

Abstract
The rates of evolution of the proteins of any organism vary across orders of magnitude. A primary factor influencing rates of protein evolution is expression. A strong negative correlation between expression levels and evolutionary rates (the so-called E–R anticorrelation) has been observed in virtually all studied organisms. This effect is currently attributed to the abundance-dependent fitness costs of misfolding and unspecific protein–protein interactions, among other factors. Secreted proteins are folded in the endoplasmic reticulum, a compartment where chaperones, folding catalysts, and stringent quality control mechanisms promote their correct folding and may reduce the fitness costs of misfolding. In addition, confinement of secreted proteins to the extracellular space may reduce misinteractions and their deleterious effects. We hypothesize that each of these factors (the secretory pathway quality control and extracellular location) may reduce the strength of the E–R anticorrelation. Indeed, here we show that among human proteins that are secreted to the extracellular space, rates of evolution do not correlate with protein abundances. This trend is robust to controlling for several potentially confounding factors and is also observed when analyzing protein abundance data for 6 human tissues. In addition, analysis of mRNA abundance data for 32 human tissues shows that the E–R correlation is always less negative, and sometimes nonsignificant, in secreted proteins. Similar observations were made in Caenorhabditis elegans and in Escherichia coli, and to a lesser extent in Drosophila melanogaster, Saccharomyces cerevisiae and Arabidopsis thaliana. Our observations contribute to understand the causes of the E–R anticorrelation.
Keywords: rates of evolution, dN/dS, expression levels, E–R anticorrelation, secreted proteins.
Introduction
The rates of evolution of the proteins of any organism differ by orders of magnitude: whereas certain proteins remain virtually unaltered over long evolutionary periods, others can quickly accumulate a substantial number of amino acid replacements (Zuckerkandl and Pauling 1965; Li et al. 1985). Rates of protein evolution are commonly used to detect natural selection and infer the functional importance of different proteins and protein regions, to construct phylogenetic trees, find orthologs among different species, etc., and understanding the reasons behind proteins’ disparate rates of evolution is a central aim of modern evolutionary biology. The neutral theory of molecular evolution (Kimura 1968, 1983) predicts that highly important proteins should evolve slowly owing to increased selective constraints (Kimura and Ohta 1974). At the turn of the 21st century, as large sequence and functional genomic datasets became available, scientists discovered a long list of factors that correlate with rates of protein evolution, including expression level, expression breadth, function, subcellular location, number of protein–protein interactions, etc. (for review, see Pál et al. 2006; Rocha 2006; Alvarez-Ponce 2014; Zhang and Yang 2015).
Intriguingly, gene expression seems to be the main correlate of rates of protein evolution. Highly expressed genes often encode slow-evolving proteins, a trend known as the Expression–Rate (E–R) anticorrelation. First described in yeasts (Pál et al. 2001), this anticorrelation has been found in virtually all organisms in which it has been investigated, from viruses to mammals (Krylov et al. 2003; Rocha and Danchin 2004; Drummond et al. 2005, 2006; Drummond and Wilke 2008; Pagán et al. 2012; Zhang and Yang 2015). Analyses in yeasts indicate that gene expression level may explain up to 34% of the variability of rates of protein evolution (Pál et al. 2001), and that a composite variable combining mRNA abundance, protein abundance and Codon Adaptation Index (CAI, which is often used as a surrogate for expression level) accounts for 41% of the variability of rates of evolution (Drummond et al. 2006).
A number of plausible hypotheses have been proposed to explain the E–R anticorrelation. First, the translational robustness hypothesis (Drummond et al. 2005; Wilke and Drummond 2006; Drummond and Wilke 2008) postulates that proteins are under selective pressures to maintain structures that are able to fold into their correct native conformation despite some translation errors, since misfolded proteins are costly to refold or degrade and resynthesize, and can even aggregate and become cytotoxic. Translation errors occur at a rate of ∼5 misincorporated amino acids per 10,000 translated codons (Parker 1989; Drummond and Wilke 2009), which means that ∼20% of average-length yeast proteins contain at least one error. Highly expressed proteins would experience increased selective pressures to remain robust to translation errors—as the costs of misfolding are proportional to the rate of translation—which would make them highly conserved during evolution. This hypothesis has been expanded to include the effect of misfolding in the absence of translation errors, giving rise to a second hypothesis, the protein misfolding avoidance hypothesis (Yang et al. 2010). The main tenet of a third hypothesis, the protein misinteraction avoidance hypothesis (Levy et al. 2012; Yang et al. 2012), is that selection would be strong against changes in protein surfaces to avoid protein–protein misinteractions. Selective constrains in this manner would be stronger in more abundant proteins, which could competitively interfere and nonspecifically interact with more proteins. Last, the mRNA folding requirement hypothesis (Park et al. 2013) posits that part of the E–R anticorrelation results from the requirement of highly abundant mRNAs to fold strongly. These hypotheses are not necessarily mutually exclusive, and each of them may explain part of the E–R anticorrelation. In particular, the protein misinteraction avoidance hypothesis may mostly explain the constraints on surface residues that come into contact with other molecules, whereas the translational robustness hypothesis may be more relevant to explain the evolutionary rate variability of structurally important core residues (Yang et al. 2012).
Building upon these hypotheses, we raise the question whether the selective pressures underlying the E–R anticorrelation are universal to all proteins, or whether there may exist classes of proteins in which such pressures (and hence the E–R anticorrelation) are relaxed. Eukaryotic cells have evolved complex mechanisms to promote and survey protein folding and deal with misfolded proteins in the endoplasmic reticulum, which specifically target secreted and membrane proteins. Newly synthesized secreted and membrane proteins are targeted to the endoplasmic reticulum. In the course of being translocated into this compartment, the nascent polypeptides encounter a unique environment where chaperones and folding enzymes recognize un/misfolded proteins and assist with their folding or degradation (Braakman and Hebert 2013) in a manner similar to their cytosolic counterparts (Buchberger et al. 2010). In addition, proteins translocated into the endoplasmic reticulum are subject to quality control of folding and maturation that prevent the exit of misfolded or aberrant proteins to their sites of function, ensuring that they remain in an environment suitable for their folding and maturation (Anelli and Sitia 2008). For instance, many secreted proteins are modified by N-linked glycosylation and/or acquire disulfide bonds, modifications that influence their stability and contribute to their native fold by highly interdependent quality control systems (Braakman and Hebert 2013). Finally, if misfolded proteins accumulate in the endoplasmic reticulum due to high secretory load, stress, or mutations that prevent proper folding, a highly efficient unfolded protein response (UPR) elicits a signaling cascade aimed at eliminating the misfolded proteins. In animals, the UPR promotes a coordinated adaptive response that expands the endoplasmic reticulum’s folding and degradative capacity, leads to selective degradation of mRNAs encoding reticulum-translocating proteins, and attenuates general translation (Walter and Ron 2011). If these measures are not sufficient to eliminate the misfolded proteins, UPR activation may lead to cell death.
Together, the endoplasmic reticulum quality control systems and the UPR are expected to decrease the rate of misfolding and the fitness costs associated with misfolding of secreted proteins, probably making the translational robustness hypothesis and the protein misfolding avoidance hypothesis less applicable to this class of proteins. In addition, once secreted proteins enter the extracellular environment, which often exhibits a lower concentration and diversity of proteins (Yerbury et al. 2005), it is relatively unlikely that they engage in misinteraction, and if misinteraction occurs its effects are expected to be milder compared with misinteraction in the intracellular space; we therefore hypothesize that the protein misinteraction avoidance hypothesis may also be less pertinent to secreted proteins than to nonsecreted ones. As a consequence, we expect a weaker E–R anticorrelation among secreted proteins than among nonsecreted ones.
To test this hypothesis, we computed rates of protein evolution in various organisms, and measured the E–R anticorrelation separately among secreted and nonsecreted proteins. In humans, nonsecreted proteins exhibit a strong E–R anticorrelation, whereas secreted proteins do not exhibit a significant correlation. The trend is independent of several potential confounding factors. Similar results were obtained in Caenorhabditis elegans and Escherichia coli, and to a lesser extent in Drosophila melanogaster, Saccharomyces cerevisiae, and Arabidopsis thaliana. In all studied organisms, the E–R anticorrelation is stronger among nonsecreted proteins than among secreted ones.
