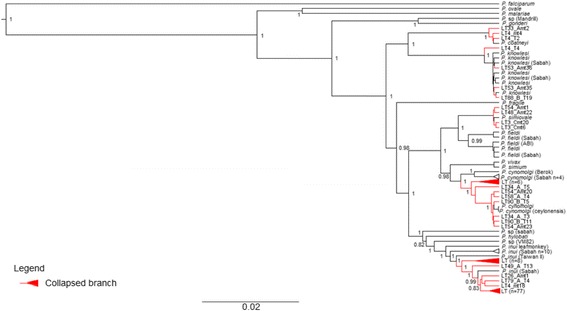
Phylogenetic analysis of Plasmodium spp. based on complete mitochondrial genomes. Maximum clade credibility phylogenetic tree highlights each corresponding segments to the relatedness of the sequences to specific species. The numbers given at the nodes represent the posterior probability values. The accession number of the sequences of Plasmodium spp. are provided in the Additional files 2 and 3
The 89 sequences that were closely related to P. inui were subjected to evolutionary and demographic analyses. These sequences were derived from samples collected in the Kapit (n = 81), Matang (n = 2) and Sarikei (n = 6) districts of Sarawak, Malaysian Borneo. Mitochondrial haplotypes from Matang and Sarikei were not genetically different than those from Kapit division. Sequence alignment revealed 560 polymorphic sites across the 5918 bp nucleotide sequences, 417 singleton variable sites and 143 parsimony informative sites. The nucleotide diversity was estimated at 0.00415, higher than the value (0.00075) observed for P. knowlesi in Kapit [18].
The time to the most recent common ancestor (TMRCA) for these P. inui-like sequences was estimated using the Bayesian coalescent approach [52]. A nucleotide substitution rate for the mitochondrial genome of 4.203 × 10− 9 (95% HPD: 2.19-6763 × 10− 9) substitutions per site per year was estimated by comparing the parasites of Asian macaques with P. gonderi (a parasite of African mangabeys) (AB434918) and Plasmodium sp. (Mandrill) (AY800112). The comparison was based on the assumption that the parasite lineages separated when Asian Old World monkeys and African Old World monkeys diverged 10 million years ago [37]. The TMRCA of the P. inui-like clades was estimated at approximately 1.5 million years before present (95% HPD: 6.76 × 105 – 2.64 × 106) (Fig. 2). In addition, the 89 sequences formed a clear paraphyletic clade showing the presence of two sub-clades, sub-clade A and sub-clade B with a posterior probability of 1.
Fig. 2.
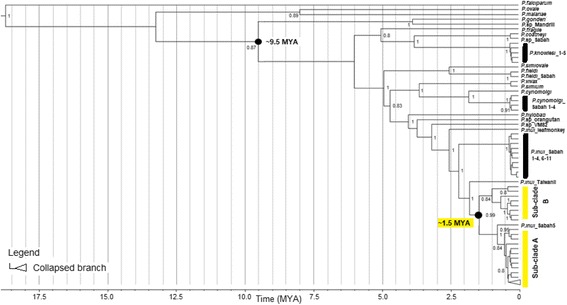
Time calibrated maximum clade credibility phylogeny based on the mtDNA of Plasmodium species of primates. The numbers given at the nodes represent the posterior probability values. TMRCA and Highest Posterior Density (HPD) for P. inui-related are indicated
The average genetic distance was estimated at 1.26 – 2.16% between the 89 sequences and the reference P. inui sequences, whereas the distance between both the sub-clades and P. inui mtDNA sequences derived from Sabah were estimated at 0.405 – 2.270%, with one (GenBank accession no. KJ569834) of the 11 haplotypes from Sabah falling within sub-clade A. These values were comparable to the genetic distance between P. vivax and P. cynomolgi, which was estimated at 1.2% (Table 1). The average genetic distance of mtDNA haplotypes of this species suggests that they belong to a lineage that is closely related to P. inui and is possibly a novel species of Plasmodium.
Table 1.
Sequence divergence values (%) for intra and inter group were calculated using a Kimura 2-parameter model of evolution and are given as percentages
| P. inui Ref (n = 3) | P. inui Sabah (n = 11) | |
|---|---|---|
| P. inui-like (n = 89) | 1.261 – 2.161 | 0.405 – 2.270 |
| Sub-clade A (n = 81) | 1.297 – 2.089 | 0.405 – 2.270 |
| Sub-clade B (n = 8) | 1.261 – 2.161 | 0.885 – 2.161 |
| P. inui Ref (n = 3) | – | 1.387 – 2.253 |
| Genetic distance (%) | ||
| Within sub-clade A (n = 81) | 0.018 – 0.955 | |
| Within sub-clade B (n = 8) | 0.618 – 1.781 | |
| Within P. inui Sabah (n = 11) | 0.018 – 1.980 | |
To further study the demographic history of these P. inui-like parasites, a median joining network of the 89 sequences was generated (Fig. 3). This network also included 14 P. inui haplotypes (from Sabah, Peninsular Malaysia and Taiwan), which had been previously characterised [10, 37, 40]. The 88 haplotypes (only two sequences shared the same haplotype) clearly formed two distinct clades that were distinct from the reference sequences. Sub-clade A (n = 81) and sub-clade B (n = 8) formed a separate group from P. inui sequences generated from macaque samples from the state of Sabah, Malaysian Borneo [40] indicating possible population structure between macaque troops in Malaysian Borneo. The star-like structure of the haplotype genealogical network of sub-clade A is indicative of an evolutionary population expansion. This was further supported by the unimodal shape of pairwise mismatch distribution (Fig. 4), and a low Harpending’s raggedness index (r = 0.0021, P = 0.993). In addition, the negative, significant neutrality test statistics also suggest that the deviation from neutrality could have been due to an expansion of this P. inui-like parasite population (Table 2). The presence of a second smaller peak in the mismatch distribution suggests that there may be more than one distinctive lineage in sub-clade A.
