

INTRODUCTION
Infiltrative cardiomyopathies are characterized by abnormal deposition or accumulation of substances in the heart. Essentially, these diseases have similarities and overlaps in echocardiographic and cardiovascular presentation, but they display a broad range of seemingly disparate extracardiac features. This review article specifically discusses the inherited infiltrative cardiomyopathies, giving an overview of the genes, molecular mechanisms, and resulting features of each disease, with emphasis on the heart. Fig. 1 summarizes the cellular mechanisms and phenotypes and Table 1 summarizes the genetic features of each disease.
Fig. 1.
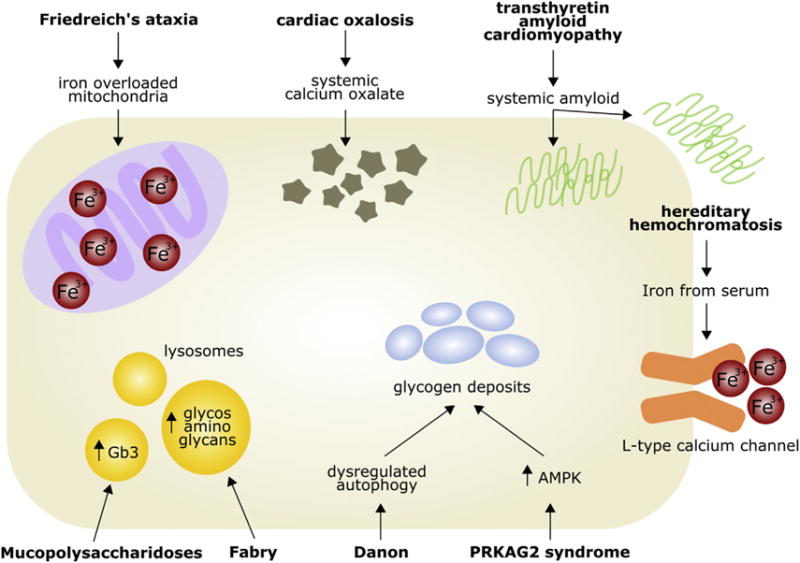
Genetic infiltrative cardiomyopathy in the cardiac myocyte. AMPK, adenosine monophosphate–activated protein kinase; Gb3, globotriaosylceramide.
Table 1.
Summary of the genetics of inherited cardiomyopathies
| Inherited Infiltrative Cardiomyopathy | Disease Type | Infiltrative Substance | Modes of Inheritance | Genes |
|---|---|---|---|---|
| Extracardiac | ||||
| Transthyretin cardiac amyloidosis | Amyloidosis | Amyloid | Autosomal dominant | TTR |
| Hereditary hemochromatosis | Disease of iron metabolism | Iron | Autosomal recessive (types 1–3); autosomal dominant (type 4) | HFE (type 1), HJV, HAMP (type 2), TFR2 (type 3), SLC40A1 (type 4) |
| Cardiac oxalosis | Disease of glyoxylate metabolism | Calcium oxalate | Autosomal recessive | AGXT (type 1), GRHPR (type 2), HOGA1 (type 3) |
| Intracardiac | ||||
| Friedreich ataxia | Disease of iron metabolism | Iron | Autosomal recessive | FXN |
| Mucopolysaccharidoses | Lysosomal storage disease | Glycosaminoglycans | Autosomal recessive (I, III, IV, VI, VII, IX); X-linked recessive (II) | IDUA (I), IDS (II), SGSH, NAGLU, HGSNAT, GNS (III), GALNS, GLB1 (IV), ARSB (VI), GUSB (VII), HYAL1 (IX) |
| Fabry disease | Lysosomal storage disease | Globotriaosylceramide | X-linked | GLA |
| Danon disease | Disease of autophagy | Glycogen | X-linked | LAMP2 |
| PRKAG2 syndrome | Glycogen storage disease | Glycogen | Autosomal dominant | PRKAG2 |
This review begins with the extracardiac diseases that primarily manifest in other organs and only reach the heart through substance accumulation in the bloodstream. Then it moves on to the intracardiac diseases that can manifest directly within the cardiac myocytes.
EXTRACARDIAC INFILTRATIVE CARDIOMYOPATHIES
Transthyretin Cardiac Amyloidosis
Amyloidosis is a general term for extracellular accumulation of amyloids, or protein aggregates, that form ß-pleated fibrous deposits. Amyloidosis can be acquired or inherited and can present with multiorgan involvement. For the purposes of this review, only the inherited form is discussed. Inherited amyloidosis is caused by mutations in TTR, which encodes transthyretin, a serum and cerebrospinal fluid transport protein secreted almost exclusively (>98%) by the liver.1 TTR transports thyroxine and retinol, from which it gets its name. Point mutations in TTR can destabilize the tetramer, increasing the likelihood that it will disassociate into amyloidogenic monomers, aggregate into fibrils, and deposit into organs. Progressive deposition of amyloid in the heart can lead to cardiac amyloidosis.