Results
The Evolutionary Rate of Human Secreted Proteins Does Not Depend on Expression Level
For each human gene, the most likely ortholog in the mouse genome was identified, and the rate of evolution of the encoded protein was estimated from the nonsynonymous to synonymous divergence ratio (ω = dN/dS). Mouse orthologs were identified for 16,581 human genes. For each of these genes, whole-body protein abundance data were obtained from the PaxDB database (Wang et al. 2012). Whole-body abundances were estimated by combining protein abundance datasets corresponding to different tissues (Wang et al. 2012). Consistent with previous results (Pál et al. 2001; Krylov et al. 2003; Rocha and Danchin 2004; Drummond et al. 2005, 2006; Drummond and Wilke 2008; Pagán et al. 2012; Zhang and Yang 2015), ω correlates negatively with protein abundance (Spearman's rank correlation coefficient, ρ = −0.204, P = 3 × 10−136). Throughout this article, we first present results for protein abundance data obtained from a consistent source database for all species analyzed (Wang et al. 2012); we then show that our results are consistent when using mRNA abundance to measure gene expression from species-specific databases (see Materials and Methods).
We classified human protein-coding genes as secreted (if at least one of the encoded proteins were known or predicted to be secreted according to the MetazSecKB database; Meinken et al. 2015) (N = 2,279) or nonsecreted (otherwise, N = 13,567). We observed striking differences in the correlation between ω and protein abundance when we analyzed the subsets of secreted and nonsecreted proteins separately (supplementary table S1, Supplementary Material online, fig. 1). A significant E–R anticorrelation was observed for nonsecreted proteins (ρ = −0.259, P = 4.56 × 10−181), but not for secreted proteins (ρ = 0.038, P = 0.084; supplementary table S1, Supplementary Material online). The differences between the secreted and nonsecreted protein correlations were highly significant (Fisher r-to-z transformation, Z = −13.43, P < 0.0001).
Fig. 1.
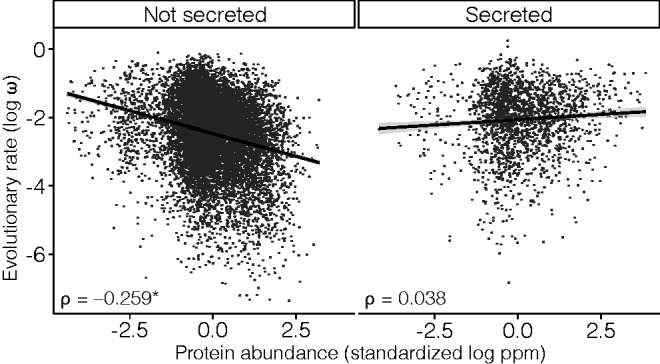
Correlation between rates of protein evolution (ω) and protein abundance for human secreted and nonsecreted proteins. The E–R anticorrelation is only significant for nonsecreted proteins (indicated by an asterisk). ρ, Spearman’s rank correlation coefficient; ppm, parts per million.
Since protein abundances are expected to have changed to some extent since the divergence of humans and mice, we repeated our analyses using human-specific evolutionary rates, which we computed using the cow genome as outgroup. We continued to observe a significant negative correlation for nonsecreted proteins (ρ = −0.234, P = 2.87 × 10−137) whereas we observed a slightly positive correlation among secreted proteins (ρ = 0.080, P = 5.39 × 10−4; Z = −12.72, P < 0.0001; supplementary table S2, Supplementary Material online).
The observed differences in E–R anticorrelations hold true when classifying proteins as secreted or nonsecreted using another three alternative databases (supplementary table S1, Supplementary Material online). In all three cases, nonsecreted proteins exhibited a significantly negative E–R correlation, whereas secreted proteins exhibited positive E–R correlations (significant in two cases; supplementary table S1, Supplementary Material online).
In order to evaluate the consistency of our observations across different tissues, we next investigated the correlation between ω and protein abundance within six individual human tissues/organs (supplementary table S3, Supplementary Material online, fig. 2). Among the nonsecreted protein class, the correlation was negative in all six tissues (significant in four). Among secreted proteins, the correlation was positive in four tissues and negative in the other two, and nonsignificant in all cases. This was also observed when using human-specific evolutionary rates (supplementary table S2, Supplementary Material online). Consistent results were obtained when we considered the correlation between ω and mRNA abundances in 32 human tissues/organs. In all 32 tissues, the correlation was significantly negative for the nonsecreted protein class (fig. 3). For the secreted protein class, the correlation was negative for 26 tissues (significant in 17) and positive in 6 tissues (significant in 1). For all 32 tissues, the correlation was always more positive (or less negative) for secreted proteins than for nonsecreted proteins (supplementary table S3, Supplementary Material online; fig. 3). Similar results were obtained when classifying secreted proteins using alternative secretion databases (supplementary table S4, Supplementary Material online).
Fig. 2.

Correlation between ω and protein abundance for secreted and nonsecreted proteins expressed at different human organs/tissues. Although nonsecreted proteins show a strong negative correlation, secreted proteins exhibit weaker correlations. Significant Spearman correlation coefficients are marked with an asterisk. ρ, Spearman’s rank correlation coefficient; ppm, parts per million.
Fig. 3.

Spearman correlation coefficients between ω and mRNA abundance in 32 human tissues/organs. Tissues are sorted according to the Spearman correlation for nonsecreted proteins, with testis having the highest (ρ = −0.140) and cerebral cortex having the lowest (ρ = −0.3235) correlations (supplementary table S3, Supplementary Material online). In all tissues, the E–R correlations are higher (less negative) for secreted proteins.
The Lack of an E–R Anticorrelation among Secreted Proteins Is Not Due to Confounding Factors
We considered the possibility that the differences between the E–R anticorrelation of secreted and nonsecreted proteins might be a by-product of confounding factors. Potentially confounding factors include those that (1) differ between secreted and nonsecreted proteins, and (2) affect the strength of the E–R anticorrelation. We evaluated the potential effects of a number of factors known to correlate with ω, including gene expression level and breadth, gene essentiality, protein length, codon usage bias, mRNA secondary structure folding strength, gene duplication, gene conservation, positive selection, presence of disulfide bonds and N-linked glycosylation, protein functional and structural categories, and protein folding stability.
Gene expression level and breadth seem to be the main determinants of rates of protein evolution (Pál et al. 2001; Drummond et al. 2005, 2006; Ingvarsson 2007; Park and Choi 2010), thus raising the potential that these factors may be behind the different E–R anticorrelations observed between secreted and nonsecreted proteins. We did not observe significant differences in protein abundance levels between secreted and nonsecreted proteins (Wilcoxon rank sums test, Z = 1.75, P = 0.080), and thus we discarded expression level as a confounding factor. We did however observe significant differences in expression breadth (defined as the number of tissues in which a gene is expressed), with nonsecreted proteins being more likely to be expressed in a greater number of tissues (Z = −5.84, P = 5.4 × 10−9), and a negative correlation between expression breadth and rates of evolution (ρ = −0.180, P = 6 × 10−121). In order to discard expression breadth as a confounding factor, we divided the dataset into two classes, proteins expressed in one to two tissues and proteins expressed in three to six tissues, and observed that in each of the classes the correlation was significantly weaker among secreted proteins than among nonsecreted ones (supplementary table S5, Supplementary Material online). We considered a second measure of protein expression breadth, τ, which takes a value between 0 (low tissue specificity) and 1 (high tissue specificity) depending on the distribution of protein expression levels across the different tissues (Yanai et al. 2004). Using this measure, we found secreted proteins to have a lower expression breadth (median for secreted proteins: τ = 0.943; median for nonsecreted proteins: τ = 0.920; Z = 8.39, P < 10−300). We divided the dataset into three approximately equally sized categories based on τ, and found that in all three cases the E–R anticorrelation was significantly stronger among nonsecreted proteins than among secreted proteins (supplementary table S5, Supplementary Material online). We therefore discarded expression breadth as a potential confounding factor. The strength of the correlation between τ and ω does not differ significantly between secreted and nonsecreted proteins (secreted proteins: ρ = 0.114, P = 2.04 × 10−7; nonsecreted proteins: ρ = 0.096, P = 2.50 × 10−26; Z = −0.74, P = 0.459).
Essential genes tend to evolve slower than nonessential ones (Hurst and Smith 1999; Liao et al. 2006; Luisi et al. 2015; Alvarez-Ponce et al. 2016). In order to discard essentiality as a confounding factor, we obtained mouse phenotypic data from the Mouse Genome Project (Eppig et al. 2015). Human genes that had a mouse ortholog with a lethal phenotype were considered essential. We observed that secreted and nonsecreted proteins were equally likely to be encoded by essential genes (Fisher’s exact test, P = 0.742), thus allowing us to discard essentiality as a confounding factor.