Fig. 3.
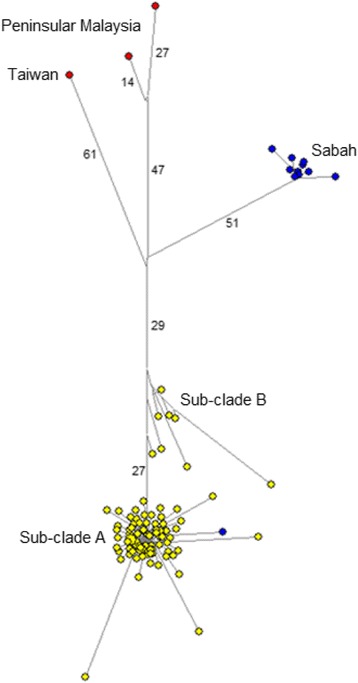
Schematic diagram of genealogical network for referral P. inui and P. inui-like haplotypes. Yellow circles represent sequences generated from this study, while, blue circles for sequences from Sabah and red for sequences from referral cases
Fig. 4.
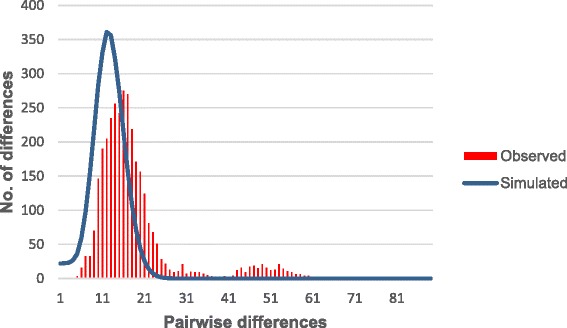
Mismatch distribution of pairwise number of differences in P. inui- like sub-clade A. The red bars represent the observed frequencies and the blue line represents the expected curve for a population that has undergone a demographic expansion
Table 2.
Neutrality tests
| Neutrality tests | All 89 sequences | Sub-clade A (n = 81) | Sub-clade B (n = 8) |
|---|---|---|---|
| Tajima’s D | −2.66949 *(P < 0.001) | − 2.8824 *(P < 0.01) | −1.1237 (P > 0.10) |
| Fu and Li’s D | −7.36191 *(P < 0.02) | −7.7211 *(P < 0.02) | −1.1250 (P > 0.10) |
| Fu and Li’s F | −6.40442 *(P < 0.02) | −6.8115 *(P < 0.02) | − 1.2561 (P > 0.10) |
| Fay and Wu’s H | −164.3309 (FW-Hn = − 2.6186) |
*reflects significant P values
To further investigate the demographic process of the P. inui-like population, changes in effective population size were estimated for sub-clade A through time using a Bayesian skyline plot [52]. The plot indicated that sub-clade A underwent a population expansion between approximately 150,000 to 250,000 years before present (Fig. 5).
Fig. 5.
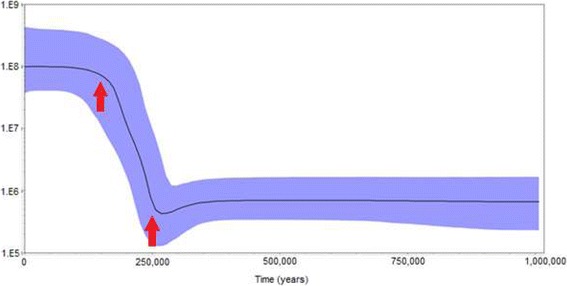
Bayesian skyline plot displaying changes in effective population size (Ne) through time for P. inui-like sub-clade A. The red arrows mark the period of the population expansion of sub-clade A
Phylogenetic analysis also revealed the presence of four haplotypes that formed a monophyletic clade with P. simiovale (Fig. 6). The genetic distance estimated between the four haplotypes from this study and the P. simiovale reference sequence (GenBank accession number: AB434920) ranged from 0.16% to 0.17%. Of the seven distinguishable point mutations observed between the present data and the published sequence of P. simiovale (GenBank accession number: AB434920), five occurred within coding regions of the P. simiovale mitochondrial genome. Only one of the five mutations resulted in an amino acid change from serine (published reference) to leucine (four sequences from the present data) in the cytochrome oxidase III gene.
Fig. 6.