Transthyretin cardiac amyloidosis is inherited in an autosomal dominant manner. Although some TTR mutations present with predictable phenotypes, affecting predominately the heart or liver, most genotype–phenotype correlations in TTR have unpredictable prognoses2 due in part to the contribution of noncoding variation in TTR regulation. Noncoding variation in TTR that affects TTR tissue-specific expression identifies clusters of TTR amyloid patients with similar phenotypes. This suggests that complex genetics contributes to the variable expressivity of TTR amyloidosis.3 The most frequent variant in TTR amyloid is TTR p.V122I, which had reported throughout the literature to be present in 3% to 4% of African Americans, though it is only present in 1.6% of the African population within the Genome Aggregation Database (gnomAD).4,5 In a recent study of 156 African Americans referred for amyloidosis, the TTR V122I (V142I) mutation was present in 23%, and most of these subjects developed TTR amyloidosis.6
Pathogenic mutations in TTR are primarily clinically characterized by progressive neuropathy (86% of cases) or cardiomyopathy (42% of cases) with some overlap.7 Cardiomyopathy presents on average in the fourth decade of life but can range significantly from less than 20 years to greater than 80 years.7 Cardiac amyloidosis typically presents as heart failure with preserved ejection fraction.8
Historically, the standard treatment of transthyretin cardiac amyloidosis has been liver transplantation, sometimes in combination with heart transplantation. However, several therapies are being developed or are in clinical trials that stabilize TTR tetramers (tafamidis, diflunisal, AG10), degrade preexisting amyloid deposits (doxycycline, anti-serum amyloid protein P [SAP]), or silence transthyretin protein production (RNA interference [RNAi]).9 Stabilizers bind to the TTR tetramer and prevent its disassociation, but the selectivity of the compounds and their ability to effectively bind various pathogenic variants of TTR are issues to consider. Degraders target amyloid protein that has already been deposited. For example, SAP is a universal constituent of amyloid protein; targeting SAP with an antibody significantly reduces amyloid deposits in animal models.10,11 Finally, silencing amyloid protein production is also a potential therapy. The largest randomized phase 3 trial to date in hereditary transthyretin amyloidosis is currently underway, using RNAi technology to target a conserved messenger RNA (mRNA) sequence across both wildtype and mutated TTR transcripts. The small interfering RNA (siRNA) is directed to the liver with the goal of suppressing TTR hepatic production and reducing circulating levels of the protein.12,13
Hereditary Hemochromatosis
Hemochromatosis is a progressive condition of iron accumulation and end-organ damage. Hemochromatosis is caused by mutations in genes encoding proteins involved in iron absorption, transportation, and storage. There are 4 types caused by mutations in their corresponding genes: type 1, HFE; type 2, HAMP or HJV; type 3, TFR2; and type 4, SLC40A1. Types I, 3, and 4, are adult-onset diseases and present in the fourth to fifth decade of life; in contrast, type 2 is a juvenile-onset disease and presents in the second or third decade. Hereditary hemochromatosis is inherited as an autosomal recessive trait, except type 4, which is autosomal dominant.14
By far the most common genotype risk for hereditary hemochromatosis is HFE p.Cys282Tyr homozygosity, which accounts for more than 80% of patients.15 The mutation that causes p.Cys282Tyr is heterozygous in 1 per 9 non-Finnish Europeans and homozygous in 1 per 538, according to gno-mAD.16 However, depending on the geographic region, the frequency, even within Europeans, can vary.15 It is also present in lower frequencies in all other ethnicities, with the lowest frequencies in South and East Asian populations.16 Homozygosity for p.Cys282Tyr demonstrates incomplete penetrance; it causes hemochromatosis in 1% to 14% of female patients and 24% to 28% of male patients.17,18 This difference is attributed in part to recurrent physiologic blood loss in women that contributes to slower iron accumulation18 but may result from other genetic or environmental factors.
Due to the heterogeneous phenotypic expression of the pC282Y/pC282Y genotype,18 it has been hypothesized that additional genetic modifiers played a role. In patients homozygous for p.Cys282Tyr, heterozygous missense or loss-of-function mutations in HJV or HAMP contribute to a more severe iron overload.19,20 Additionally, a combination of heterozygous missense mutations in HAMP and HFE can also cause hemochromatosis.19 Although HFE causal genotypes lead to adult-onset hemochromatosis, homozygous loss-of-function in HAMP21 and homozygous missense mutations in HJV22 can cause juvenile-onset hereditary hemochromatosis.