Furthermore, secreted proteins are shorter (protein length: Z = −9.80, P < 10−300), more likely to use optimal codons (frequency of optimal codons, Fop: Z = 13.00, P = 10−38; CAI: Z = 8.45, P < 10−300) and more likely to have stronger mRNA secondary structures (folding strength: Z = 8.66, P = 5 × 10−18; minimum free energy [MFE]: Z = 2.04, P = 0.017) than nonsecreted proteins, and protein length, codon bias, and mRNA folding strength correlate with rates of protein evolution (Pál et al. 2006; Rocha 2006; Park et al. 2013; Alvarez-Ponce 2014). For each of the three variables, we divided the dataset into three groups (low, intermediate and high, each with roughly the same number of genes) and investigated the differences in the E–R anticorrelations of secreted and nonsecreted proteins within each group (supplementary table S5, Supplementary Material online). In all cases, the E–R anticorrelation was significantly weaker among secreted proteins, allowing us to discard these factors as potential confounding variables.
Next, we investigated whether gene duplication may be a confounding factor. Secreted proteins were more likely to be encoded by duplicated genes than nonsecreted ones (Fisher’s exact test, P = 2.95 × 10−8) and the E–R anticorrelation was weaker among duplicated genes (ρ = −0.181, P = 2 × 10−76) compared with nonduplicated genes (ρ = −0.320, P = 7 × 10−103). However, when we analyzed duplicated and nonduplicated genes separately, we continued to observe that the E–R anticorrelation was significantly weaker among secreted genes (supplementary table S5, Supplementary Material online). Therefore, we concluded that the higher duplicability of genes encoding secreted proteins was not the reason why secreted proteins do not exhibit a significant E–R anticorrelation.
Secreted proteins are known to evolve faster on average than nonsecreted ones (Julenius and Pedersen 2006; Cui et al. 2009; Liao, Weng and Zhang 2010; Nogueira et al. 2012). To discard the possibility that certain conserved proteins among the nonsecreted class may be driving the E–R anticorrelation in nonsecreted proteins but not in secreted proteins, we investigated the E–R correlation among a subset of nonsecreted proteins with a nearly identical distribution of evolutionary rates to that of the secreted protein class. For each secreted protein, a nonsecreted protein with an identical or very similar ω was randomly chosen (either the one immediately up in the ranking, or that immediately down) (median ω for secreted proteins: 0.1529; median ω for selected nonsecreted proteins: 0.1528; Z = −0.077, P = 0.939). Among this subset of nonsecreted proteins, we continued to observe a strong negative correlation (ρ = −0.229, P = 6.00 × 10−25, N = 1,973). We performed a scan of positive selection comparing the genomes of 10 mammalian species (see Materials and Methods). Out of the 10,539 genes for which positive selection could be tested (those with orthologs in all 10 genomes), positive selection was inferred in 1,043 (168 when correcting for multiple testing). We found that secreted proteins are more likely to be encoded by genes under positive selection than nonsecreted proteins (fraction of genes under positive selection for secreted and nonsecreted proteins: 13.93% and 9.11%, respectively; Fisher’s exact test, P = 8.49 × 10−8). These differences were less pronounced when correcting for multiple testing (2.19% and 1.48%, respectively; Fisher’s exact test, P = 0.06). In addition, we found that, among secreted proteins, those encoded by genes under positive selection are more abundant than those encoded by genes with no signatures of positive selection (Z = 3.97, P = 0.0001; correcting for multiple testing: Z = 2.00, P = 0.046). This observation, together with the fact that positive selection accelerates protein evolution (e.g., Arbiza et al. 2006), might potentially explain why secreted proteins do not exhibit a significant E–R anticorrelation. Nonetheless, when we removed proteins encoded by genes with signatures of positive selection, we observed that the E–R anticorrelation was still strong for nonsecreted proteins and nonsignificant among secreted proteins (nonsecreted: ρ = –0.236, P = 3.00 × 10−90; secreted: ρ = 0.061, P = 0.044; Z = –9.23, P < 0.0001). We therefore discarded positive selection as a confounding factor.
Disulfide bonds are almost exclusive to secreted and membrane proteins (Sevier and Kaiser 2002), and it has been suggested that proteins with disulfide bonds evolve faster than those with no disulfide bonds (Hegyi and Bork 1997). It is thus possible that disulfide bonds are behind the differences between secreted and nonsecreted proteins described here. In order to discard this possibility, we repeated our analyses after removing proteins with disulfide bonds. Mirroring our observations for the entire dataset, the E–R anticorrelation was strong for nonsecreted proteins (ρ = −0.271, P = 2.76 × 10−184) and nonsignificant among secreted proteins (ρ = −0.015, P = 0.635), which led us to discard the presence of disulfide bonds as the reason for this difference. Likewise, many secreted proteins undergo N-glycosylation, a modification that generally increases their solubility and stability (Shental-Bechor and Levy 2008; Hanson et al. 2009; Lee et al. 2015) and enables a tight control of protein folding (Xu and Ng 2015). Nonetheless, when we removed proteins with known N-linked glycosylation sites from the secreted protein dataset (N = 533), the E–R anticorrelation remained nonsignificant (ρ = −0.030, P = 0.170).
We next considered the possibility that the lack of an E–R anticorrelation among secreted proteins might be due to their enrichment in certain functional categories. In order to discard this possibility, we classified human proteins into 24 different functional categories and investigated the E–R anticorrelation within each category. Among nonsecreted proteins, we observed significant negative correlations in 18 out of the 24 KOG categories, whereas only 3 significant negative correlations were observed among secreted proteins (supplementary table S6, Supplementary Material online). In all three cases, where significant correlations were observed among secreted proteins, correlations were weaker than for nonsecreted proteins. We therefore discarded protein functional categories as a confounding factor.
We classified proteins into structural categories using SCOPe 2.03 (Fox et al. 2014). We only considered SCOPe categories that had >50 proteins in both the secreted and nonsecreted classes. Among nonsecreted proteins, all six SCOPe classes exhibited significant negative E–R correlations. Among secreted proteins, only class “a” (all alpha proteins) had a significant negative correlation (supplementary table S7, Supplementary Material online), although the E–R anticorrelation was not significantly different between secreted (ρ = −0.362, P = 0.001, N = 86) and nonsecreted (ρ = −0.289, P = 1.28 × 10−13, N = 633) proteins (Z = –0.7, P = 0.484). In all other investigated categories, nonsecreted proteins showed significantly stronger E–R anticorrelations. We therefore discarded protein structural categories as a potential confounding factor.
Finally, we considered whether proteins’ structural robustness to translation errors might differ between secreted and nonsecreted proteins. By protecting secreted proteins from misfolding, a higher robustness of these proteins might potentially account for the weaker E–R anticorrelation observed in this class. However, we did not observe evidence for a higher robustness of secreted proteins. For each human protein with a well-resolved three-dimensional structure, we obtained the median ΔΔG value (median across all possible amino acid changes) from Faure and Koonin (2015). We found that secreted proteins had significantly higher ΔΔG values (median for secreted proteins: 2.15 kcal/mol, median for nonsecreted proteins: 2.00 kcal/mol; Z = 4.01, P = 0.0001), suggesting lower robustness. It should be noted, however, that ΔΔG values represent incomplete predictions of the effect of amino acid changes on protein structures: highly stable proteins (those with low ΔG) may remain stable despite highly destabilizing amino acid changes (changes inducing a large ΔΔG), and lowly stable proteins may be destabilized even by changes with slightly positive ΔΔG. Nonetheless, ΔG values are very hard to predict computationally, and experimentally determined ΔG values are available for only a handful of proteins (Kumar et al. 2006; Yang et al. 2010). Therefore, a complete comparison of robustness to translation errors will only be possible once large ΔG datasets become available. In order to discard ΔΔG as a confounding factor, we divided proteins into two categories according to their median ΔΔG and investigated the E–R anticorrelation within each class. We observed that secreted proteins exhibited a weaker and nonsignificant anticorrelation in both classes (supplementary table S6, Supplementary Material online).