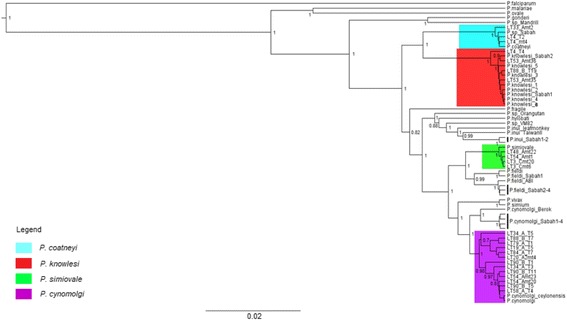
Maximum clade credibility phylogeny based on mtDNA of Plasmodium species of primates for P. simiovale (green), P. cynomolgi (purple), P. knowlesi (red) and P. coatneyi (blue) haplotypes. The numbers given at the nodes represent the posterior probability values
Partial apicoplast ClpM gene
A total of 71 partial apicoplast ClpM gene sequences (677 bp) of Plasmodium sp. were generated from 12 long-tailed macaques from Kapit, while 18 sequences were obtained from three long-tailed macaques from Sarikei and Matang divisions in Sarawak. In addition, four sequences of P. knowlesi partial ClpM genes were also generated from the blood samples of two patients from Kapit hospital in Sarawak collected during a previous study by Lee et al. [18].
Overall, Bayesian inference showed that 37 of the 89 partial ClpM gene sequences were of P. knowlesi, 39 were P. inui-like, five were likely P. coatneyi, four were P. cynomolgi and four were P. simiovale (Fig. 7). The paraphyletic nature of the CLpM P. inui-like haplotypes supports the mtDNA findings. Furthermore, partial sequences of the ClpM genes have also confirmed the presence of P. simiovale among the long-tailed macaques of Malaysian Borneo.
Fig. 7.
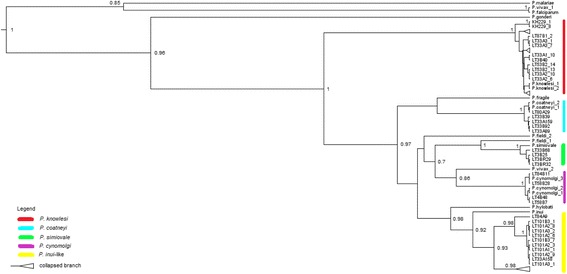
Phylogenetic analysis of Plasmodium sp. based on partial ClpM genes. Maximum clade credibility phylogenetic tree highlights each corresponding segments to the relatedness of the sequences to specific species; yellow for P. inui, purple for P. cynomolgi, green for P. simiovale, blue for P. coatneyi and red for P. knowlesi. The numbers given at nodes represent the posterior probability values. The accession number of the sequences of Plasmodium sp. are provided in the Additional files 4 and 5
Morphological characteristics of the P. inui-like parasites
Only one of the 43 macaques (LT7) was identified with a single-species infection of Plasmodium, based on examination with nested PCR assays and sequencing of the mitochondrial genome and ClpM gene. The morphological characteristics of the P. inui-like parasites from macaque LT7 were compared to those of P. inui described by Coatney et al. [1]. The early stages appeared in a form of a ring with single and fairly large nucleus (Fig. 8a-c). Just as for P. inui, the size of the vacuole increased as the parasites grew (Fig. 8d). Growing trophozoites had dense cytoplasm with stippling and pigmentation (Fig. 8e-f), which is also characteristic of P. inui. No obvious host cell enlargement was observed as the trophozoites grew. Immature and mature trophozoites occupied half to two-thirds of the infected red blood cell and showed a serrated and irregular periphery (Fig. 8g-i). As the parasites developed into schizonts, stippling was sparse and the cytoplasm appeared to be irregular in shape (Fig. 8j). Mature schizonts had yellowish brown pigment mass and no cell enlargement was observed. The number of merozoites per schizont varied between 4 to 12 (Fig. 8k-l). The cytoplasm of the gametocytes stained blue and pink (Fig. 8m-n). All the characteristics above are similar to those described for P. inui [1].
Fig. 8.
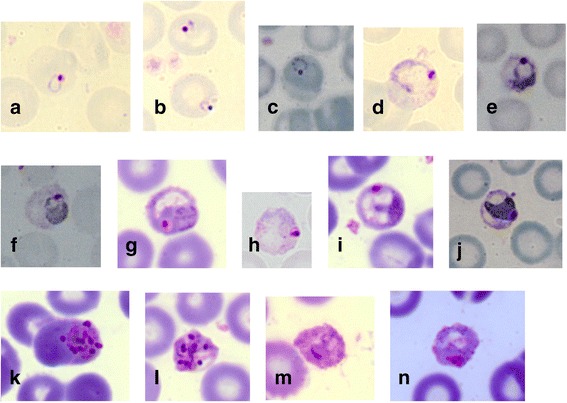
Giemsa-stained blood films of P. inui-like parasite from the long-tailed macaque, LT7. All development stages were observed; early trophozoites or ring forms (a-c), developing trophozoites (d-j), schizonts (k and l) and gametocytes (m and n)
Discussion
The ‘Red Queen hypothesis’ [53, 54] was formulated to describe an “arms race” relationship of a host-parasite co-evolution. Ecological factors that alter the gene frequencies within a host population alternately cause significant changes in the organisms’ evolutionary trends over a substantial period of time [55, 56]. For a parasitic organism, the host represents a prime environment. Therefore, a varying environment increases the likelihood of diversification for the parasites [57]. According to Poulin [58], parasitic organisms exhibit remarkable adaptive radiation because of their narrow generation times and large population sizes [59]. Hence, the evolutionary pathway of parasites can be linked with the hosts to better understand the evolutionary histories of parasites.
