

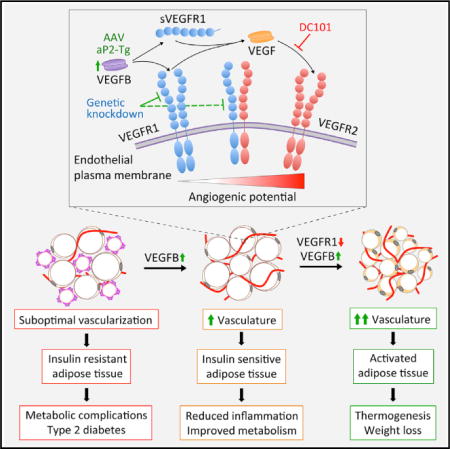
SUMMARY
Impaired angiogenesis has been implicated in adipose tissue dysfunction and the development of obesity and associated metabolic disorders. Here, we report the unexpected finding that vascular endothelial growth factor B (VEGFB) gene transduction into mice inhibits obesity-associated inflammation and improves metabolic health without changes in body weight or ectopic lipid deposition. Mechanistically, the binding of VEGFB to VEGF receptor 1 (VEGFR1, also known as Flt1) activated the VEGF/VEGFR2 pathway and increased capillary density, tissue perfusion, and insulin supply, signaling, and function in adipose tissue. Furthermore, endothelial Flt1 gene deletion enhanced the effect of VEGFB, activating the thermogenic program in subcutaneous adipose tissue, which increased the basal metabolic rate, thus preventing diet-induced obesity and related metabolic complications. In obese and insulin-resistant mice, Vegfb gene transfer, together with endothelial Flt1 gene deletion, induced weight loss and mitigated the metabolic complications, demonstrating the therapeutic potential of the VEGFB/VEGFR1 pathway.
Graphical abstract
In Brief: Robciuc et al. show that the VEGFB/VEGFR1 pathway can be employed to engage the VEGF/VEGFR2 angiogenic pathway in adipose tissue. This mechanism increases adipose tissue vascularity without pathological side effects and provides a safe therapeutic option for restoring insulin sensitivity and promoting weight loss in obesity.
INTRODUCTION
Obesity is of serious concern, because it increases the incidence of type 2 diabetes and many other chronic diseases. There are no effective treatments available for the general population, and lifestyle interventions are difficult to sustain and ineffective in patients with established diabetes (Wing et al., 2013). Therefore, treatments that prevent obesity from progressing to cardiovascular and metabolic disease are desperately needed.
Vascular endothelial growth factors (VEGFs) and their receptors (VEGFRs) control the growth and remodeling of the vasculature, and recent evidence has highlighted the role of these processes in obesity and insulin resistance. The VEGF family consists of five secreted glycoproteins in mammals: VEGF (or VEGFA), VEGFB, VEGFC, VEGFD, and placenta growth factor (PlGF) (Holmes and Zachary, 2005; Shibuya, 2013). The VEGF ligands bind with differing specificities to three endothelial tyrosine kinase receptors: VEGFR1/fms-like tyrosine kinase 1 (Flt1), VEGFR2/human kinase insert domain receptor (KDR)/mouse fetal liver kinase 1 (Flk1), and VEGFR3/fms-like tyrosine kinase 4 (Flt4) (Koch and Claesson-Welsh, 2012; Shibuya, 2013). Neuropilin (NRP)-1 and NRP-2 are co-receptors for the various VEGFRs in endothelial cells (Pellet-Many et al., 2008). VEGF signaling via VEGFR2 is the bona fide angiogenic pathway in normal and pathological processes (Chung and Ferrara, 2011). VEGF has a 10-fold lower affinity to VEGFR2 than to VEGFR1, which serves mainly as a decoy receptor without effective signal transduction in the endothelium (Koch et al., 2011). A soluble form of VEGFR1 is also produced, which regulates angiogenesis by blocking VEGF activity (Shibuya, 2013). PlGF and VEGFB bind only to VEGFR1, but unlike PlGF, VEGFB does not induce VEGFR1 downstream signaling (Anisimov et al., 2013). Instead, VEGFB is considered to function by displacing VEGF from VEGFR1 to VEGFR2, thus activating VEGFR2 (Bry et al., 2014; Kivelä et al., 2014; Shibuya and Claesson-Welsh, 2006).
Induction of angiogenesis in adipose tissue via VEGF/VEGFR2 has been shown to activate of the thermogenic program in brown and white adipose tissue and to protect mice from obesity and associated metabolic complications (Elias et al., 2012; Sun et al., 2012, 2014; Sung et al., 2013). Although VEGFB shows great potential for improving tissue vascularization, this is thought to be limited to cardiac tissue, which has the highest endogenous expression of VEGFB (Bry et al., 2010; Li et al., 2008a). Studies by Hagberg et al. (2010, 2012) proposed that VEGFB induces fatty acid transport across the endothelium in heart, skeletal muscle, and brown adipose tissue and that VEGFB inhibition could provide a novel treatment for type 2 diabetes. Blockade of VEGFB prevented lipid deposition in extra-adipose tissues, increased peripheral glucose uptake, maintained fasting and postprandial glucose levels, and improved glucose tolerance and insulin sensitivity. VEGFR1 and its tyrosine kinase activity, as well as NRP-1, were needed for the VEGFB-mediated effects on fatty acid transport across the endothelium. This was surprising, because VEGFR1 is a weak tyrosine kinase and VEGFB does not induce efficient VEGFR1 dimerization or signaling (Anisimov et al., 2013). Moreover, we found no difference in fatty acid uptake among the hearts of VEGFB transgenic, gene-deleted, and wild-type rats (Kivelä et al., 2014). Replication of high-fat diet (HFD) studies in VEGFB gene-deleted mice did not reproduce the data published by Hagberg et al. (2010, 2012) (Dijkstra et al., 2014).