Separate Analysis of Buried and Surface Amino Acid Residues
The trend described here (a nonexistent, or reduced, E–R anticorrelation among secreted proteins) may be due to either (1) the fact that secreted proteins are folded in the presence of folding catalysts and undergo stringent quality control, thus reducing the likelihood and/or the deleterious effects of misfolding, or (2) the fact that these proteins act in the intracellular space, thus reducing the likelihood and/or the deleterious effects of misinteraction (see Discussion). In an attempt to distinguish between both scenarios, we analyzed separately the parts of the CDSs encoding buried amino acid residues and those encoding exposed residues. For each human protein, we used ACCPro, release 5.2 in the software SCRATCH 1-D, release 1.1 (Magnan and Baldi 2014) to predict buried and surface amino acid residues. From each of our human–mouse CDS alignments, we generated two subalignments: one containing the codons encoding buried residues, and another containing the codons encoding exposed residues. A separate ω was computed for each subalignment. For both classes of amino acids, the E–R anticorrelation was significantly negative for nonsecreted proteins, and nonsignificantly positive for secreted proteins (buried nonsecreted: ρ = –0.260, P = 6.50 × 10−183; buried secreted: ρ = 0.013, P = 0.556; Z = –11.63, P < 0.0001; exposed nonsecreted: ρ = –0.259, P = 3.29 × 10−181; exposed secreted: ρ = 0.038, P = 0.089; Z = –12.62, P < 0.0001). These results support a scenario in which the reduced E–R anticorrelation may be due to a combination of both effects (strong quality control and extracellular location).
Human Membrane Proteins Partially Defy the E–R Anticorrelation
Similar to secreted proteins, membrane proteins are synthesized in the endoplasmic reticulum and follow the secretory pathway, and end up partially exposed to the extracellular space. Therefore, we investigated whether the E–R anticorrelation was also reduced in membrane proteins. For that purpose, we used the LOCATE database (Sprenger et al. 2008) to classify all human proteins as either secreted (N = 1,627), membrane (N = 3,641) and other proteins (N = 11,355). Similar to our observations for secreted proteins (fig. 1, supplementary table S1, Supplementary Material online), the correlation between ω and whole-body protein abundance (Wang et al. 2012) was weak and nonsignificant among membrane proteins (membrane proteins: ρ = −0.028, P = 0.119; secreted: ρ = 0.040, P = 0.126; others: ρ = −0.294, P = 7 × 10−197). Using expression data from individual tissues, we found that the rates of evolution of membrane proteins showed significant negative correlations with protein abundance in 3 out of 6 tissues, and with mRNA abundance in 25 out of 32 tissues. Consistent with what is observed in secreted proteins, most of the correlations in individual tissues were weaker for membrane proteins compared with their nonsecreted (28 out of 32 tissues) or cytoplasmic (31 out of 32 tissues) counterparts (supplementary table S4, Supplementary Material online).
Next, we further classified membrane proteins as either type I (those with the N-terminus on the extracellular space; N = 1,391), type II (with the N-terminal domain in the intracellular space; N = 81), or multi-pass (with two or more transmembrane domains; N = 1,685) membrane proteins. The correlation between ω and whole-body protein abundance (Wang et al. 2012) was significant only for multi-pass membrane proteins, and in all cases it was substantially weaker than that for cytoplasmic or nonsecreted proteins (type I: ρ = −0.037, P = 0.164; type II: ρ = −0.128, P = 0.256; multi-pass: ρ = −0.037, P = 0.164) (supplementary table S1, Supplementary Material online). We further investigated the E–R correlation among membrane proteins in individual tissues, using both protein and mRNA expression data. Multi-pass and type I membrane proteins shared similar characteristics with the secreted protein class analyzed above, having partially positive correlations, and generally higher correlations than either nonsecreted or cytoplasmic proteins (supplementary table S4, Supplementary Material online).
Secreted Proteins in Other Organisms Also Exhibit a Weaker E–R Anticorrelation
In order to establish whether the trend was universal or specific to certain organisms, we considered whether secreted proteins also exhibited a nonsignificant, or a weaker E–R anticorrelation in other organisms, including C. elegans, D. melanogaster, S. cerevisiae, A. thaliana and E. coli. These organisms differ in their secretory pathways and effective population sizes.
For each C. elegans gene, ω was computed from comparison to its most likely ortholog in the C. briggsae genome. Out of the 13,121 genes for which orthologs were identified, 10,677 were classified as nonsecreted and 2012 were classified as secreted. Using whole-organism protein abundance data (Wang et al. 2012), we found that the E–R anticorrelation was observed in nonsecreted proteins (N = 5,165, ρ = −0.092, P = 3 × 10−11) but not in secreted proteins (N = 957, ρ = 0.070, P = 0.030; Z = –4.61, P < 0.0001) (fig. 4). This was also observed when we considered branch-specific evolutionary rates for C. elegans, which we estimated using C. japonica as an outgroup (supplementary table S2, Supplementary Material online). Similar results were obtained when genes with signatures of positive selection (as inferred from comparison of five Caenorhabditis genomes) were removed from the analyses (nonsecreted proteins: N = 3,678, ρ = −0.095, P = 9.2 × 10−9; secreted proteins: N = 611, ρ = 0.063, P = 0.122; Z = –3.62, P = 0.0001). However, when positive selection tests are corrected for multiple testing, the correlation among secreted proteins becomes significantly positive, with significant differences between the E–R correlations of secreted and nonsecreted proteins (nonsecreted proteins: N = 4,044, ρ = −0.094, P = 2.1 × 10−9; secreted proteins: N = 670, ρ = 0.090, P = 0.023; Fisher r-to-z transformation test, Z = −4.42, P < 0.0001). Using mRNA expression data corresponding to 17 developmental stages of C. elegans (Kolesnikov et al. 2015), we found E–R correlations to be negative in all 17 developmental stages for nonsecreted proteins (significantly in 16), whereas for secreted proteins only 7 developmental stages exhibited negative correlations (none of which were significant) and the remaining 10 developmental stages showed positive correlations (4 significant). In all cases in which negative correlations were observed among secreted proteins, Spearman correlations were weaker in secreted proteins than in nonsecreted proteins (supplementary table S8, Supplementary Material online, fig. 5).
Fig. 4.

Correlation between rates of protein evolution and protein abundance for secreted and nonsecreted proteins in C. elegans, D. melanogaster, S. cerevisiae, A. thaliana, and E. coli. Significant correlation coefficients are marked with an asterisk. ρ, Spearman’s rank correlation coefficient; ppm, parts per million.
Fig. 5.

Correlation between rates of protein evolution and mRNA abundance for secreted and nonsecreted proteins in C. elegans, D. melanogaster, S. cerevisiae, A. thaliana, and E. coli. For C. elegans and D. melanogaster, expression datasets corresponding to the whole adult body were used. For A. thaliana, a dataset corresponding to the whole plant (entry ATGE 23) was used. Significant correlation coefficients are marked with an asterisk. ρ, Spearman’s rank correlation coefficient.
We computed ω ratios for 11,714 pairs of D. melanogaster–D. yakuba orthologs, of which 9,220 were classified as nonsecreted and 2,100 were classified as secreted. Using whole-body protein abundance data, the correlations were significantly negative in both secreted (N = 836, ρ = −0.143, P = 3.1 × 10−5) and nonsecreted (N = 4,808, ρ = −0.300, P = 9.0 × 10−101) classes (fig. 4). However, consistent with our observations in human and C. elegans, secreted proteins exhibited a significantly weaker anticorrelation than nonsecreted proteins (Fisher r-to-z transformation test, Z = −4.41, P < 0.0001). This was also observed when using D. melanogaster branch-specific evolutionary rates, which we estimated using D. ananassae as an outgroup (supplementary table S2, Supplementary Material online). Similar results were obtained when genes with signatures of positive selection (as inferred from comparison of 6 Drosophila genomes) were removed from the analyses (nonsecreted proteins: ρ = −0.264, P = 6.00 × 10−33; secreted proteins: ρ = −0.110, P = 0.047; Z = −2.25, P = 0.008), and when we corrected positive selection inferences for multiple testing (nonsecreted proteins: ρ = −0.260, P = 3.35 × 10−35; secreted proteins: ρ = −0.101, P = 0.055; Z = –2.86, P = 0.0042). Using whole-fly mRNA abundance data, we observed different E–R anticorrelations between secreted and nonsecreted proteins (nonsecreted proteins: N = 9,214, ρ = −0.276, P = 3 × 10−161; secreted proteins: N = 2,097, ρ = −0.086, P = 1.00 × 10−5; Z = –8.14, P < 0.0001; fig. 5). Using mRNA abundance data for 26 individual fly tissues, we found negative E–R correlations in all tissues for both nonsecreted and secreted proteins (significant in all 26 tissues for nonsecreted proteins, and in 25 tissues for secreted ones). Nonetheless, the E–R anticorrelations were weaker for secreted proteins in 25 of the 26 tissues (supplementary table S9, Supplementary Material online).
