
Figure 2.
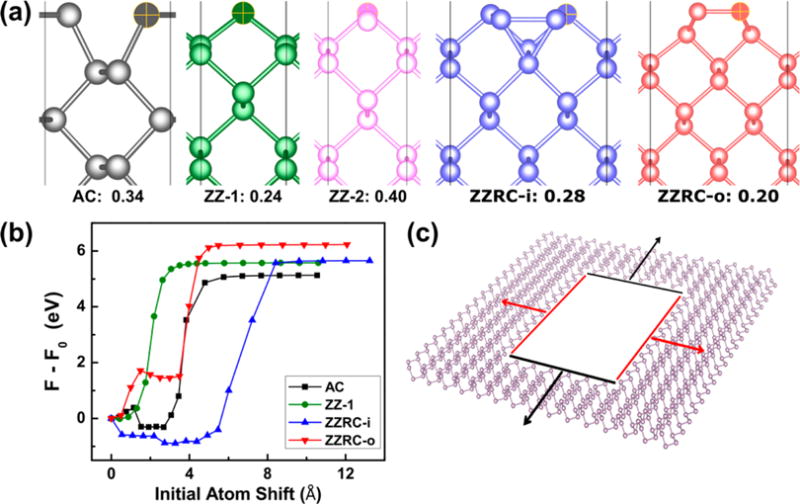
Structure and energetics of the phosphorene edges as computed using density functional theory. (a) Overhead views of the armchair (AC), zigzag single termination (ZZ-1), zigzag double termination (ZZ-2), inner-shifted reconstruction of the ZZ-2 edge (ZZRC-i),51 and outer-shifted reconstruction of the ZZ-2 edge (ZZRC-o)32 phosphorene edges. The corresponding edge energies for the supercells used are given in eV/Å to quantify the thermodynamic stability. (b) Energy landscapes for removing a single atom from phosphorene edges via the NEB method. (c) Schematic illustrating the observation of an elliptical nanopore in phosphorene. The armchair edges (black) recede faster than the most stable zigzag edge (ZZRC-o) due to the greater energy barrier for removing atoms from the latter edge, which results in relatively longer nanopore dimensions in the zigzag (200) direction (Figure 1f–i).