Here, we report on a comprehensive interrogation of the role of VEGFB and VEGFR1 in obesity and insulin resistance. Unexpectedly, we found improved glucose metabolism, insulin sensitivity, and reduced inflammation in adipose tissue of Vegfb-transduced mice and in a Vegfb transgenic mouse model with increased VEGFB expression in adipose tissue. Mechanistically, we demonstrate that VEGFB binding to VEGFR1 increases adipose tissue vascularity and blood perfusion by activating the VEGF/VEGFR2 pathway. The vascular remodeling induced by VEGFB in obese mice improved the delivery, signaling, and function of insulin, thereby alleviating the metabolic complications of obesity. When fully engaged, the VEGFB/VEGFR1 pathway promoted the thermogenic program in subcutaneous adipose tissue and enhanced the basal metabolic rate, rendering mice resistant to diet-induced obesity and related metabolic complications. Thus, the VEGFB/VEGFR1 mechanism overcomes the well-established toxicity of VEGFA, which disrupts the normal patterning of the microvasculature.
RESULTS
VEGFB Improves Obesity-Related Metabolic Complications
We showed recently that VEGFB is a potent inducer of cardiac vascular remodeling, which protects the heart from ischemic damage but has no effect on cardiac fatty acid uptake (Kivelä et al., 2014). Because discordant results were reported from HFD studies in Vegfb−/− mice, we replicated these studies in our laboratory using the same protocol that was used in the previous two reports (Dijkstra et al., 2014; Hagberg et al., 2012). We observed no differences in body weight, body composition, fasting glucose, or glucose tolerance test between Vegfb−/− and wild-type mice fed a standard diet or a HFD (Figure S1).
To determine whether increased VEGFB levels affect obesity and insulin sensitivity, we performed HFD studies in mice transduced with recombinant adeno-associated virus vector (AAV) producing VEGFB186 (AAV-B186) or containing a scrambled control sequence (AAV-Ctrl) (Figures S2A–S2D). Although AAV-B186 did not affect body weight, body composition, or food consumption, it significantly improved the metabolic health of the obese mice (Figures 1A and 1B; Table S1). The AAV-B186 mice had no significant changes in glucose levels but had lower fasting levels of triglycerides and insulin in serum (Figures 1C–1E) and an improved response in intraperitoneal glucose and insulin tolerance tests (IP-GTT and IP-ITT, respectively) (Figures 1F–1H). There were no changes in triglyceride levels in the liver or heart or in lipid class profiles in the liver (Figures 1I–1K; Figures S3A–S3C), contrasting with the proposed role of VEGFB in ectopic lipid deposition (Hagberg et al., 2010, 2012). No significant changes in glucose-induced insulin secretion, basal metabolic rate, substrate utilization, or circadian activity were observed in the AAV-B186 mice (data not shown).
Figure 1. Improved Lipid and Glucose Metabolism but No Changes in Ectopic Lipid Deposition in HFD-Fed AAV-B186-Expressing Mice.
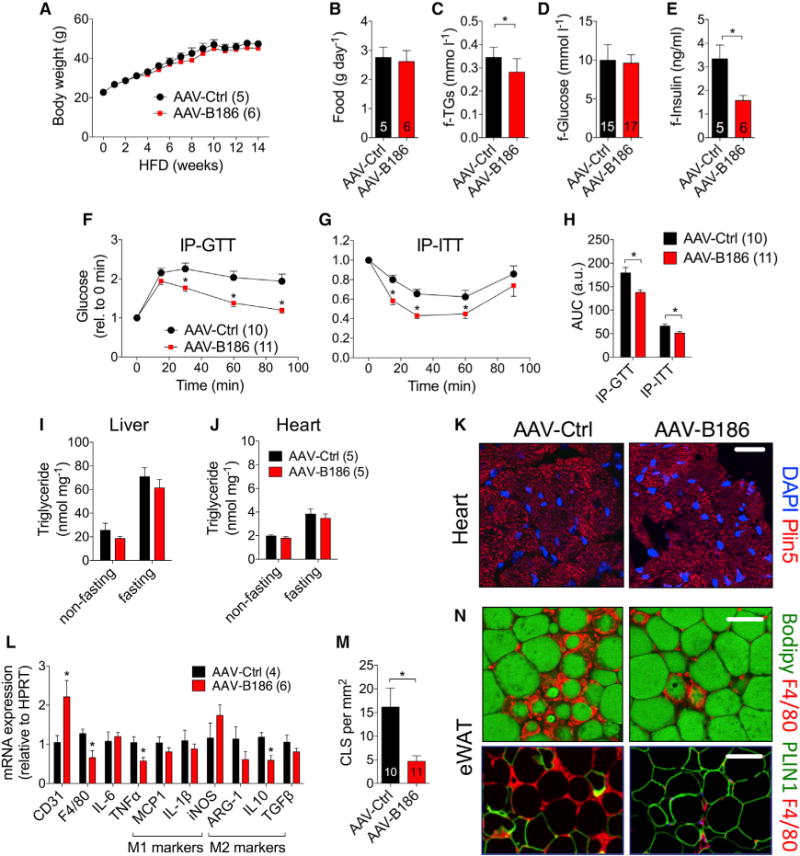
C57BL/6JOlaHsd males transduced with AAVs and fed a HFD were used for the experiments.