For each S. cerevisiae gene, we computed ω from comparison to its most likely ortholog in S. paradoxus (N = 5,603). A total of 5,423 genes were classed as nonsecreted and 125 were classed as secreted. Correlations between ω and protein abundance (Wang et al. 2012) were significantly negative among both secreted (ρ = −0.422, P = 1.5 × 10−6, N = 120) and nonsecreted (ρ = −0.515, P < 10−300, N = 5,241) protein classes (fig. 4). The correlation was weaker, but not significantly weaker, for secreted proteins (Z = −1.28, P = 0.200). This was also observed when using S. cerevisiae lineage-specific rates, which we estimated using S. mikatae as an outgroup (supplementary table S2, Supplementary Material online). Similar results were obtained when genes with signatures of positive selection (as inferred from comparison of 5 Saccharomyces genomes) were removed from the analyses (nonsecreted proteins: ρ = −0.484, P = 1 × 10−59; secreted proteins: ρ = −0.247, P = 0.214; Z = −1.33, P = 0.18), and when correcting our tests of positive selection for multiple testing (nonsecreted proteins: ρ = −0.480, P = 2 × 10−63; secreted proteins: ρ = −0.215, P = 0.255). Using mRNA expression levels (Nagalakshmi et al. 2008), we observed significantly negative correlations among both secreted (N = 111, ρ = −0.315, P = 0.0007 and nonsecreted proteins (N = 4,533, ρ = −0.427, P < 2 × 10−200; Z = 1.33, P = 0.183; fig. 5). Similar results were obtained when genes with signatures of positive selection were removed from the analyses (nonsecreted proteins: ρ = −0.389, P = 7 × 10−33; secreted proteins: ρ = −0.108, P = 0.599; Z = −1.4, P = 0.153).
Smaller differences between the E–R anticorrelations of secreted and nonsecreted proteins were observed in Arabidopsis. For each A. thaliana gene, we computed the ω ratio from comparison with its A. lyrata ortholog (N = 15,472). A total of 14,393 genes were classed as nonsecreted and 893 were classed as secreted. Negative correlations between protein expression and evolutionary rate were observed in both secreted (N = 636, ρ = −0.218, P = 2.9 × 10−8) and nonsecreted proteins (N = 11,438, ρ = −0.295, P = 9 × 10−228), with a significantly weaker correlation in secreted proteins (Z = 2.02, P = 0.043; fig. 4). Similar observations where made when using branch-specific rates of evolution for A. thaliana, which we estimated using Thellungiella parvula as an outgroup (supplementary table S2, Supplementary Material online). These differences, however, were less pronounced when we investigated mRNA expression levels. When we analyzed mRNA expression data for 79 individual A. thaliana expression datasets (Schmid et al. 2005), the E–R correlation was always significantly negative in all datasets for nonsecreted proteins, and for secreted proteins it was negative in 78 of the datasets (significant in 71). In 68 of the datasets the E–R anticorrelation was weaker among secreted than among nonsecreted proteins (supplementary table S10, Supplementary Material online).
To see whether these general observations also held in bacteria, we calculated evolutionary rates between E. coli and Salmonella enterica pairs of orthologs. E. coli proteins were classified into 6 subcellular locations using the LocTree 3 server (Goldberg et al. 2014) (supplementary table S11, Supplementary Material online; fig. 4). Using protein abundance data (Wang et al. 2012), we observed the strongest E–R anticorrelation among cytoplasmic proteins (N = 1,417, ρ = −0.240, P = 6 × 10−20), whereas the correlation was nonsignificant for secreted proteins (N = 141, ρ = −0.164, P = 0.051). The two correlations, however, were not significantly different (Z = −0.89, P = 0.373). This was also observed when we used E. coli-specific evolutionary rates, estimated using Yersinia pestis as an outgroup (supplementary table S2, Supplementary Material online). Similar results were obtained using mRNA abundance data (Covert et al. 2004): the strongest E–R anticorrelation was observed among cytoplasmic proteins (N = 1,398, ρ = −0.157, P = 3.6 × 10−9), whereas secreted proteins exhibited a nonsignificant anticorrelation (N = 131, ρ = −0.039, P = 0.660; Z = −1.29, P = 0.20; fig. 5).
Discussion
We have found that, among human secreted proteins, the correlation between protein abundances and rates of evolution is nonexistent (figs. 1and 2). Consistent trends have been observed using expression data (protein and mRNA abundance) of different human tissues/organs (fig. 3), and trends are robust to controlling for several potentially confounding factors (supplementary tables S4 and S5, Supplementary Material online).
Following the translational robustness hypothesis (Drummond et al. 2005; Wilke and Drummond 2006; Drummond and Wilke 2008) and the more general protein misfolding avoidance hypothesis (Yang et al. 2010), the strength of the E–R anticorrelation should be directly related to the fitness costs of misfolding (re-folding, degradation, re-synthesis, and in extreme cases cytotoxic aggregation; Drummond and Wilke 2009; Geiler-Samerotte et al. 2011). (It should be noted, nonetheless, that protein aggregation can be reversible in some cases; Wallace et al. 2015.) At least two lines of evidence suggest that the E–R anticorrelation is indeed a result of these costs. First, evolutionary simulations in which misfolding costs are removed result in no E–R anticorrelation (Drummond and Wilke 2008). Second, the E–R anticorrelation is particularly strong in neural tissues, which has been attributed to these tissues being less capable of dealing with misfolding (Drummond and Wilke 2008). According to the protein misinteraction avoidance hypothesis (Levy et al. 2012; Yang et al. 2012), the strength of the E–R anticorrelation should directly depend on the fitness costs of misinteraction. Indeed, computer simulations in which these costs are suppressed result in no E–R anticorrelation (Yang et al. 2012).
Therefore, the lack of an E–R anticorrelation among secreted proteins described here may be the result of either reduced rates of misfolding or misinteraction, or reduced fitness costs of misfolding or misinteraction affecting secreted proteins. In other words, our observations are compatible with a scenario in which considering two hypothetical identical proteins, one being secreted and another not being secreted, the one that is secreted either: (1) is less likely to misfold; (2) is less likely to misinteract; (3) if it misfolds, misfolding entails lower fitness costs; or (4) if it misinteracts, misinteraction entails lower fitness costs. All of these possibilities seem plausible. First, newly synthesized secreted proteins fold in the presence of chaperones and folding catalysts, and are subject to quality control of folding and maturation. For instance, the highly abundant endoplasmic reticulum chaperone BiP (also known as GRP78 and Kar2) binds to the majority of proteins that traverse the endoplasmic reticulum (Braakman and Hebert 2013). This chaperone not only protects newly translocated and misfolded proteins from aggregation and assists their folding, but it is also required for the protein translocation process itself. BiP deletion leads to very early embryonic lethality, consistent with its multiple functions and variety of known client proteins (Anelli and Sitia 2008). In addition, a number of observations suggest that secreted proteins may be less prone to translation errors. Our analyses indicate that secreted proteins tend to be encoded by a higher fraction of optimal codons and by more stable mRNAs, and tend to be shorter than nonsecreted proteins (median length of secreted proteins: 411; median length of nonsecreted proteins: 456; Z = −7.98; P = 1.0 × 10−15). Optimal codons are more likely to be translated accurately (Akashi 1994; Stoletzki and Eyre-Walker 2007; Hershberg and Petrov 2008). Stable mRNAs reduce the speed of translation, also reducing translation errors (Yang et al. 2014). Assuming a constant likelihood of mistranslation per translated codon (e.g., ∼5 misincorporated amino acids per 10,000 translated codons; Parker 1989; Drummond and Wilke 2009), short proteins are less likely to accumulate translation errors than long ones. In addition, many secreted proteins exhibit disulfide bonds (Sevier and Kaiser 2002) and/or N-linked glycosylation (Xu and Ng 2015). By increasing protein stability, disulfide bonds may reduce the destabilizing effects of translation errors, and N-linked glycans provide structural information to instruct folding and to enable a rigorous quality control of glycoprotein folding (Xu and Ng 2015). Nonetheless, when we classified genes into different classes of codon bias, mRNA stability, or length, or when proteins with disulfide bonds or N-linked glycosylation were removed from our analyses, the E–R anticorrelation was always lower among secreted proteins than among nonsecreted ones (supplementary table S5, Supplementary Material online), indicating that these factors alone are not responsible for the lack of an E–R anticorrelation among secreted proteins. Second, secreted proteins are released to the extracellular space. This space may often exhibit a lower concentration and diversity of proteins—particularly signaling proteins—than the intracellular environment (Yerbury et al. 2005), which is expected to reduce the likelihood of misinteraction. In fact, secreted proteins seem to have, on average, a lower number of protein–protein interactions than nonsecreted ones (Kim et al. 2007). Third, secreted proteins are synthesized and folded in the endoplasmic reticulum, where, in addition to chaperones that facilitate folding, the UPR may reduce the costs of misfolding by efficiently dealing with misfolded proteins (Walter and Ron 2011). Fourth, misinteraction in the extracellular space may in general have less deleterious effects than misinteraction in the crowded intracellular environment. Separate analysis of buried amino acid residues (whose rate of evolution may be largely explained by the protein misfolding avoidance hypothesis) and exposed residues (whose rate of evolution may be mostly explained by the misinteraction avoidance hypothesis), shows that the E–R anticorrelation among secreted proteins is nonsignificant in both classes, thus suggesting that the applicability of both hypotheses is reduced for this class of proteins.