The iron overload that results from hemochromatosis can cause dilated or restrictive cardiomyopathy14 and can manifest with conduction abnormalities and tachyarrhythmia.14 During iron overload, L-type calcium channels in cardiomyocytes can uptake excess iron, which elicits harmful free radical production in the cell.23 Calcium channel blockers have been used effectively in mice to decrease cardiac iron accumulation but have not been studied in humans.24,25 Cellular injury from iron overload-induced free radical production may be exacerbated in patients receiving doxorubicin chemotherapy, due to formation of doxorubicin-iron complexes in the mitochondria.26,27 This can lead to increased risk of doxorubicin-induced cardiotoxicity in hemochromatosis patients, resulting in diastolic dysfunction.26,28 Therapy for hereditary hemochromatosis includes cardiac transplantation,29 phlebotomy, and iron chelation.30
Cardiac Oxalosis
Primary hyperoxaluria (PH) is an autosomal recessive disease of glyoxylate metabolism that causes oxalate overproduction. PH is divided into 3 types, each caused by mutations in different genes that encode enzymes involved in glyoxylate metabolism: type I, alanine-glyoxylate aminotransferase (AGXT); type II, glyoxylate reductase/hydroxypyruvate reductase (GRHPR); type III, 4-hydroxy-2-oxoglutarate aldolase (HOGA1). PH type 1, which accounts for almost 80% of all cases,31 has a prevalence of 1 to 3 per 1 million people.32,33
In PH, glyoxylate accumulates and is converted to oxalate, which cannot be metabolized. Recurrent urolithiasis and progressive nephrocalcinosis lead to kidney damage, consequent reductions in excreted oxalate, increased systemic levels of oxalate, and subsequent calcium oxalate deposition in tissues.34 The organs affected include skin, bone, the retina, vessels, and the myocardium.35
After the kidneys become damaged and oxalate begins to deposit in the tissues, it often results in end-stage renal disease, but cardiac symptoms can also occur. However, PH is an extremely rare disease and not all patients experience cardiac dysfunction. In a review study of cardiac phenotypes in 38 PH subjects, approximately 11% experienced dyspnea, 9% experienced chest pain and palpitation, and 3% experienced syncope. The most common echocardiographic findings were increased left ventricular mass (29%) and left atrium enlargement (21%), and abnormal electrocardiogram findings included left ventricular hypertrophy (5%), bundle branch block (9%), and atrioventricular block (5%).36 There have also been reports of PH patients presenting with ventricular tachycardia,37 mitral valve regurgitation,38 restrictive cardiomyopathy39 and heart failure.38 Combined liver and kidney transplantation has been shown to reverse cardiac dysfunction and oxalate deposits in the heart.40
INTRACARDIAC INFILTRATIVE CARDIOMYOPATHIES
Friedreich Ataxia
Friedreich ataxia is primarily a neurodegenerative disorder but is also characterized by other multisystemic effects. Progressive neurologic features include poor balance, muscle weakness, loss of motor skills, and visual and hearing impairment. Other nonneurological complications include scoliosis, diabetes mellitus, and hypertrophic cardiomyopathy (HCM).41
Friedreich ataxia is autosomal recessive with an estimated prevalence of 3 to 4 per 100,000.42 It is caused by unstable expansion of a trinucleotide repeat in the first intron of FXN that inhibits transcription elongation. Repeats in the nascent transcript form tertiary structures, called R-loops, with the genomic DNA, leading to reduced expression.43,44 FXN encodes frataxin, a mitochondrial protein involved in iron homeostasis. Frataxin deficiency is associated with dysregulated iron trafficking leading to mitochondrial iron aggregation.45–48 Unaffected persons typically have less than 12 repeats, but they can range from 12 to 59.42 Friedreich ataxia patients have 60 to 1500.41 Whereas 97% of patients are homozygous for this expansion,41 the remaining individuals are compound heterozygous; on 1 allele they have the expansion and on the other a loss-of-function point mutation.41,49 All reported patients have at least 1 expanded allele, and animal models suggest that homozygous loss-of-function is embryonic lethal.50
The age of onset can range significantly, from less than 1 year to 70 years, with a mean age of 12 to 16 years and a mean age of death of 37 years.41,51 Young age of onset, disease severity, and degree of diastolic dysfunction are generally predicted by higher numbers of trinucleotide repeats.41,51,52 However, the age of onset only accounts for 36% of the variability in trinucleotide repeats, suggesting additional genetic modifiers may play a role.41 One study, for example, showed that FXN methylation can predict frataxin expression and clinical outcome.53
Although cardiomyopathy is the presenting finding in only 5% of patients,41 cardiac dysfunction from congestive heart failure or arrhythmia accounts for an estimated 59% of death.51 Individuals with homozygous expansions are more likely to develop cardiomyopathy.53 Cardiac dysfunction includes left ventricular hypertrophy, systolic dysfunction, and diastolic dysfunction.52 Current treatment options that address the cardiac issues in Friedreich ataxia are limited to antioxidants; iron chelation; and, uncommonly, cardiac transplantation in patients with heart failure.46,54 Many reports of antioxidant treatment42 or combined antioxidant and iron chelation55 decreased left ventricular mass and improved cardiac function, but other studies have found no changes after therapy.56
Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) is a group of lysosomal storage disorders that result from deficient glycosaminoglycan-degrading enzymes. Glycosaminoglycans build up in the lysosomes of cells, leading to progressive tissue and organ dysfunction. Deficiency in 11 different enzymes is known to cause 7 different MPS phenotypes that are inherited in either an autosomal recessive (MPS I, III, IV, VI, VII, IX) or X-linked recessive (MPS II) fashion. There are also several reported cases in which MPS II has occurred in female patients via skewed lionization.57–59
MPS are quite rare, with an overall prevalence of 1 per 22,000.60 Among these, MPS 1, among the relatively more common MPS, has a reported prevalence of 1 per 35,000.61 Depending on the phenotype and the degree of enzyme deficiency, typical life expectancy can range from infancy to the fifth decade. Cardiac abnormalities have been reported for all MPS types, particularly I, II, and IV, and occur in most of the subjects studied (60%–100%, depending on the study).62 The most common cardiac phenotypes in MPS are hypertrophy and valve disease. In MPS I, II, and VI, 50% of patients have increased left ventricular mass due to either concentric (I and II) or eccentric hypertrophy (VI). Approximately 50% to 60% of patients have valvular regurgitation in at least 1 valve, and all patients have abnormal valves. Valve replacement in MPS is common but can be challenging given concomitant respiratory compromise seen in many MPS patients.63,64 Impaired systolic function is less common but present.65,66 Other MPS VI patients have experienced sinus tachycardia, left atrial dilation, and congestive heart failure.16,66
Intravenous enzyme replacement therapy is a strategy to treat MPS and is currently available for MPS I, II, and VI. Due to its progressive nature, early enzyme replacement therapy may stabilize or slow disease progression, including cardiac dysfunction, and sibling control studies have suggested that the earlier the intervention, the better.67,68 Therapy decreased left ventricular mass in MPS I, II, and VI,65 and decreased intraventricular septal hypertrophy in MPS VI,69 but it did not affect physiologic valvular regurgitation.65,69 Another study in MPS VI showed stabilization but not improvement of cardiac function.70
Fabry Disease
Similar to the MPS, Fabry disease is also a lysosomal storage disorder, but it results in progressive accumulation of a type of fat, particularly globotriaosylceramide (Gb3), in lysosomes. Fabry disease is an X-linked trait resulting from partial or complete deficiency of α-galactosidase A, a lysosomal enzyme encoded by GLA. Due to the ubiquitous nature of lysosomes, Fabry results in progressive dysfunction of multiple organs, including the kidneys, brain, and heart. Enzyme activity is inversely related to both age of onset and disease severity; some variants that maintain 2% to 20% enzymatic function result in attenuated phenotypes that present much later in life with fewer symptoms.71,72 Typically, female heterozygotes present with a milder form of the disease and later in life than male patients, but severity of symptoms may increase with skewed lyonization.73
Historically, the prevalence estimates for Fabry have suggested it is a relatively rare disease; however, the newest estimates, based on recent newborn screening data, suggest it is more common than previously thought.72 Typically, the newborn screening process will assay α-galactosidase A enzyme activity in blood spots followed by confirmatory genetic testing. Results from these studies support a prevalence ranging approximately 1 per 1400 to 7800 in male patients.61,74,75
Typically, Fabry disease presents in early childhood with an array of symptoms, including hypohidrosis (reduced or absent sweating); autonomic nervous system dysfunction, leading to gastrointestinal issues; and acroparesthesia pains in the extremities, especially in the setting of viral illnesses or fevers.72,76 They can also present in childhood with cardiac findings. The most common are increased left ventricular mass and valvular dysfunction, but reduced heart rate variability and electrocardiograph changes such as T-wave inversion and PR prolongation can also occur.76–78 Nonclassical Fabry disease is more variable and may manifest only with cardiac dysfunction.71 As children with Fabry disease age, their cardiac manifestations progress into more severe cardiac problems, including hypertrophic or dilated cardiomyopathy phenotype, arrhythmia, heart failure, and sudden cardiac death.79,80 Fabry disease may be responsible for 12% of late-onset HCM in female patients, suggesting that the condition may be underreported in female patients.81 Enzyme replacement therapy is clinically available for these patients and, although not able to cure Fabry disease, it has been shown to slow disease progression in some studies, particularly when administered at an earlier stage.82–85
Danon
Danon disease is an X-linked dominant disease that predominantly affects the heart but is also characterized by skeletal myopathy and intellectual disability. Danon is caused by mutations in LAMP2, which encodes the lysosome-associated membrane glycoprotein 2. The earliest histopathological studies of muscle biopsies from Danon subjects demonstrated glycogen deposits in lysosomes, leading to the conclusion that it was primarily a glycogen storage disease.86 However, continued genetic and cellular studies have suggested that it is instead a disease of deficient autophagy that leads to glycogen accumulation.87,88