(A–E) Comparison of (A) body weight, (B) food consumption, and fasting levels of (C) triglycerides, (D) glucose, and (E) insulin.
(F–H) IP-GTT (F), IP-ITT (G), and area under the curce (AUC) (H).
(I and J) Triglyceride concentration in the (I) liver and (J) heart.
(K) Visualization of lipid droplets in Plin5-stained heart sections.
(L) eWAT gene expression analysis by qPCR.
(M and N) Crown-like structure (CLS) quantification (M) from F4/80 (macrophage, red)-stained eWAT sections (N). Adipocytes were stained green for lipids (Bodipy) or perilipin 1 (PLIN1).
Scale bars, (K) 50 μm and (N) 100 μm. The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated with two-tailed unpaired t test.
VEGFB Induces Expansion of Adipose Tissue Vasculature
VEGFB186 promotes cardiac hypertrophy, inducing an athletelike heart without conversion to cardiomyopathy (Kivelä et al., 2014). Necropsy analyses showed that the VEGFB186-transduced mice had larger hearts after HFD feeding (Table S2). The epididymal white adipose tissue (eWAT) mass was also increased, even though other fat pads, whole body weight, and body composition were not changed (Tables S2 and S1, respectively). Gene expression analysis of eWAT showed reduced expression of inflammatory markers in the AAV-B186 mice but no changes in the ratio of M1/M2 macrophage polarization markers (Figure 1L). The increased eWAT mass in the AAV-B186 mice was likely due to reduced death of adipocytes that accumulate in crown-like structures (Figures 1M and 1N). CD31 gene expression analysis indicated an increase in vascular density in the eWAT of AAV-B186 mice, which was confirmed by whole-mount staining (Figure 1L; Figure S4A–S4D).
The increased vascular density in adipose tissue was a surprising finding, because the vascular effects of VEGFB were thought to be restricted mostly to cardiac tissue. The VEGFB effects on adipose tissue vasculature have not been investigated previously, although adipose tissue expresses the highest level of VEGFB mRNA after cardiac and skeletal muscle (Bry et al., 2014).
To determine whether the increased vascular density in the Vegfb-transduced mice is secondary to reduced inflammation in adipose tissue or is a primary effect of VEGFB, we analyzed adipose tissue vasculature at earlier time points after AAV administration and on a standard diet. Just 2 weeks after AAV administration, VEGFB186 induced a robust increase in the capillary network density and vessel size, causing reddening of the eWAT (Figures 2A–2F). Microvasculature in the VEGFB186-transduced adipose tissue had a regular pattern in contrast to VEGF164-transduced tissue, which displayed capillary abnormalities even though the AAV-VEGFA164 dose was 5-fold lower (Figures 2A and 2B). The VEGFB167 isoform had similar effects as VEGFB186 on adipose tissue microvasculature (data not shown). Capillary blood flow, blood vessel coverage with mural cells, and permeability to Evans blue or 100 nm nanoparticles was not altered in the Vegfb-transduced mice (Figures S5A–S5D) (Bry et al., 2010). Furthermore, unlike with VEGF, the vascular remodeling was not accompanied by an increased infiltration of inflammatory cells (Figure S5E). Increased vasculature remodeling was also observed in brown adipose tissue (BAT) from rats transduced with AAV encoding human VEGFB186 (Figures S6A and S6B) and in inguinal white adipose tissue (iWAT) of female mice (Figure S6C).
Figure 2. VEGFB Transduction Leads to Adipose Tissue Capillary Bed Expansion.