Interestingly, membrane proteins exhibit a weak E–R anticorrelation (supplementary table S1, Supplementary Material online), but not as weak as that of secreted proteins. On the one hand, membrane proteins follow the secretory pathway and part of their residues end up exposed to the extracellular space. On the other hand, they have domains on both the cytosolic and lumenal faces of the endoplasmic reticulum membrane, and their folding status must be monitored by the coordinated action of quality control systems of both compartments, thus posing particular problems to the quality control of folding (Houck and Cyr 2012). Several proteins have been implicated in monitoring the assembly of transmembrane domains and in facilitating their degradation by autophagy (Houck and Cyr 2012). Finally, membrane proteins exported from the endoplasmic reticulum are subject to peripheral quality control along the late secretory pathway and at the plasma membrane (Okiyoneda et al. 2011).
Similar to what is observed in humans, a lack of an E–R anticorrelation is also observed among the secreted proteins of C. elegans and E. coli, and a very reduced (but significant) anticorrelation was observed among the secreted proteins of D. melanogaster (figs. 4and 5). Smaller differences were observed among the secreted and nonsecreted proteins of S. cerevisiae and A. thaliana (figs. 4and 5). Although several aspects of the endoplasmic reticulum quality control and UPR are conserved among eukaryotes, the mechanisms to cope with accumulation of misfolded proteins vary among yeasts, metazoans and plants, which may explain, at least in part, the differences that we observed between the studied organisms. Metazoans have evolved complex mechanisms to respond to accumulation of misfolded proteins, deployed by activation of three endoplasmic reticulum stress sensors (IRE1, ATF6, and PERK). In contrast, fungi rely on IRE1 as the single endoplasmic reticulum stress sensor. Harding et al. (2002) proposed that the complexity of intercellular communications in metazoans and the evolution of long-lived differentiated cells imposed a need to respond to rapid fluctuations in protein load and the challenge of dealing with accumulation of potentially cytotoxic misfolded proteins that may accumulate over time. Furthermore, the fitness cost of accumulating misfolded proteins may be reduced by partitioning them into daughter cells in rapidly dividing unicellular eukaryotes (Babour et al. 2010). In plants, no functional ortholog of the gene encoding PERK has been identified, but a component of the G protein complex AGB1 acts as an endoplasmic reticulum stress sensor (Ruberti and Brandizzi 2014). In addition, in A. thaliana part of the E–R anticorrelation is explained by exon edge conservation, rather than by translational selection or misinteraction avoidance (Bush et al. 2015). Escherichiacoli secreted proteins are folded and assembled in the periplasm under quality control afforded by chaperones, protein folding catalysts, and proteases (Miot and Betton 2004). In addition, unicellular organisms often live in very protein-poor environments (or at least environments with a low concentration of their own proteins), which may minimize the likelihood and the harmful effects of misinteraction.
We considered the possibility that the lack of an E–R anticorrelation (or a reduction thereof) in secreted proteins could be due to a high incidence of positive selection among highly expressed secreted proteins. Indeed, genes encoding secreted proteins with signatures of positive selection are more highly expressed than genes encoding secreted proteins with no signatures of positive selection (median protein abundances: 0.67 and 0.49, respectively, Z = 2.4; P = 0.01). This could potentially reduce the, or even result in a positive, E–R anticorrelation. However, when we removed genes with positive selection from our analyses, the E–R anticorrelation was still nonexistent (or significantly reduced, depending on the species) among secreted proteins. It should be noted, however, that positive selection is not always detectable; nonetheless, the number of species used in our scan of positive selection (10 mammalian species) should guarantee an acceptable statistical power (e.g., Anisimova et al. 2001).
According to the mRNA folding requirement hypothesis (Park et al. 2013), the lack of an E–R anticorrelation among secreted proteins may also be the result of a reduced correlation between expression level and mRNA stability among secreted proteins. However, we found that the correlation between mRNA abundance (averaged across the 32 human tissues) and predicted mRNA stability was nonsignificant among either secreted (ρ = 0.009, P = 0.677) and nonsecreted (ρ = 0.007, P = 0.450) proteins. In addition, the partial correlation between the MFE and mRNA abundance controlling for the length of the coding regions is indistinguishable between secreted and nonsecreted proteins (secreted: ρ = 0.091, P = 2.46 × 10−5; nonsecreted: ρ = 0.099, P = 2.82 × 10−29; Z = −0.35, P = 0.726). Finally, when we classified genes into three categories according to either their mRNA stability or MFE, the E–R anticorrelation was always significantly less negative among secreted proteins than among nonsecreted proteins (supplementary table S5, Supplementary Material online), allowing us to discard folding requirements as the driving factor behind our observations.
Materials and Methods
Secreted Proteins Datasets
Secretion data used throughout most of this work was obtained from MetazSecKB (Meinken et al. 2015) (for human, D. melanogaster and C. elegans), FunSecKB2 (Lum and Min 2011) (for S. cerevisiae) and PlantSecKB (Lum et al. 2014) (for A. thaliana). In this family of databases, proteins are classified as secreted either by manual curation or by computational prediction. Proteins predicted to be secreted are further classified into “highly likely” or “likely” secreted. We deemed proteins as secreted if they were either curated as secreted or in the “highly likely” predicted category. Proteins were classified as nonsecreted if they were neither classified as “curated”, “highly likely”, or “likely” secreted. For humans, additional secretion data were obtained from the LOCATE (Sprenger et al. 2008), SPD (Chen et al. 2005), and UniProt (UniProt 2015) databases, to verify consistency among different databases. Because secretion data for bacteria were not available in any of the databases used for the other species, we obtained a list of E. coli secreted proteins from the LocTree 3 server (Goldberg et al. 2014).
Protein and mRNA Abundance Data
Protein abundance data for all species were obtained from the PaxDB database v3.0 (Wang et al. 2012). The whole organism integrated datasets, which include collated data from numerous proteomics studies, were used. For human, additional integrated datasets were obtained from the same database for brain, heart, liver, lung, plasma, and platelet.
For each human gene, mRNA abundance data for 32 different tissues/organs, measured by RNA sequencing experiments, was obtained from HumanAtlas (Uhlen et al. 2015). Only genes with an FPKM value of 1 or higher were included in the analysis for each individual tissue. Messenger RNA abundance data for D. melanogaster and C. elegans was obtained from FlyBase (Attrill et al. 2016) and the modENCODE base experiments from the EBI Expression Atlas (accession number E-MTAB-2812) (Petryszak et al. 2015), respectively. For D. melanogaster we discarded a minority of values which could not be mapped to a single gene. Saccharomycescerevisiae gene expression data were obtained from Nagalakshmi et al. (2008). Seventy-nine datasets corresponding to different A. thaliana tissues, developmental stages and conditions were obtained from Schmid et al. (2005) and processed as in Alvarez-Ponce and Fares (2012). Escherichiacoli expression data were obtained from Covert et al. (2004). For each gene, mRNA abundances were averaged across three biological replicates.
Evolutionary Rate Estimation
All human protein and coding (CDS) sequences were obtained from the Ensembl database, release 62 (Cunningham et al. 2015). For each gene, the longest encoded protein was selected for analysis. Human–mouse pairs of orthologs were identified using a Reciprocal Best Hits approach. Each human protein sequence was used as query in a BLASTP search against the mouse proteome (with an E-value cut-off of 10−10). The best hit was used as query in a second search against the human proteome (using the same E-value cut-off). If the best hit identified in the second search was the original human protein, then the encoding human and mouse genes were considered as orthologs. Human genes for which mouse orthologs could not be detected were excluded from our analyses. For each pair of orthologs, the protein sequences were aligned using ProbCons 1.12 (Do et al. 2005), and the resulting alignment was used to guide the alignment of the CDS sequences using in-house software. For each of the resulting alignments, we estimated the nonsynonymous to synonymous divergence ratio (ω = dN/dS) using the M0 model implemented in the codeml program from the PAML package, version 4.4 (Yang 2007). The same methods were used to identify and analyze pairs of D. melanogaster–D. yakuba, C. elegans–C. briggsae, S. cerevisiae–S. paradoxus, A. thaliana–A. lyrata, and E. coli (strain K-12, substrain MG1655)–S. enterica enterica (serovar Typhimurium strain LT2) orthologs.