The long-tailed macaques of Borneo, which have been geographically isolated from those of Peninsular Malaysia and mainland Asia over thousands of years [60–62] may potentially harbour unique Plasmodium species. The utility of two sets of molecular markers to track ancestry and to study the evolutionary histories of the Plasmodium species infecting the long-tailed macaques revealed the presence of a potentially novel species of Plasmodium. The uniparentally inherited mitochondrial genome was specifically used to estimate the divergence time of all the Plasmodium species within the macaques. Due to the lack of fossil records, a host-parasite co-divergence point was selected for calibration [37]. The Bayesian analysis was calibrated using the split of Plasmodium spp. of Asian macaques from P. gonderi/ Plasmodium sp. (Mandrill) of African origin which took place approximately 6 to 14.2 million years ago which strongly coincides with the geographical isolation of the hosts [32, 37, 39]. The use of this calibration point by Lee and co-workers in a previous study [18] to estimate the time of the most recent common ancestor for P. knowlesi also indicated that the divergence of P. falciparum-P. reichenowi occurred approximately 5-7 million years before present, thereby suggesting the consistency in the parasite’s mutation rates based on these two host-parasite speciation events. However, it should be noted that the validity of our current observed divergence time for the P. inui-like parasites is strongly dependent on the validity of the assumption of strict cospeciation patterns.
Phylogenetic analyses of the mitochondrial genome and partial ClpM gene sequences clearly showed that the macaques harboured a species of Plasmodium that is phylogenetically distinct but closely related to P. inui, herein referred to as P. inui-like. The structure of the haplotype network and the significant genetic distance observed between the P. inui (reference) and the P. inui-like haplotypes have provided further evidence that the P. inui-like population is a novel species. Our estimated TMRCA of P. inui-like parasite was approximately 1.5 million years before present (637,200 - 2,492,000 years ago) indicating that this species of Plasmodium was derived from an ancestral parasitic species population of P. inui. The divergence of the Plasmodium species can be related to the dispersal of the hosts. The emergence of macaques in Asia occurred some 5 million years ago [63]. The long-tailed macaques alongside Sumatran surilis (Presbytis melalophos) and banded leaf monkeys (P. femoralis) colonised Borneo, Sumatra and Natuna Islands approximately 1.8 million years ago [61]. The mitochondrial diversification of the common ancestor of P. inui and P. inui-like species could have taken place during the migration period or soon after the colonization of Borneo by long-tailed macaques. Therefore, the spatial isolation of the host species from the macaque troops from mainland Asia resulted in the diversification of the parasites [64] via the disruption of gene flow between geographically isolated Plasmodium species [15].
The haplotype network for the P. inui-like population along with reference sequences of P. inui from Taiwan, Peninsular Malaysia and Sabah (map of origins shown in Fig. 9) clearly illustrate distant relationships among these species. In addition, the star-like pattern of the haplotype network supported a population expansion event. The mtDNA sequences derived from the long-tailed macaques of Sarawak did not overlap with the majority of the published sequences from Sabah except for one that was derived from a pig-tailed macaque [40]. Sequences of P. inui and P. inui-like parasites formed separate clades in the mtDNA tree (Fig. 2). A couple of hypotheses can be proposed to describe the separation of the Sabah and Sarawak P. inui-like populations. The separation between the clades could be due to the restricted mobility of the macaques across the dense forests which resulted in reduced or no gene flow between the parasite species. For long-tailed macaques, females remain in their natal habitats while only the males migrate to adjacent troops [65]. The clear separation between the haplotypes from Sarawak and Sabah strongly suggests that the dispersal of the macaques might have been impeded by geographic barriers, risk of predation, restricted food sources or resistance from residents [65–70]. Hence, constrained mobility resulted in restricted gene flow among the macaques and malaria parasites within a troop. Another hypothesis is that two independent introductions or dispersal waves of macaques occurred in Borneo. Of the two commonly found macaques in Borneo, the pig-tailed macaques are classified under the silenus-sylvanus lineage while the long-tailed macaques fall under the fascicularis lineage [62, 71]. The dispersal of both these macaques is speculated to have occurred in two separate waves [61, 62]. Hence, both lineages of macaques could have brought along their malaria parasites during colonization, which then co-evolved with these hosts as they diversified themselves.
Fig. 9.