LAMP2 encodes 3 isoforms via differential splicing, with LAMP2-A and LAMP2-B significantly more expressed than LAMP2-C in the heart.88 Although most described mutations in LAMP2 affect all 3 isoforms, there are reports of mutations that only affect the LAMP2-B isoform, suggesting that deficiency of this isoform is necessary and sufficient to cause disease.87,88 For a complete review of the molecular biology of LAMP2 in Danon, see the recent review by Rowland and colleagues88 (2016). Generally, loss-of-function mutations are associated with an earlier age of onset compared with missense mutations.87 Due to homozygosity and skewed X-inactivation, Danon affects male patients earlier and more severely than female patients. Male patients typically experience first symptom, cardiac transplantation, and death at ages 12, 18, and 19, respectively. These experiences are delayed approximately 15 years in female patients who manifest the disease.89–91
Danon disease primarily affects the heart. Male patients present more often with hypertrophy (HCM phenotype) and female patients with dilation (dilated cardiomyopathy phenotype).89,90 Cardiomyopathy manifests as left ventricular systolic dysfunction; ventricular preexcitation associated with T-wave inversion; palpitations; conduction abnormalities and Wolff-Parkinson-White syndrome; and rapid, progressive heart failure, resulting in transplantation or death.89,91,92 Although there are no specific evidence-driven guidelines for management of Danon, there are published recommendations. Due to the rapid, progressive nature of Danon, early monitoring of electrophysiology and early consideration of implantable cardioverter-defibrillator (ICD) implantation is suggested, particularly in patients with symptomatic arrhythmias, moderate to severe hypertrophy, fibrosis, or a positive family history for sudden cardiac death.87 The molecular mechanism by which defective or deficient LAMP2 causes Danon remains elusive, but activation of autophagy via activation of the Aktm-TORC1 pathway has been an effective therapy in a mouse model of HCM, and may be a potential target for Danon.93
PRKAG2 Syndrome
Like Danon, PRKAG2 syndrome also results in the accumulation of glycogen in the cardiomyocyte. PRKAG2 encodes the gamma-2 subunit of adenosine monophosphate–activated protein kinase (AMPK). AMPK is a cellular fuel gauge that is constantly sensing and responding to the energy needs of the cell, particularly in cardiomyocyte metabolism.94 PRKAG2 syndrome is inherited in an autosomal dominant manner with high penetrance. Mutations in PRKAG2 alter the binding affinity and enzymatic activity of AMPK, activating AMPK activity and glycogen accumulation.95 Pathologic analysis of human tissue from patients with PRKAG2 mutations suggests that cardiac hypertrophy and conduction system disease are the manifesting symptoms of a glycogen storage disease. Patient tissue demonstrated myocyte enlargement and minimal interstitial fibrosis, consistent with hypertrophy, but also revealed the presence of vacuoles containing glycogen-derivatives within myocytes.96
Clinically, PRKAG2 syndrome leads to cardiac hypertrophy, supraventricular arrhythmias, and conduction abnormalities such as preexcitation, particularly in the context of Wolff-Parkinson-White Syndrome.97 Patients often present with palpitations and syncope.98 Reports indicating the onset of PRKAG2 syndrome range widely from infantile-fatal to the fifth decade of life with a mean age of onset of 30 years,92,99,100 though an infantile-fatal mutation has been reported.100 Similar to Danon, there are no specific guidelines for PRKAG2 syndrome; however, suggested guidelines include assessment of risk factors for sudden death (symptomatic arrhythmias, family history) and early consideration for pacemaker or ICD implantation.99 Because AMPK is activated in PRKAG2 syndrome, a small molecule inhibitor of AMPK may be an effective therapy, but this has not been tested.
SUMMARY
The genetic infiltrative cardiomyopathies have overlapping presentations in the heart but are characterized by a diversity of diseases. These diseases manifest in multisystemic mechanisms with cardiomyopathy as either a primary or secondary feature. Each disease has distinct genes and mechanisms, but can adversely affect the heart in similar ways.
KEY POINTS.
Infiltrative cardiomyopathies result from progressive buildup of abnormal substances in the heart.
Several infiltrative cardiomyopathies are inherited and have known genetic mechanisms.
Each inherited infiltrative cardiomyopathy has distinct extracardiac manifestations.
Although the pathologic mechanisms, type of infiltrative substances, and extracardiac presentations differ, many of these cardiomyopathies have similar or overlapping cardiac presentations.
Footnotes
Disclosures: There are no conflicts of interest or financial disclosures associated with this work.
References
- 1.Lachmann HJ. A new era in the treatment of amyloidosis? N Engl J Med. 2013;369(9):866–8. doi: 10.1056/NEJMe1308768. [DOI] [PubMed] [Google Scholar]
- 2.Zeldenrust SR. Genotype–phenotype correlation in FAP. Amyloid. 2012;19(Suppl 1):22–4. doi: 10.3109/13506129.2012.665400. [DOI] [PubMed] [Google Scholar]
- 3.Iorio A, De Lillo A, De Angelis F, et al. Non-coding variants contribute to the clinical heterogeneity of TTR amyloidosis. Eur J Hum Genet. 2017;25(9):1055–60. doi: 10.1038/ejhg.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26:16–23. doi: 10.1016/j.gde.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466–73. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 6.Connors LH, Prokaeva T, Lim A, et al. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158(4):607–14. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Coelho T, Maurer MS, Suhr OB. THAOS - The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi: 10.1185/03007995.2012.754348. [DOI] [PubMed] [Google Scholar]