We also calculated lineage-specific ω ratios (specific to human, D. melanogaster, C. elegans, S. cerevisiae, A. thaliana and E. coli). For that purpose, we added outgroup species to our alignments (respectively, cow, D. ananassae, C. japonica, S. mikatae, T.parvula and Y.pestis subsp. pestis) and used the free-ratios model of codeml to estimate a separate ω for each branch.
Positive Selection Analyses
For each human gene, we identified orthologs in nine mammalian genomes (chimpanzee, gorilla, orangutan, macaque, mouse, rat, cow, dog, and opossum) using the best reciprocal hit approach described above. These genomes were chosen because the currently available assemblies have a particularly high coverage. We did not include more distant species in our analyses in order to avoid saturation of synonymous sites. We could identify orthologs in all 9 species for 10,702 human genes. Protein sequences were aligned using ProbCons 1.12 (Do et al. 2005). Because genome annotations contain a substantial amount of errors (Devos and Valencia 2001; Alvarez-Ponce et al. 2011; Tu et al. 2012), and positive selection detection methods are very sensitive to such errors (Schneider et al. 2009; Fletcher and Yang 2010; Markova-Raina and Petrov 2011; Jordan and Goldman 2012), we filtered our alignments using two methods: Gblocks (Castresana 2000) with default parameters, and a sliding window approach. Our sliding window approach identified and removed (1) alignment regions of 15 amino acids in which one of the sequences contained 10 or more amino acids that were unique to that species (singleton nonsynonymous mutations) and (2) regions of 5 amino acids in which one of the sequences contained 5 amino acids unique to that species. The protein alignments, the results of the two filtering techniques, and the CDS sequences were used to generate filtered CDS alignments. Each of the resulting alignments were used in a M7 vs. M8 test of positive selection (Yang 2000) using the codeml program from the PAML package (Yang 2007). This test allows detecting the presence of a class of codons with ω > 1. All PAML computations were repeated three times, using different starting ω values (ω = 0.04, 0.4, and 4), in order to alleviate the problem of local optima. We considered genes with a P-value lower than 0.05 to be positively selected. We repeated our analyses after controlling our tests of positive selection for multiple testing using a false discovery rate of q < 0.1 (Benjamini and Hochberg 1995).
The fraction of removed residues was slightly higher for secreted than for nonsecreted proteins, consistent with their higher rates of evolution (median for secreted proteins: 33.47%, median for nonsecreted proteins: 31.24%, Z = 2.63, P = 0.0085). In order to discard this as a confounding factor, we evaluated the E–R anticorrelation among: (1) secreted proteins with a 10-species alignment available and (2) a group of nonsecreted proteins with a nearly identical distribution of fraction of filtered residues in the 10-species alignment. For each secreted protein, a nonsecreted protein with an identical or very similar percent of filtered residues was randomly chosen (either the one immediately up in the ranking, or that immediately down). Again, a negative E–R correlation was observed for nonsecreted proteins (ρ = −0.252, P = 2.32 × 10−19), but not for secreted ones (ρ = −0.081, P = 0.004; Z = –8.44, P < 0.0001).
The same methods were used to detect positive selection in six Drosophila species (D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, and D. ananassae), five Caenorhabditis species (C. elegans, C. brenneri, C. remanei, C. briggsae, and C. japonica), and five Saccharomyces species (S. cerevisiae, S. paradoxus, S. mikatae, S. kudriavzevii, and S. bayanus).
Additional Gene Information
For each human gene, the CAI (Sharp and Li 1987) was computed using the cai program in the EMBOSS package (Rice et al. 2000). The frequency of optimal codons (Fop; Ikemura 1981) in humans was calculated using the codon frequencies recorded in the Codon Usage Database (Nakamura et al. 2000). Genes were deemed as duplicated if Ensembl’s BioMart contained annotated paralogs, and as singleton otherwise. For each human gene, we estimated its mRNA folding strength using RNAfold in the ViennaRNA 2.1.9 package (Lorenz et al. 2011) at a temperature of 30 °C. A sliding window with a length of 150 and a step size of 10 nucleotides was used to calculate the average number of bonds in the mRNA secondary structure, similar to the approach used by Park et al. (2013). We also calculated the MFE over the entire sequence using the same package. Genes were classified into 24 different functional categories using the euKaryotic Orthologous Groups (KOG) classification defined in EggNOG version 4.5 (Huerta-Cepas et al. 2016). Proteins that were assigned to more than one category were omitted in functional category analyses. A list of proteins with disulfide bonds was derived from UniProt (UniProt 2015), and a list of proteins with N-linked glycosylation was obtained from the Unipep database (Zhang et al. 2006).
Statistical Analyses
Nonparametric statistical analyses, including Wilcoxon tests and Spearman rank correlation tests, were conducted using R (http://www.r-project.org) and JMP (SAS Institute, Inc., NC, USA). Correlation coefficients were compared using Fisher r-to-z transformation tests (Myers and Sirois 2004).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors are grateful to Mario Fares and Sandip Chakraborty for helpful discussion and for reading the manuscript critically, to Guilhem Faure and Eugene V. Koonin for sharing their protein ΔΔG data, and to two anonymous referees for helpful comments. We are also grateful to the University of Nevada, Reno Information Technology Operations for computational resources. This work was supported by funds from the University of Nevada, Reno awarded to D.A.P. P.M.B. was partially supported by the National Institutes of Health (grant RR024210/GM103554 from the COBRE Program of the National Center for Research Resources).
Footnotes
Associate editor: Csaba Pal
References
- Akashi H. 1994. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D. 2014. Why proteins evolve at different rates: the determinants of proteins’ rates of evolution In: Fares MA, editor. Natural selection: methods and applications. London: CRC Press (Taylor & Francis; ). p. 126–178. [Google Scholar]
- Alvarez-Ponce D, Aguadé M, Rozas J. 2011. Comparative genomics of the vertebrate insulin/TOR signal transduction pathway: a network-level analysis of selective pressures. Genome Biol Evol. 3:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D, Fares MA. 2012. Evolutionary rate and duplicability in the Arabidopsis thaliana protein-protein interaction network. Genome Biol Evol. 4:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D, Sabater-Muñoz B, Toft C, Ruiz-González MX, Fares MA. 2016. Essentiality is a strong determinant of protein rates of evolution during mutation accumulation experiments in Escherichia coli. Genome Biol Evol. 8(9): 2914–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T, Sitia R. 2008. Protein quality control in the early secretory pathway. EMBO J. 27:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 18:1585–1592. [DOI] [PubMed] [Google Scholar]
- Arbiza L, Dopazo J, Dopazo H. 2006. Positive selection, relaxation, and acceleration in the evolution of the human and chimp genome. PLoS Comput Biol. 2:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attrill H, Falls K, Goodman JL, Millburn GH, Antonazzo G, Rey AJ, Marygold SJ, FlyBase c. 2016. FlyBase: establishing a gene group resource for Drosophila melanogaster. Nucleic Acids Res. 44:D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babour A, Bicknell AA, Tourtellotte J, Niwa M. 2010. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell 142:256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57:289–300. [Google Scholar]
- Braakman I, Hebert DN. 2013. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 5:a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T. 2010. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40:238–252. [DOI] [PubMed] [Google Scholar]
- Bush SJ, Kover PX, Urrutia AO. 2015. Lineage-specific sequence evolution and exon edge conservation partially explain the relationship between evolutionary rate and expression level in A. thaliana. Mol Ecol. 24:3093–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Yin Y, Gao G, Li S, Jiang Y, Gu X, Luo J. 2005. SPD—a web-based secreted protein database. Nucleic Acids Res. 33:D169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. 2004. . Integrating high-throughput and computational data elucidates bacterial networks. Nature 429:92–96. [DOI] [PubMed] [Google Scholar]
- Cui Q, Purisima EO, Wang E. 2009. Protein evolution on a human signaling network. BMC Syst Biol. 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2015. Ensembl 2015. Nucleic Acids Res 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Valencia A. 2001. Intrinsic errors in genome annotation. Trends Genet 17:429–431. [DOI] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. 2005. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA 102:14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO. 2006. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 23:327–337. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2009. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 10:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, The Mouse Genome Database Group. 2015. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res 43(Database issue):D726–D736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, Koonin EV. 2015. Universal distribution of mutational effects on protein stability, uncoupling of protein robustness from sequence evolution and distinct evolutionary modes of prokaryotic and eukaryotic proteins. Phys Biol. 12(3): 035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher W, Yang Z. 2010. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol Biol Evol. 27:2257–2267. [DOI] [PubMed] [Google Scholar]