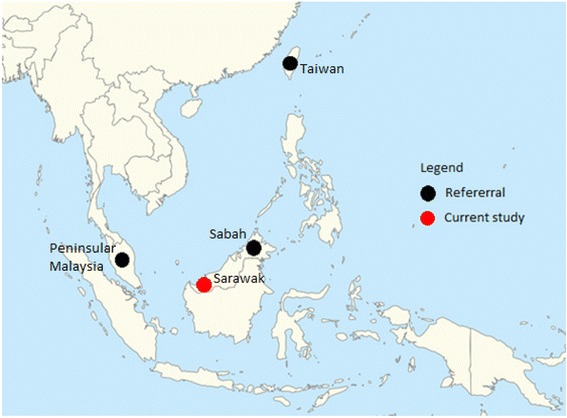
Location of long-tailed macaque blood sample collection sites (current study) and the origins of P. inui references used in the phylogenetic analyses
The genetic distance of mtDNA sequences of P. inui derived from macaques of Sabah and P. inui-like from Sarawak was high compared with the reference mtDNA sequences from Taiwan and Peninsular Malaysia (Table 2). Although a specific genetic distance value has not been set to Plasmodium species, the values tabulated in Table 2 exceeds that of the distance between P. cynomolgi and P. vivax, 1.2% [38]. This suggests that the P. inui-like haplotypes from Sarawak and the P. inui haplotypes from Sabah could most likely be a sister species of P. inui.
The population expansion of the P. inui-like parasite population is concordant with the colonization, survival adaptation and a stabilised population growth of the long-tailed macaques in Malaysian Borneo [60, 61]. This estimated expansion event preceded the population growth of P. knowlesi derived from the long-tailed and pig-tailed macaques of Kapit division of Sarawak [18]. Hence, the expansion of P. knowlesi could not be due to a similar expansion in the long-tailed macaque population since a similar pattern was not observed among the P. inui-like lineage.
Traditional taxonomy classifies Plasmodium species based on morphological and morphometric features [1, 72]. Although researchers have begun using molecular techniques for parasite taxonomy and to infer phylogenies, morphological characterization is still considered important in confirming novel species. Only macaque LT7 had a single species infection of P. inui-like parasites. Based on the morphological characteristics of P. inui-like parasites observed from Giemsa-stained blood films of macaque LT7, there was no significant morphological variation compared with those described by Coatney et al. [1] for P. inui. All blood stages were identified with trophozoites being the most abundant blood stage observed. However, morphological similarities between P. inui and P. inui-like parasites do not rule out the possibility that these parasites are different species, since indistinguishable morphological characteristics have previously been observed for Plasmodium, such as for P. knowlesi and P. malariae [6].
The toque macaque which is endemic to Sri Lanka, is known to be the natural host for P. simiovale. It is also known to be the host for P. cynomolgi, P. fragile and P. shortii [73, 74]. The low genetic distances between the haplotypes in the current study and reference sequences suggested that the haplotypes in this current study are P. simiovale based on the phylogenetic analyses of the mtDNA and ClpM genes. Therefore, our study demonstrated that long-tailed macaques of Sarawak are new hosts of P. simiovale.
Conclusions
Analysis of the molecular data generated in this study indicates that the long-tailed macaques of Malaysian Borneo harbour a parasite that is novel and is phylogenetically distinct from P. inui. The results also demonstrate these macaques as new hosts for P. simiovale. The isolation of macaques from mainland Asia may have caused an evolutionary adaptation of malaria parasites in new niches resulting in new species of Plasmodium.
Additional files
14 internal primers used to sequence the complete mitochondrial genomes of Plasmodium sp. (DOCX 12 kb)
List of referral Plasmodium mtDNA gene sequences from GenBack used in the phylogenetic analyses. (DOCX 22 kb)
The GenBank accession numbers of mtDNA gene sequences generated in this study. (DOCX 23 kb)
List of referral Plasmodium ClpM gene sequences from GenBack used in the phylogenetic analyses. (DOCX 15 kb)
The GenBank accession numbers of ClpM gene sequences generated in this study. (DOCX 29 kb)
Acknowledgements
We thank the Sarawak Forestry Department, Sarawak Biodiversity Council and the Medical Ethics Committee of Universiti Malaysia Sarawak for permission to collect blood samples from long-tailed macaques and to conduct the study. We also thank all personnel involved in the collection of macaque blood samples.
Funding
This study was supported by a grant (number: FRGS/ST03(05)/966/2013(07)) from the Fundamental Research Grant Scheme from the Ministry of Higher Education, Malaysia and they also awarded TNR a MyPhD scholarship under their MyBrain15 program.
Availability of data and materials
Sequence data that support the findings of this study have been deposited in GenBank with the accession codes provided in Additional files 3 and 5.
Abbreviations
- ClpM
caseinolytic protease M
- csp
circumsporozoite protein
- ESS
Effective Sample Size
- MCMC
Markov Chain Monte Carlo
- mtDNA
mitochondrial DNA
- rRNA
ribosomal RNA
- SSUrRNA
Small subunit ribosomal RNA
- TMRCA
Time to Most Recent Common Ancestor
Authors’ contributions
BS and RZ conceived the study. TNR and THH, supervised by BS, undertook the experimental work. TNR, BS, RZ, KSL and SLP analysed the results and wrote the paper. All authors provided comments on the manuscript and approved the final manuscript.