- 8.Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113–22. doi: 10.1016/j.jchf.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castano A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163–78. doi: 10.1007/s10741-014-9462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–7. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pepys MB, Herbert J, Hutchinson WL, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417(6886):254–9. doi: 10.1038/417254a. [DOI] [PubMed] [Google Scholar]
- 12.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. New Engl J Med. 2013;369(9):819–29. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 13.Adams D, Suhr OB, Dyck PJ, et al. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017;17(1):181. doi: 10.1186/s12883-017-0948-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011;124(20):2253–63. doi: 10.1161/CIRCULATIONAHA.111.050773. [DOI] [PubMed] [Google Scholar]
- 15.European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. doi: 10.1016/j.jhep.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Azevedo AC, Schwartz IV, Kalakun L, et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet. 2004;66(3):208–13. doi: 10.1111/j.1399-0004.2004.00277.x. [DOI] [PubMed] [Google Scholar]
- 17.Gallego CJ, Burt A, Sundaresan AS, et al. Penetrance of Hemochromatosis in HFE Genotypes Resulting in p.Cys282Tyr and p.[Cys282Tyr];[Hi-s63Asp] in the eMERGE network. Am J Hum Genet. 2015;97(4):512–20. doi: 10.1016/j.ajhg.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. New Engl J Med. 2008;358(3):221–30. doi: 10.1056/NEJMoa073286. [DOI] [PubMed] [Google Scholar]
- 19.Jacolot S, Le Gac G, Scotet V, et al. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood. 2004;103(7):2835–40. doi: 10.1182/blood-2003-10-3366. [DOI] [PubMed] [Google Scholar]
- 20.Le Gac G, Scotet V, Ka C, et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum Mol Genet. 2004;13(17):1913–8. doi: 10.1093/hmg/ddh206. [DOI] [PubMed] [Google Scholar]
- 21.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–2. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 22.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36(1):77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 23.Tsushima RG, Wickenden AD, Bouchard RA, et al. Modulation of iron uptake in heart by L-type Ca2+ channel modifiers: possible implications in iron overload. Circ Res. 1999;84(11):1302–9. doi: 10.1161/01.res.84.11.1302. [DOI] [PubMed] [Google Scholar]
- 24.Oudit GY, Sun H, Trivieri MG, et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med. 2003;9(9):1187–94. doi: 10.1038/nm920. [DOI] [PubMed] [Google Scholar]
- 25.Ludwiczek S, Theurl I, Muckenthaler MU, et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat Med. 2007;13(4):448–54. doi: 10.1038/nm1542. [DOI] [PubMed] [Google Scholar]
- 26.Lipshultz SE, Lipsitz SR, Kutok JL, et al. Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer. 2013;119(19):3555–62. doi: 10.1002/cncr.28256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panjrath GS, Patel V, Valdiviezo CI, et al. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J Am Coll Cardiol. 2007;49(25):2457–64. doi: 10.1016/j.jacc.2007.02.060. [DOI] [PubMed] [Google Scholar]
- 28.Penel N, Adenis A, Mailliez A, et al. Silent hereditary hematochromatosis as a susceptibility factor of doxorubicin-induced acute cardiac failure. Ann Oncol. 2010;21(11):2293–4. doi: 10.1093/annonc/mdq567. [DOI] [PubMed] [Google Scholar]
- 29.Schofield RS, Aranda JM, Jr, Hill JA, et al. Cardiac transplantation in a patient with hereditary hemochromatosis: role of adjunctive phlebotomy and erythropoietin. J Heart Lung Transplant. 2001;20(6):696–8. doi: 10.1016/s1053-2498(00)00212-6. [DOI] [PubMed] [Google Scholar]
- 30.Cancado R, Melo MR, de Moraes Bastos R, et al. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: results of a 1-yr Phase 2 study. Eur J Haematol. 2015;95(6):545–50. doi: 10.1111/ejh.12530. [DOI] [PubMed] [Google Scholar]
- 31.Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290–6. doi: 10.1159/000086360. [DOI] [PubMed] [Google Scholar]
- 32.Cochat P, Deloraine A, Rotily M, et al. Epidemiology of primary hyperoxaluria type 1. Societe de nephrologie and the societe de nephrologie pediatrique. Nephrol Dial Transplant. 1995;10(Suppl 8):3–7. doi: 10.1093/ndt/10.supp8.3. [DOI] [PubMed] [Google Scholar]
- 33.van Woerden CS, Groothoff JW, Wanders RJ, et al. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transpl. 2003;18(2):273–9. doi: 10.1093/ndt/18.2.273. [DOI] [PubMed] [Google Scholar]
- 34.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8(8):467–75. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 35.Salido E, Pey AL, Rodriguez R, et al. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012;1822(9):1453–64. doi: 10.1016/j.bbadis.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Mookadam F, Smith T, Jiamsripong P, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. 2010;74(11):2403–9. doi: 10.1253/circj.cj-10-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quan KJ, Biblo LA. Type I primary hyperoxaluria: an unusual presentation of ventricular tachycardia. Cardiol Rev. 2003;11(6):318–9. doi: 10.1097/01.crd.0000065421.50549.21. [DOI] [PubMed] [Google Scholar]
- 38.Van Driessche L, Dhondt A, De Sutter J. Heart failure with mitral valve regurgitation due to primary hyperoxaluria type 1: case report with review of the literature. Acta Cardiol. 2007;62(2):202–6. doi: 10.2143/AC.62.2.2020243. [DOI] [PubMed] [Google Scholar]