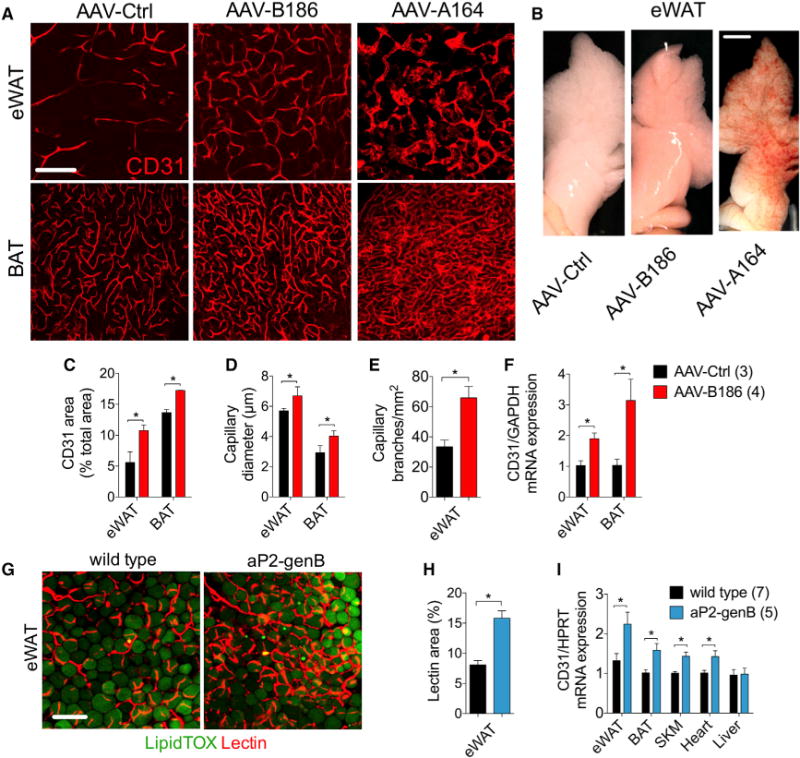
C57BL/6JOlaHsd males on a standard diet, transduced for 2 to 4 weeks with the indicated AAVs or transgenic for VEGFB were used for the experiments.
(A) Representative confocal projections of adipose tissue whole-mount staining for CD31.
(B) Macroscopic images of eWAT.
(C–E) CD31 area (C), capillary diameter (D), and capillary branch density (E) were quantified from images represented by the panels in (A).
(F) CD31 mRNA expression in adipose tissue.
(G and H) Representative confocal projections of adipose tissue whole-mount staining from mice injected intravenously with lectin (G) and lectin area quantification (H).
(I) CD31 mRNA expression in various tissues.
Scale bar, (A and G) 100 μm and (B) 3 μm. The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated with two-tailed unpaired t test.
Because the liver is effectively transduced by AAVs (Figure S2B) and VEGF is known to induce a strong response in the liver, we analyzed the livers from our AAV-transduced mice. VEGFB had no effect on liver capillary density, sinusoidal endothelial cell fenestrae, macrophage recruitment, or metabolic zonation in the liver (Figures S7A–S7D). Furthermore, global transcriptomics analysis of the liver did not reveal significant changes in gene expression or pathway analysis (data not shown).
To further validate these results, we generated transgenic mice with aP2/FABP4 promoter-driven expression of the mouse Vegfb gene (aP2-genB), which encodes both VEGFB isoforms. VEGFB levels were increased in adipose tissue and serum from the aP2-genB mice (Figures S8A–S8E), which also showed increased vessel perfusion in eWAT (Figures 2G and 2H) and increased CD31 mRNA levels in tissues expressing the transgene (Figure 2I). Although the VEGFB protein levels were significantly increased in skeletal muscles of the aP2-genB mice, the skeletal muscle endothelial cells markers were not altered (Figure S8F). HFD feeding of aP2-genB mice resulted in a similar phenotype to that in the AAV-VEGFB-transduced mice, although there was no clear reduction of eWAT inflammation (Figures S9A–S9G). HFD-fed aP2-genB mice gained less weight (Figure S9A) and had a significantly reduced fat mass when compared to wild-type littermates (Figure S9H). The reduction in fat mass was associated with upregulation of the thermogenic program in the subcutaneous adipose tissue (Figure S9I).
VEGFB-Induced Vascular Remodeling in Adipose Tissue Requires VEGF/VEGFR2
VEGFB binding to VEGFR1 does not trigger cellular signals directly; instead, VEGFB is considered to function by displacing VEGF from VEGFR1 to VEGFR2 (Anisimov et al., 2013; Bry et al., 2014; Kivelä et al., 2014; Shibuya and Claesson-Welsh, 2006). To test this proposed mechanism in adipose tissue, we deleted Flt1 in endothelial cells of Flt1-fl/fl;Pdgfb-CreERT2 (Flt1-ECKO) mice by tamoxifen administration (Figures S10A–S10D). This resulted in reduced levels of VEGFR1, with the remaining protein in adipose tissue and serum likely produced by non-endothelial cells such as macrophages. The Flt1-ECKO mice had increased adipose tissue capillary density and tissue perfusion, which was boosted by Vegfb gene transfer (Figure 3A). To determine whether VEGF binding to VEGFR2 is responsible for the increased adipose tissue vascularity in the VEGFB-transduced mice, we blocked VEGF/VEGFR2 signaling using the anti-VEGFR2 antibody DC101. DC101 treatment completely prevented the expansion of adipose tissue capillary network induced by VEGFB (Figure 3B), while VEGFR2 deletion was incomplete and therefore not conclusive (Figure S11). Elevated levels of VEGFB or VEGFR1 deletion did not affect PlGF levels in adipose tissue (Figures S12A–S12D). These data indicate that in vivo, elevated levels of VEGFB and reduced levels of VEGFR1 can induce adipose tissue angiogenesis by engaging the VEGF/VEGFR2 angiogenic pathway.
Figure 3. VEGFR1 Deletion Boosts, whereas VEGF/VEGFR2 Blockage Abolishes the Effect of VEGFB on Adipose Tissue Vasculature.