Ethics approval and consent to participate
Sarawak has no legislation on the use of animals in research. Nevertheless, this study was carried out in strict accordance with the recommendations by the Sarawak Forestry Department for the capture, use and release of wild macaques. A veterinarian took blood samples from macaques following anaesthesia and all efforts were made to minimize suffering by collecting blood from macaques at the trap sites and releasing the animals immediately after the blood samples had been obtained. The Sarawak Forestry Department approved the study protocol for capture, collection of blood samples and release of wild macaques (Permits Numbers: NPW.907.4.2-32, NPW.907.4.2-97, NPW.907.4.2-98, 57/2006 and 70/2007). A permit to access and collect macaque blood samples for the purpose of research was also obtained from the Sarawak Biodiversity Council (Permit Number: SBC-RP-0081-BS). This study was approved by the Medical Ethics Committee of Universiti Malaysia Sarawak.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1170-9) contains supplementary material, which is available to authorized users.
Contributor Information
Thamayanthi Nada Raja, Email: thayan4@yahoo.com.
Ting Huey Hu, Email: t.hhu@hotmail.com.
Ramlah Zainudin, Email: zramlah@unimas.my.
Kim Sung Lee, Email: lks9@np.edu.sg.
Susan L. Perkins, Email: perkins@amnh.org
Balbir Singh, Phone: +6 082 581000, Email: bsingh@unimas.my.
References
- 1.Coatney GR, Collins WE, Warren M, Contacos PG. The primate malarias. Washington: Government Printing Office; 1971. [Google Scholar]
- 2.Ramasamy R. Zoonotic malaria-global overview and research and policy needs. Front Pub Hlth. 2014;2(123):1–7. doi: 10.3389/fpubh.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collins WE. Major animal models in malaria research: simian. In: malaria: principles and practice of malariology. Edinburgh: Churchill Livingstone; 1988. [Google Scholar]
- 4.Fooden J. Malaria in macaques. Int J Primatol. 1994;15:573–596. doi: 10.1007/BF02735972. [DOI] [Google Scholar]
- 5.Galinski MR, Barnwell JW. Monkey malaria kills four humans. Trends Parasitol. 2009;25:200–204. doi: 10.1016/j.pt.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Singh B, Lee KS, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363(9414):1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 7.Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbial Rev. 2013;26(2):165–184. doi: 10.1128/CMR.00079-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prugnolle F, Durand P, Neel C, Ollomo B, Ayala FJ, Arnathau C, et al. African great apes are natural hosts of multiple related malaria species, including Plasmodium falciparum. Proc Natl Acad Sci U S A. 2010;107:1458–1463. doi: 10.1073/pnas.0914440107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, Nkoghe D, et al. A new malaria agent in African hominids. PLoS Pathog. 2009;5:1–5. doi: 10.1371/journal.ppat.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krief S, Escalante AA, Pacheco MA, Mugisha L, André C, Halbwax M, et al. On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from bonobos. PLoS Pathog. 2010;6:1–12. doi: 10.1371/journal.ppat.1000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, Malenke JA, et al. African origin of the malaria parasite Plasmodium vivax. Nat Commun. 2014;5:3346. doi: 10.1038/ncomms4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF. Origin of the human parasite Plasmodium falciparum in gorillas. Nature. 2010;467(7314):420–425. doi: 10.1038/nature09442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duval L, Nerrienet E, Rousset D, Mba SAS, Houze S, Fourment M, et al. Chimpanzee malaria parasites related to Plasmodium ovale in Africa. PLoS One. 2009; 10.1371/journal.pone.0005520. [DOI] [PMC free article] [PubMed]
- 14.Leclerc MC, Hugot JP, Durand P, Renaud F. Evolutionary relationships between 15 Plasmodium species from new and old world primates (including humans): a 18S rDNA cladistic analysis. Parasitology. 2004;129:1–8. doi: 10.1017/S0031182004006146. [DOI] [PubMed] [Google Scholar]
- 15.Nadler SA, Leon GPPD. Integrating molecular and morphological approaches for characterizing parasitic cryptic species: implications for parasitology. Parasitology. 2011;138:1688–1609. doi: 10.1017/S003118201000168X. [DOI] [PubMed] [Google Scholar]
- 16.Garnham PCC. Malaria parasites and other haemosporidia. Oxford: Blackwell Scientific Publications; 1966. [Google Scholar]
- 17.Whelan NV. Species tree inference in the age of genomics. Trends Evol Biol. 2011;3(1):23. doi: 10.4081/eb.2011.e5. [DOI] [Google Scholar]
- 18.Lee KS, Divis PCS, Zakaria SK, Matusop A, Julin RA, Conway DJ, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7(4):1–11. doi: 10.1371/journal.ppat.1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makanga B, Yangari P, Rahola N, Rougeron V, Elguero E, Boundenga L, et al. Ape malaria transmission and potential for ape-to-human transfers in Africa. Proc Natl Acad Sci U S A. 2016;113(19):5329–5334. doi: 10.1073/pnas.1603008113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barthelémy N, Larson B, Arnathau C, Mombo IM, Durand P, Tsoumbou T-A, et al. The host specificity of ape malaria parasites can be broken in confined environments. Int J Parasitol. 2016;46(11):737–744. doi: 10.1016/j.ijpara.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Liedigk R, Kolleck J, Böker KO, Meijaard E, Md-Zain BM, Abdul-Latiff MAB, et al. Mitogenomic phylogeny of the common long-tailed macaque (Macaca fascicularis fascicularis) BMC Genomics. 2015;16:222. doi: 10.1186/s12864-015-1437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Divis PCS. Identification and molecular characterization of malaria parasites of macaques in Kapit, Sarawak. (MSc thesis) Universiti Malaysia Sarawak; 2007.