- 39.Schulze MR, Wachter R, Schmeisser A, et al. Restrictive cardiomyopathy in a patient with primary hyperoxaluria type II. Clin Res Cardiol. 2006;95(4):235–40. doi: 10.1007/s00392-006-0362-2. [DOI] [PubMed] [Google Scholar]
- 40.Detry O, Honore P, DeRoover A, et al. Reversal of oxalosis cardiomyopathy after combined liver and kidney transplantation. Transpl Int. 2002;15(1):50–2. doi: 10.1007/s00147-001-0364-y. [DOI] [PubMed] [Google Scholar]
- 41.Reetz K, Dogan I, Costa AS, et al. Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol. 2015;14(2):174–82. doi: 10.1016/S1474-4422(14)70321-7. [DOI] [PubMed] [Google Scholar]
- 42.Schulz JB, Boesch S, Burk K, et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol. 2009;5(4):222–34. doi: 10.1038/nrneurol.2009.26. [DOI] [PubMed] [Google Scholar]
- 43.Punga T, Buhler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med. 2010;2(4):120–9. doi: 10.1002/emmm.201000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groh M, Lufino MM, Wade-Martins R, et al. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014;10(5):e1004318. doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitnall M, Suryo Rahmanto Y, Huang ML, et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc Natl Acad Sci U S A. 2012;109(50):20590–5. doi: 10.1073/pnas.1215349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitnall M, Suryo Rahmanto Y, Sutak R, et al. The MCK mouse heart model of Friedreich’s ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc Natl Acad Sci United States America. 2008;105(28):9757–62. doi: 10.1073/pnas.0804261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puccio H, Simon D, Cossee M, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27(2):181–6. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 48.Michael S, Petrocine SV, Qian J, et al. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum. 2006;5(4):257–67. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 49.Campuzano V, Montermini L, Molto MD, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423–7. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 50.Cossee M, Puccio H, Gansmuller A, et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9(8):1219–26. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- 51.Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307(1–2):46–9. doi: 10.1016/j.jns.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 52.Regner SR, Lagedrost SJ, Plappert T, et al. Analysis of echocardiograms in a large heterogeneous cohort of patients with friedreich ataxia. Am J Cardiol. 2012;109(3):401–5. doi: 10.1016/j.amjcard.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 53.Evans-Galea MV, Carrodus N, Rowley SM, et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann Neurol. 2012;71(4):487–97. doi: 10.1002/ana.22671. [DOI] [PubMed] [Google Scholar]
- 54.McCormick A, Shinnick J, Schadt K, et al. Cardiac transplantation in Friedreich Ataxia: Extended follow-up. J Neurol Sci. 2017;375:471–3. doi: 10.1016/j.jns.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 55.Elincx-Benizri S, Glik A, Merkel D, et al. Clinical Experience With Deferiprone Treatment for Friedreich Ataxia. J Child Neurol. 2016;31(8):1036–40. doi: 10.1177/0883073816636087. [DOI] [PubMed] [Google Scholar]
- 56.Lagedrost SJ, Sutton MS, Cohen MS, et al. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA) Am Heart J. 2011;161(3):639–45.e1. doi: 10.1016/j.ahj.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 57.Tuschl K, Gal A, Paschke E, et al. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32(4):270–2. doi: 10.1016/j.pediatrneurol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Cudry S, Tigaud I, Froissart R, et al. MPS II in females: molecular basis of two different cases. J Med Genet. 2000;37(10):E29. doi: 10.1136/jmg.37.10.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sukegawa K, Matsuzaki T, Fukuda S, et al. Brother/sister siblings affected with Hunter disease: evidence for skewed X chromosome inactivation. Clin Genet. 1998;53(2):96–101. doi: 10.1111/j.1399-0004.1998.tb02654.x. [DOI] [PubMed] [Google Scholar]
- 60.Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105(1–2):151–6. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- 61.Scott CR, Elliott S, Buroker N, et al. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J Pediatr. 2013;163(2):498–503. doi: 10.1016/j.jpeds.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183–97. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goksel OS, El H, Tireli E, et al. Combined aortic and mitral valve replacement in a child with mucopolysaccharidosis type I: a case report. J Heart Valve Dis. 2009;18(2):214–6. [PubMed] [Google Scholar]
- 64.Butman SM, Karl L, Copeland JG. Combined aortic and mitral valve replacement in an adult with Scheie’s disease. Chest. 1989;96(1):209–10. doi: 10.1378/chest.96.1.209. [DOI] [PubMed] [Google Scholar]
- 65.Brands MM, Frohn-Mulder IM, Hagemans ML, et al. Mucopolysaccharidosis: cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI. J Inherit Metab Dis. 2013;36(2):227–34. doi: 10.1007/s10545-011-9444-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kampmann C, Lampe C, Whybra-Trumpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269–76. doi: 10.1007/s10545-013-9649-4. [DOI] [PubMed] [Google Scholar]
- 67.Leal GN, de Paula AC, Morhy SS, et al. Advantages of early replacement therapy for mucopolysaccharidosis type VI: echocardiographic follow-up of siblings. Cardiol Young. 2014;24(2):229–35. doi: 10.1017/S1047951113000152. [DOI] [PubMed] [Google Scholar]