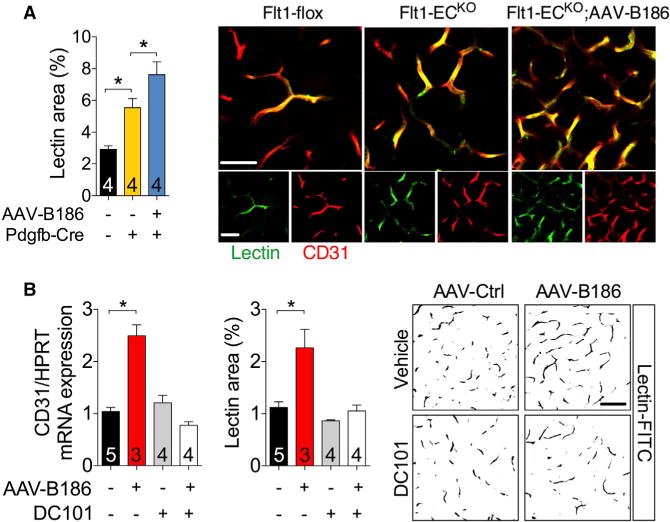
Mice were transduced with the indicated AAVs 2 weeks before analysis.
(A) Quantification of lectin-perfused vessel areas and the representative optical sections of eWAT from Flt1-flox and Flt1-ECKO mice.
(B) CD31 mRNA levels, quantification of lectinperfused areas, and the representative optical sections of eWAT from C57Bl/6JOlaHsd male mice that received DC101 or vehicle.
Scale bar, (A) 50 μm and (B) 100 μm. The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated using the one-way ANOVA, Holm-Sidak’s multiple comparisons test.
VEGFB-Induced Vascular Remodeling Improves Insulin Supply, Signaling, and Function in Obese Mice
To determine whether the VEGFB-induced capillary expansion is necessary for the improvement of glucose metabolism, we tested acute and chronic effects of VEGFB. Injection of VEGFB protein did not affect the insulin-induced decrease of blood glucose (Figure S13A); however, improved glucose tolerance was evident 4 weeks after the injection of AAV-B186 (Figure S13B) suggesting that vascular remodeling is required for the changes in glucose metabolism induced by VEGFB.
To gain further mechanistic insight on how increased capillary density induced by VEGFB is related to the improved glucose metabolism, we tested insulin supply, signaling, and function at the tissue level. We found that the AAV-B186 mice fed a HFD for 5 weeks had increased insulin levels and signaling in the eWAT, heart, and to some extent, liver (Figures 4A–4C). Accordingly, adipose tissue insulin sensitivity was retained in AAV-B186 mice, as indicated by a significant insulin-induced inhibition of the adipose tissue lipolytic rate, as evidenced by reduced serum glycerol (Figure 4D).
Figure 4. Elevated VEGFB Levels Increase Insulin Delivery, Signaling, and Function and Improve Insulin Sensitivity in HFD-Fed Mice.
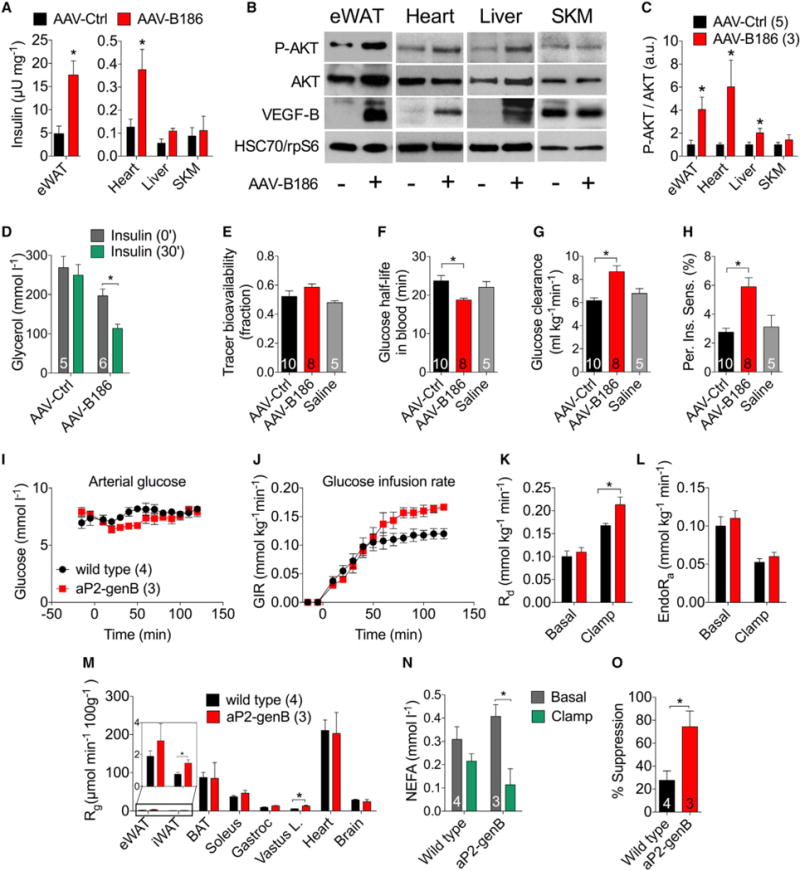
C57Bl/6JOlaHsd (A–F) or apoE*3Leiden;hCETP-Tg (G–K) males transduced with AAVs or Vegfb transgenic males (I–O) were fed a HFD and used for the experiments. Mice were injected with 0.75 U/kg insulin and analyzed after 10 min (A–C) or 30 min (D–F).