- 23.Corredor V, Enea V. Plasmodial ribosomal RNA as phylogenetic probe: a cautionary note. Mol Biol Evol. 1993;10:924–926. doi: 10.1093/oxfordjournals.molbev.a040039. [DOI] [PubMed] [Google Scholar]
- 24.Hughes MK, Hughes AL. Natural selection on Plasmodium surface proteins. Mol Biochem Parasitol. 1995;71:99–113. doi: 10.1016/0166-6851(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 25.Zilversmit M, Perkins S. “Plasmodium”. Tree of Life Web Project. http://tolweb.org/Plasmodium/68071. Accessed 30 December 2016.
- 26.Rathore D, Wahl AM, Sullivan M, McCutchan TF. A phylogenetic comparison of gene trees constructed from plastid, mitochondrial and genomic DNA of Plasmodium species. Mol Biochem Parasitol. 2001;114(1):89–97. doi: 10.1016/S0166-6851(01)00241-9. [DOI] [PubMed] [Google Scholar]
- 27.Moriyama Y, Kawano S. Rapid, selective digestion of mitochondrial DNA in accordance with the matA hierarchy of multiallelic mating types in the mitochondrial inheritance of Physarum polycephalum. Genetics. 2003;164(3):963–975. doi: 10.1093/genetics/164.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waller RF, McFadden GI. The Apicoplast: a review of the derived plastid of apicomplexan parasites. Curr Issues Mol Biol. 2005;7:57–80. [PubMed] [Google Scholar]
- 29.Kriwinski J, Wilkerson RC, Besansky NJ. Evolution of mitochondrial and ribosomal gene sequences in anophelinae (Diptera: Culicidae): implications for phylogeny reconstruction. Mol Phylogenet Evol. 2001;18(3):479–487. doi: 10.1006/mpev.2000.0894. [DOI] [PubMed] [Google Scholar]
- 30.Zink RM, Borrowclough GF. Mitochondrial DNA under siege in avian phylogeography. Mol Ecol. 2008;17(9):2107–2121. doi: 10.1111/j.1365-294X.2008.03737.x. [DOI] [PubMed] [Google Scholar]
- 31.El-Bakkouri M, Pow A, Mulichak A, Cheung KLY, Artz JD, Amani M, et al. The Clp chaperones and proteases of the human malaria Plasmodium falciparum. J Mol Biol. 2010;404(3):456–477. doi: 10.1016/j.jmb.2010.09.051. [DOI] [PubMed] [Google Scholar]
- 32.Galtier N, Nabholz B, Glémin S, Hurst GD. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol. 2009;18(22):4541–4550. doi: 10.1111/j.1365-294X.2009.04380.x. [DOI] [PubMed] [Google Scholar]
- 33.Arisue N, Hashimoto T. Phylogeny and evolution of apicoplasts and apicomplexan parasites. Parasitol Int. 2015;64(3):254–259. doi: 10.1016/j.parint.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Swafford DL. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, Massachusetts: Sinnauer Associates; 2002. [Google Scholar]
- 36.Bradley RD, Baker RJ. A test of the genetic species concept: cytochrome-b sequences and mammals. J Mammal. 2001;82(4):960–973. doi: 10.1644/1545-1542(2001)082<0960:ATOTGS>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricklefs RE. Host-pathogen co-evolution, secondary sympatry and species diversification. Philios Trans R Soc Lond B Biol Sci. 2010;365(1543):1139–1147. doi: 10.1098/rstb.2009.0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pacheco MA, Cranfield M, Cameron K, Escalante AA. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J. 2013;12:328. doi: 10.1186/1475-2875-12-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, et al. Accelerated diversification of non-human primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2014;32(2):422–439. doi: 10.1093/molbev/msu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 41.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999;152(3):1079–1089. doi: 10.1093/genetics/152.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol. 1999;66(4):591–600. [PubMed] [Google Scholar]
- 44.Tajima F, Nei M. Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol. 1984;1(3):269–285. doi: 10.1093/oxfordjournals.molbev.a040317. [DOI] [PubMed] [Google Scholar]
- 45.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155(3):1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, et al. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011;11:299. doi: 10.1186/1471-2148-11-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Posada D, Crandell KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 50.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4(5):699–710. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aris-Brosou S, Excoffier L. The impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol. 1996;13(3):494–504. doi: 10.1093/oxfordjournals.molbev.a025610. [DOI] [PubMed] [Google Scholar]
- 52.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22(5):1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 53.Heylinghen F. The Red Queen Principle. http://pespmc1.vub.ac.be/REDQUEEN.html. Accessed 30 September 2016.