- 68.McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age–a sibling control study. Clin Genet. 2010;77(5):492–8. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 69.Braunlin E, Rosenfeld H, Kampmann C, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis. 2013;36(2):385–94. doi: 10.1007/s10545-012-9481-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)–10-year follow-up of patients who previously participated in an MPS VI survey study. Am J Med Genet A. 2014;164A(8):1953–64. doi: 10.1002/ajmg.a.36584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elliott PM. Fabry disease: a rare condition emerging from the darkness. Circ Cardiovasc Genet. 2017;10(4) doi: 10.1161/CIRCGENETICS.117.001862. [pii:e001862] [DOI] [PubMed] [Google Scholar]
- 72.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44–54. doi: 10.1111/cge.12613. [DOI] [PubMed] [Google Scholar]
- 74.Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2(5):450–6. doi: 10.1161/CIRCGENETICS.109.862920. [DOI] [PubMed] [Google Scholar]
- 75.Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laney DA, Peck DS, Atherton AM, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med. 2015;17(5):323–30. doi: 10.1038/gim.2014.120. [DOI] [PubMed] [Google Scholar]
- 77.Kampmann C, Wiethoff CM, Whybra C, et al. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008;97(4):463–9. doi: 10.1111/j.1651-2227.2008.00700.x. [DOI] [PubMed] [Google Scholar]
- 78.Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–5. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- 79.Patel V, O’Mahony C, Hughes D, et al. Clinical and genetic predictors of major cardiac events in patients with Anderson-Fabry disease. Heart. 2015;101(12):961–6. doi: 10.1136/heartjnl-2014-306782. [DOI] [PubMed] [Google Scholar]
- 80.Linhart A, Kampmann C, Zamorano JL, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28(10):1228–35. doi: 10.1093/eurheartj/ehm153. [DOI] [PubMed] [Google Scholar]
- 81.Chimenti C, Pieroni M, Morgante E, et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation. 2004;110(9):1047–53. doi: 10.1161/01.CIR.0000139847.74101.03. [DOI] [PubMed] [Google Scholar]
- 82.Arends M, Wijburg FA, Wanner C, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. 2017;121(2):157–61. doi: 10.1016/j.ymgme.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 83.Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119(4):524–9. doi: 10.1161/CIRCULATIONAHA.108.794529. [DOI] [PubMed] [Google Scholar]
- 84.Weidemann F, Niemann M, Stork S, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274(4):331–41. doi: 10.1111/joim.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146(2):77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 86.Danon MJ, Oh SJ, DiMauro S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31(1):51–7. doi: 10.1212/wnl.31.1.51. [DOI] [PubMed] [Google Scholar]
- 87.D’Souza RS, Levandowski C, Slavov D, et al. Danon disease: clinical features, evaluation, and management. Circ Heart Fail. 2014;7(5):843–9. doi: 10.1161/CIRCHEARTFAILURE.114.001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rowland TJ, Sweet ME, Mestroni L, et al. Danon disease - dysregulation of autophagy in a multi-system disorder with cardiomyopathy. J Cell Sci. 2016;129(11):2135–43. doi: 10.1242/jcs.184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boucek D, Jirikowic J, Taylor M. Natural history of Danon disease. Genet Med. 2011;13(6):563–8. doi: 10.1097/GIM.0b013e31820ad795. [DOI] [PubMed] [Google Scholar]
- 90.Sugie K, Yamamoto A, Murayama K, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology. 2002;58(12):1773–8. doi: 10.1212/wnl.58.12.1773. [DOI] [PubMed] [Google Scholar]
- 91.Maron BJ, Roberts WC, Arad M, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301(12):1253–9. doi: 10.1001/jama.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. New Engl J Med. 2005;352(4):362–72. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 93.Singh SR, Zech ATL, Geertz B, et al. Activation of autophagy ameliorates cardiomyopathy in Mybpc3-targeted knockin mice. Circ Heart Fail. 2017;10(10) doi: 10.1161/CIRCHEARTFAILURE.117.004140. [pii:e004140] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Horman S, Beauloye C, Vanoverschelde JL, et al. AMP-activated protein kinase in the control of cardiac metabolism and remodeling. Curr Heart Fail Rep. 2012;9(3):164–73. doi: 10.1007/s11897-012-0102-z. [DOI] [PubMed] [Google Scholar]
- 95.Hinson JT, Chopra A, Lowe A, et al. Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep. 2017;19(11):2410. doi: 10.1016/j.celrep.2017.05.038. [DOI] [PubMed] [Google Scholar]
- 96.Arad M, Benson DW, Perez-Atayde AR, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109(3):357–62. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gollob MH, Green MS, Tang AS, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. New Engl J Med. 2001;344(24):1823–31. doi: 10.1056/NEJM200106143442403. [DOI] [PubMed] [Google Scholar]
- 98.Mehdirad AA, Fatkin D, DiMarco JP, et al. Electrophysiologic characteristics of accessory atrioventricular connections in an inherited form of Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol. 1999;10(5):629–35. doi: 10.1111/j.1540-8167.1999.tb00239.x. [DOI] [PubMed] [Google Scholar]
- 99.Porto AG, Brun F, Severini GM, et al. Clinical Spectrum of PRKAG2 Syndrome. Circ Arrhythm Electrophysiol. 2016;9(1):e003121. doi: 10.1161/CIRCEP.115.003121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burwinkel B, Scott JW, Buhrer C, et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet. 2005;76(6):1034–49. doi: 10.1086/430840. [DOI] [PMC free article] [PubMed] [Google Scholar]