(A) Human insulin concentration in mouse tissues measured by an ultrasensitive ELISA.
(B and C) Western blot images (B) and quantifications using ImageJ (C). HSC70 or rpS6 were used as loading controls.
(D) Serum levels of glycerol.
(E–H) Bioavailability of [U-13C]-glucose (E) and analysis of tracer half-life (F), glucose clearance rate (G), and peripheral insulin sensitivity (H).
(I and J) Arterial glucose (I) and GIR (J) during the IC.
(K and L) Rd (K) and EndoRa (L) during the IC.
(M) Rg after the IC in the eWAT, iWAT, BAT, soleus, gastrocnemius, vastus lateralis, heart, and brain.
(N) Non-esterified fatty acid (NEFA) levels in the basal and hyperinsulinemic (clamp) states.
(O) Calculated suppression of adipose tissue lipolysis by hyperinsulinemia.
The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated with two-tailed unpaired t test (A, C, K, M, and O), two-tailed paired t test (D and N), or one-way ANOVA, Holm-Sidak’s multiple comparisons test (E–H).
To analyze the effect of VEGFB on glucose metabolism and insulin action in more detail, we performed experiments using a glucose tracer in an established preclinical model of the metabolic syndrome, the apoE*3Leiden;hCETP-Tg mouse (van den Hoek et al., 2014). Similar to the results obtained using wildtype mice, the apoE*3Leiden;hCETP-Tg mice transduced with VEGFB had significantly lower fasting insulin levels and improved glucose tolerance (Figures S14A–S14C). We then injected [U-13C]-glucose to estimate whole-body glucose metabolism with minimal perturbation of the steady-state metabolism (van Dijk et al., 2013). The bioavailability of the isotope was similar among the three groups, but glucose half-life was significantly reduced in the VEGFB-transduced mice (Figures 4E and 4F). Moreover, VEGFB increased glucose clearance rate and peripheral insulin sensitivity (Figures 4G and 4H).
A more comprehensive measurement of the insulin sensitivity can be achieved by employing the hyperinsulinemic-euglycemic clamp (IC) with isotopic glucose tracers. For these studies, we used HFD-fed aP2-genB and wild-type littermates according to an established protocol (Ayala et al., 2011; Ayala et al., 2006). Basal and clamp glucose levels were similar in wild-type and aP2-genB mice (Figure 4I). The aP2-genB mice were more sensitive to insulin than were their wild-type littermates, as evidenced by the 30% higher glucose infusion rate (GIR) (Figure 4J). The increased GIR during the clamp was due to an increase in glucose disappearance (Rd) calculated using arterial [3-3H] glucose dilution (Figure 4K). The rate of endogenous glucose appearance (endoRa) was not affected by aP2-driven expression of VEGFB (Figure 4L). The increased Rd was supported by the increased glucose metabolic index (Rg) in muscle and inguinal fat calculated from intravenous 2-[14C]deoxyglucose (2DG) and the tissue accumulation of phosphorylated 2DG (Figure 4M). Similar to the observations in AAV-B186-transduced mice, the adipose tissue insulin sensitivity was greatly improved, because hyperinsulinemia effectively reduced the non-esterified fatty acid levels in the serum (Figures 4N and 4O).
In summary, these results demonstrate that the insulin sensitizing effects of VEGFB are reproducible in three independent tests of insulin action in three independent laboratories.
Endothelial Deletion of VEGFR1 Enhances the Metabolic Effects of VEGFB
VEGFB binds to VEGFR1 and to the NRP-1 co-receptor (Bry et al., 2014), whereas the VEGFB exon 1-5 (Bex1–5) encoded protein lacks NRP-1 binding (Makinen et al., 1999). To analyze whether NRP-1 binding by VEGFB is required for the observed phenotypes, we performed a HFD study comparing AAV-B186, AAV-B167, and AAV-Bex1–5. After 6 weeks of HFD, the mice in all three groups had a significant reduction in fasting insulin and improved glucose tolerance (Figures S15A–S15F). Furthermore, AAV-Bex1–5-transduced mice had increased vessel density and reduced inflammation in adipose tissue (Figures S15G–S15K), indicating that NRP-1 binding was not required.
Mice with partial deletion of VEGFR1 induced by three different Cre lines showed improved glucose tolerance, which was potentiated by Vegfb gene transfer (Figure 5A). These findings prompted us to study the metabolic effects of VEGFR1 deletion with or without VEGFB transduction. Partial VEGFR1 deletion rendered mice resistant to diet-induced obesity, and transduction of these mice with AAV-B186 boosted the phenotype (see Figure 5B and Table S3 for body composition analyses). The phenotype was not explained by changes in food consumption or lipid excretion (Figure 5C; Figure S14A). Although fasting serum glucose was only modestly reduced in the Flt1-ECKO mice (Figure S16B), there was robust reduction of fasting serum insulin and improved tolerance to glucose and insulin (Figures 5D–5F; Figures S16B–S16D). Moreover, adipose tissue response to insulin was preserved in the Flt1-ECKO mice transduced with AAV-B186 (Figure 5G). Necropsy showed that Flt1-ECKO;AAV-B186 mice had less abdominal adipose tissue and liver fat than control mice (Figure 5H). This was confirmed using biochemical and histological analysis (Figures 5I and 5J; Figure S16E). Furthermore, serum lipid analysis showed a significant reduction of cholesterol and triglycerides in the Flt1-ECKO;AAV-B186 mice (Figures S16F and S16G).