- 54.Rabajante JF, Tubay JM, Ito H, Uehara T, Kakishima S, Morita S, et al. Host-parasite red queen dynamics with phase-locked rare genotypes. Sci Adv. 2016;2(3):1–7. doi: 10.1126/sciadv.1501548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nadler SA. Microevolution and the genetic structure of parasite populations. J Parasitol. 1995;81(3):395–403. doi: 10.2307/3283821. [DOI] [PubMed] [Google Scholar]
- 56.Clayton DH, Johnson KP. Linking coevolutionary history to ecological processes: doves and lice. Evolution. 2003;57(10):2335–2341. doi: 10.1111/j.0014-3820.2003.tb00245.x. [DOI] [PubMed] [Google Scholar]
- 57.De Meeûs T, Michalakis Y, Renaud F. Santa Rosalia revisited: ‘or why are there so many kinds of parasites in the garden of early delights?’. Parasitol Today. 1998;14(1):10–13. doi: 10.1016/S0169-4758(97)01163-0. [DOI] [PubMed] [Google Scholar]
- 58.Poulin R. Evolutionary biology of parasites. New Jersey: Princeton University Press; 1980.
- 59.Kochin BF, Bull JJ, Antia R. Parasite evolution and life history theory. PLoS Biol. 2010;8(10):1–4. doi: 10.1371/journal.pbio.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. Big bang in the evolution of extant malaria parasites. Mol Biol Evol. 2008;25(10):2233–2239. doi: 10.1093/molbev/msn171. [DOI] [PubMed] [Google Scholar]
- 61.Harrison T, Krigbaum J, Manser J. Primate biogeography. New York: Springer; 2006. Primate biogeography and ecology on the Sunda shelf islands: a paleontological and zooarchaeological perspective. [Google Scholar]
- 62.Fooden J. Provisional classification and key to living species of macaques (Primates: Macaca) Folia Primatol. 1976;25:225–236. doi: 10.1159/000155715. [DOI] [PubMed] [Google Scholar]
- 63.Ziegler T, Abegg C, Meijaard E, Perwitasari-Farajallah D, Walter L, Hodges JK, et al. Molecular phylogeny and evolutionary history of southeast Asian macaques forming the Macaca silenus group. Mol Phylogenet Evol. 2007;42(3):807–816. doi: 10.1016/j.ympev.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 64.Sweigart AL, Willis JH. Patterns of nucleotide diversity in two species of Mimulus are affected by mating system and asymmetric introgression. Evolution. 2003;57(11):2490–2406. doi: 10.1111/j.0014-3820.2003.tb01494.x. [DOI] [PubMed] [Google Scholar]
- 65.de Ruiter JR, Geffen E. Relatedness of matrilines, dispersing males and social groups in long-tailed macaques (Macaca fascicularis) Proc Biol Sci. 1998;265(1391):79–87. doi: 10.1098/rspb.1998.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alberts SC, Altmann J. Balancing costs and opportunities: dispersal in male baboons. Am Nat. 1995;145:279–306. doi: 10.1086/285740. [DOI] [Google Scholar]
- 67.Pärt J. The importance of local familiarity ans search cost for age- and sex-biased philopatry in the collared flycatcher. Animal Behav. 1995;49:1029–1038. doi: 10.1006/anbe.1995.0132. [DOI] [Google Scholar]
- 68.Kanthaswamy S, Satkosko J, George D, Kou A, Erickson BJ-A, Smith DG. Interspecific hybridization and the stratification of nuclear genetic variation of rhesus (Macaca mulatta) and long-tailed macaques (Macaca fascicularis) Int J Primatol. 2008;29(5):1295–1311. doi: 10.1007/s10764-008-9295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meiri S, Meijaard E, Wich SA, Groves CP, Helgen KM. Mammals of Borneo- small size on a large island. J Biogeogr. 2008;35:1087–1094. doi: 10.1111/j.1365-2699.2008.01897.x. [DOI] [Google Scholar]
- 70.Marty PR, Hodges K, Agil M, Engelhardt A. Determinants of immigration strategies in male crested macaques (Macaca nigra) Sci Rep. 2016;6(32028):1–8. doi: 10.1038/srep32028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Groves CP. Primate taxonomy. Washington: Smithsonian Institution Press; 2001. [Google Scholar]
- 72.Wheeler QD. The new taxonomy. Florida: CRC Press, Boca Raton; 2008. [Google Scholar]
- 73.Dissanaike AS, Nelson S, Garnham PCC. Plasmodium simiovale sp. nov., a new simian malaria parasite from Ceylon. Ceylon J Med Sci. 1965;14:27–32. [Google Scholar]
- 74.Dissanaike AS. Simian malaria parasites in Ceylon. Geneva: WHO; 1964. [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
14 internal primers used to sequence the complete mitochondrial genomes of Plasmodium sp. (DOCX 12 kb)
List of referral Plasmodium mtDNA gene sequences from GenBack used in the phylogenetic analyses. (DOCX 22 kb)
The GenBank accession numbers of mtDNA gene sequences generated in this study. (DOCX 23 kb)
List of referral Plasmodium ClpM gene sequences from GenBack used in the phylogenetic analyses. (DOCX 15 kb)
The GenBank accession numbers of ClpM gene sequences generated in this study. (DOCX 29 kb)
Data Availability Statement
Sequence data that support the findings of this study have been deposited in GenBank with the accession codes provided in Additional files 3 and 5.