- Fox NK, Brenner SE, Chandonia JM. 2014. SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 42:D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. 2011. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 108:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg T, Hecht M, Hamp T, Karl T, Yachdav G, Ahmed N, Altermann U, Angerer P, Ansorge S, Balasz K, et al. 2014. LocTree3 prediction of localization. Nucleic Acids Res. 42:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SR, Culyba EK, Hsu TL, Wong CH, Kelly JW, Powers ET. 2009. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci USA. 106:3131–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. 2002. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 18:575–599. [DOI] [PubMed] [Google Scholar]
- Hegyi H, Bork P. 1997. On the classification and evolution of protein modules. J Protein Chem. 16:545–551. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2008. Selection on codon bias. Annu Rev Genet. 42:287–299. [DOI] [PubMed] [Google Scholar]
- Houck SA, Cyr DM. 2012. Mechanisms for quality control of misfolded transmembrane proteins. Biochim Biophys Acta 1818:1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, et al. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44:D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Smith NG. 1999. Do essential genes evolve slowly? Curr Biol. 9:747–750. [DOI] [PubMed] [Google Scholar]
- Ikemura T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 151:389–409. [DOI] [PubMed] [Google Scholar]
- Ingvarsson PK. 2007. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol. 24:836–844. [DOI] [PubMed] [Google Scholar]
- Jordan G, Goldman N. 2012. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol Biol Evol. 29:1125–1139. [DOI] [PubMed] [Google Scholar]
- Julenius K, Pedersen AG. 2006. Protein evolution is faster outside the cell. Mol Biol Evol. 23:2039–2048. [DOI] [PubMed] [Google Scholar]
- Kim PM, Korbel JO, Gerstein MB. 2007. Positive selection at the protein network periphery: evaluation in terms of structural constraints and cellular context. Proc Natl Acad Sci USA. 104:20274–20279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217:624–626. [DOI] [PubMed] [Google Scholar]
- Kimura M. 1983. The neutral theory of molecular evolution. Cambridge: Cambridge University Press. [Google Scholar]
- Kimura M, Ohta T. 1974. On some principles governing molecular evolution. Proc Natl Acad Sci USA. 71:2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov N, Hastings E, Keays M, Melnichuk O, Tang YA, Williams E, Dylag M, Kurbatova N, Brandizi M, Burdett T, et al. 2015. ArrayExpress update–simplifying data submissions. Nucleic Acids Res. 43:D1113–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov DM, Wolf YI, Rogozin IB, Koonin EV. 2003. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 13:2229–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MS, Bava KA, Gromiha MM, Prabakaran P, Kitajima K, Uedaira H, Sarai A. 2006. ProTherm and ProNIT: thermodynamic databases for proteins and protein–nucleic acid interactions. Nucleic Acids Res. 34:D204–D206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Qi Y, Im W. 2015. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci Rep. 5:8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED, De S, Teichmann SA. 2012. Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. Proc Natl Acad Sci USA. 109:20461–20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Wu CI, Luo CC. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol. 2:150–174. [DOI] [PubMed] [Google Scholar]
- Liao BY, Scott NM, Zhang J. 2006. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 23:2072–2080. [DOI] [PubMed] [Google Scholar]
- Liao BY, Weng MP, Zhang J. 2010. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol. 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisi P, Alvarez-Ponce D, Pybus M, Fares MA, Bertranpetit J, Laayouni H. 2015. Recent positive selection has acted on genes encoding proteins with more interactions within the whole human interactome. Genome Biol Evol. 7:1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum G, Meinken J, Orr J, Frazier S, Min XJ. 2014. PlantSecKB: the Plant Secretome and Subcellular Proteome KnowledgeBase. Comput Mol Biol 4:1–17. [Google Scholar]
- Lum G, Min XJ. 2011. FunSecKB: the Fungal Secretome KnowledgeBase. Database (Oxford) 2011:bar001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan CN, Baldi P. 2014. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 30(18): 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova-Raina P, Petrov D. 2011. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res 21:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinken J, Walker G, Cooper CR, Min XJ. 2015. MetazSecKB: the human and animal secretome and subcellular proteome knowledgebase. Database (Oxford) 2015:bav077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miot M, Betton JM. 2004. Protein quality control in the bacterial periplasm. Microb Cell Fact 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers L, Sirois MJ. 2004. Spearman correlation coefficients, differences between In: Encyclopedia of Statistical Sciences. New York: John Wiley & Sons, Inc; p. 7901–7903. [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, Ikemura T. 2000. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira T, Touchon M, Rocha EP. 2012. Rapid evolution of the sequences and gene repertoires of secreted proteins in bacteria. PLoS One 7:e49403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Apaja PM, Lukacs GL. 2011. Protein quality control at the plasma membrane. Curr Opin Cell Biol. 23:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagán I, Holmes EC, Simon-Loriere E. 2012. Level of gene expression is a major determinant of protein evolution in the viral order Mononegavirales. J Virol. 86:5253–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst LD. 2001. Highly expressed genes in yeast evolve slowly. Genetics 158:927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Lercher MJ. 2006. An integrated view of protein evolution. Nat Rev Genet. 7:337–348. [DOI] [PubMed] [Google Scholar]
- Park C, Chen X, Yang JR, Zhang J. 2013. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 110:E678–E686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Choi SS. 2010. Expression breadth and expression abundance behave differently in correlations with evolutionary rates. BMC Evol Biol. 10:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. 1989. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 53:273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Fullgrabe A, Fuentes AM, Jupp S, Koskinen S, et al. 2015. Expression Atlas update-an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44:D746–D752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rocha EP. 2006. The quest for the universals of protein evolution. Trends Genet. 22:412–416. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. 2004. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 21:108–116. [DOI] [PubMed] [Google Scholar]
- Ruberti C, Brandizzi F. 2014. Conserved and plant-unique strategies for overcoming endoplasmic reticulum stress. Front Plant Sci. 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU. 2005. A gene expression map of Arabidopsis thaliana development. Nat Genet. 37:501–506. [DOI] [PubMed] [Google Scholar]
- Schneider A, Souvorov A, Sabath N, Landan G, Gonnet GH, Graur D. 2009. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol Evol. 1:114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. 2002. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 3:836–847. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shental-Bechor D, Levy Y. 2008. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci USA. 105:8256–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger J, Lynn Fink J, Karunaratne S, Hanson K, Hamilton NA, Teasdale RD. 2008. LOCATE: a mammalian protein subcellular localization database. Nucleic Acids Res. 36:D230–D233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. 2007. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 24:374–381. [DOI] [PubMed] [Google Scholar]
- Tu Q, Cameron RA, Worley KC, Gibbs RA, Davidson EH. 2012. Gene structure in the sea urchin Strongylocentrotus purpuratus based on transcriptome analysis. Genome Res. 22:2079–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347:1260419. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EW, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, et al. 2015. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162:1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. 2012. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics 11:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke CO, Drummond DA. 2006. Population genetics of translational robustness. Genetics 173:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Ng DT. 2015. Glycosylation-directed quality control of protein folding. Nat Rev Mol Cell Biol. 16:742–752. [DOI] [PubMed] [Google Scholar]
- Yanai I, Banjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2004. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21:650–659. [DOI] [PubMed] [Google Scholar]
- Yang JR, Chen X, Zhang J. 2014. Codon-by-codon modulation of translational speed and accuracy via mRNA folding. PLoS Biol. 12(7):e1001910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JR, Liao BY, Zhuang SM, Zhang J. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci USA. 109:E831–E840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JR, Zhuang SM, Zhang J. 2010. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol Syst Biol. 6:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2000. Maximum likelihood estimation on large phylogenies and analysis of adaptive evolution in human influenza virus A. J Mol Evol. 51:423–432. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yerbury JJ, Stewart EM, Wyatt AR, Wilson MR. 2005. Quality control of protein folding in extracellular space. EMBO Rep. 6:1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Loriaux P, Eng J, Campbell D, Keller A, Moss P, Bonneau R, Zhang N, Zhou Y, Wollscheid B, et al. 2006. UniPep–a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 7:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang JR. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L. 1965. Evolutionary divergence and convergence in proteins In: Bryson V, Vogel H, editors. Evolving genes and proteins. New York: Academic Press; p. 97–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.