Figure 5. Resistance to Diet-Induced Obesity and Associated Metabolic Complications in Flt1-ECKO Mice Transduced with AAV-B186.
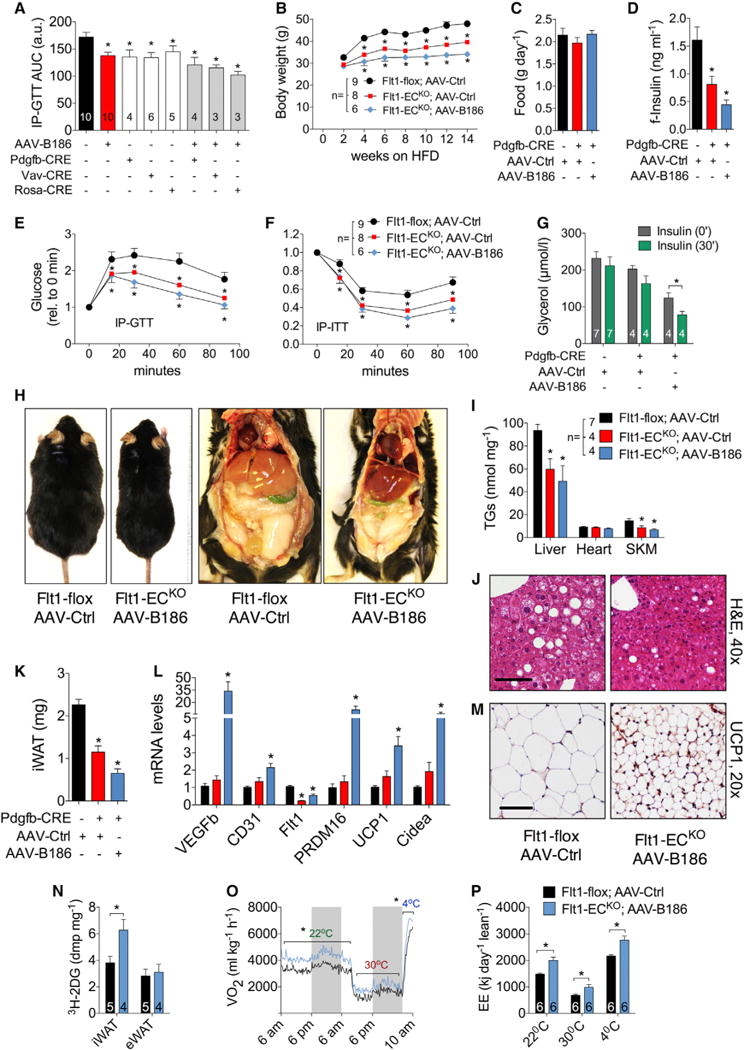
Flt1-flox male mice on a standard diet (A) or HFD (B–P) were used for the experiments. The mice in control groups were transduced with AAV-Ctrl.
(A) Area under curve from IP-GTT performed in the indicated Flt1-flox;Cre transgenic mouse lines treated with tamoxifen and transduced with AAV-Ctrl or AAV-B186.
(B and C) Body weight (B) and food consumption (C) measurements.
(D–F) Fasting serum insulin (D), IP-GTT (E), and IP-ITT (F) for 6–7 weeks of HFD.
(G) Adipose tissue response to insulin.
(H) Representative necropsy images.
(I) Triglyceride levels in tissues.
(J) H&E staining of paraffin sections from the liver.
(K–M) iWAT weight (K), gene expression (L), and UCP-1 immunohistochemistry (M).
(N) Insulin stimulated 2DG uptake in iWAT and eWAT.
(O and P) Oxygen consumption (O) and energy expenditure (P) assessed using metabolic cages.
Scale bar, 50 μm. The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated with one-way ANOVA, Holm-Sidak’s multiple comparisons test (A–H and J) or two-tailed paired t test (I) when compared to the untreated group (insulin, 0 min).
These analyses revealed that the Flt1-ECKO;AAV-B186 mice fed a HFD are lean and metabolically healthy, but they did not explain the mechanism behind their resistance to diet-induced obesity. The most striking reduction in fat pad weight occurred in subcutaneous WAT (iWAT), which had small adipocytes and increased vascular density (Figure 5K; Figures S16H and S16I). Gene expression analysis revealed that besides the increased expression of the endothelial marker CD31, there was strong induction of thermogenic genes, such as PRDM16, UCP-1, and Cidea (Figure 5L). Increased UCP-1 expression was evident also by immunohistochemistry (Figure 5M). Furthermore, Flt1-ECKO;AAV-B186 mice had a significantly increased 2DG uptake in iWAT and heart (Figure 5N; Figure S16J). To determine whether these changes increased the whole-body basal metabolic rate, we performed indirect calorimetry analyses after 3 weeks of HFD feeding, before overt changes in body weight occurred. The Flt1-ECKO;AAV-B186 mice showed increased O2 consumption, CO2 production, and energy expenditure independent of the ambient temperature, without changes in the respiratory exchange ratio or locomotor activity (Figures 5O and 5P; Figures S16K–S16M). Increased metabolic activity was also reflected by increased body temperature in Flt1-ECKO;AAV-B186 mice at room temperature and after acute cold exposure (Figure S16N).
These results demonstrate that the VEGFB/VEGFR1 pathway can be employed to induce adipose tissue angiogenesis via activation of the VEGF/VEGFR2 pathway and that this can act as a fat-burning switch that increases the basal metabolic rate and protects mice from diet-induced obesity and related metabolic complications.
Demonstration of the Therapeutic Potential of the VEGFB/VEGFR1 Pathway in Obese Mice
Next, we studied whether the VEGFB gene transfer can be used as a therapy for insulin-resistant obese mice. For this, we used mice fed a HFD for 7 weeks that were obese and glucose and insulin intolerant. We transduced the mice with AAV-Ctrl or AAV-B186, continued HFD feeding for an additional 6 weeks, and reassessed their weight gain and response to IP-GTT and IP-ITT (Figure 6A). Vegfb transduction had no effect on obesity but reverted the glucose intolerance and delayed the progression of the insulin intolerance (Figures 6B–6F).
Figure 6. Therapeutic Potential of the VEGFB/VEGFR1 Pathway in Obesity.
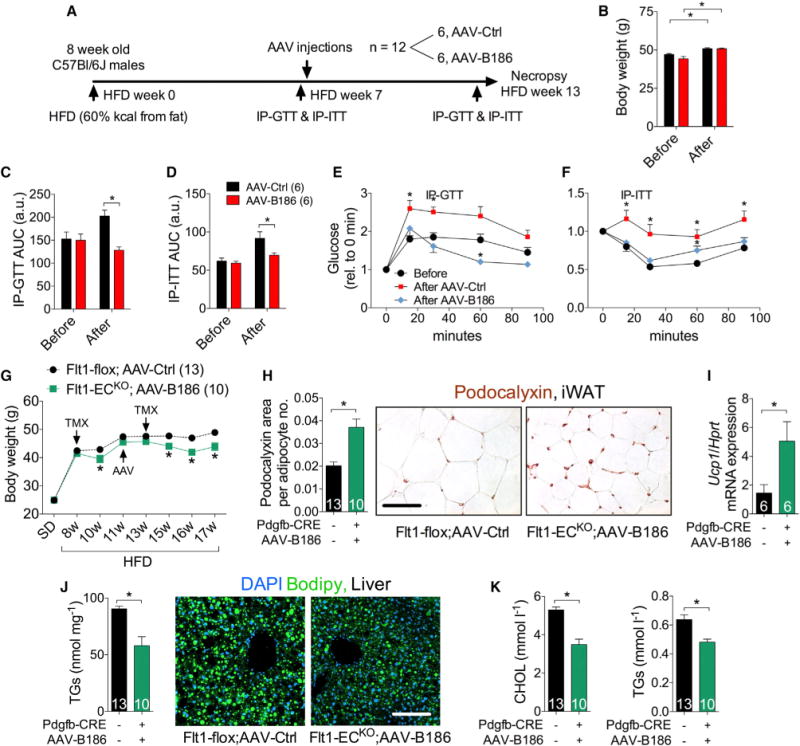
HFD-fed C57BL/6JOlaHsd (A–F) and Flt1-flox (G–I) males were used for the experiments.
(A) Experimental design to assess the therapeutic potential of AAV-B186.
(B) Body weight before and 6 weeks after AAV injections.
(C–F) AUC and IP-GTT and IP-ITT
(G) Body weight monitoring and therapeutic strategy. Tamoxifen treatment (TMX) was administered during two periods, as indicated.
(H) iWAT capillary density analysis by podocalyxin staining. The podocalyxin area was normalized to the number of adipocytes per field to account for the reduction in adipocyte size.
(I) Relative iWAT UCP-1 mRNA levels measured by RT-PCR.
(J) Quantification of liver triglyceride levels and images of liver lipid droplets stained with Bodipy.
(K) Serum levels of cholesterol (CHOL) and triglycerides (TGs).
Scale bar, 50 μm. The number of mice is indicated in the figure. Data are represented as mean ± SEM. *p < 0.05, calculated with two-tailed paired t test in (B, E, and F) and two-tailed unpaired t test in (C, D, and G–K).
The availability of the Flt1-flox mice allowed us to determine whether the thermogenic capacity of white adipose tissue can be induced in obese mice via the VEGFB/VEGFR1 pathway. Two sets of tamoxifen treatment were necessary to achieve a sustainable reduction of the body weight in the Flt1-flox mice, likely because of their older age and obesity (Figure 6G). The weight loss was accompanied by a significant increase in iWAT vascularity and UCP-1 mRNA expression (Figures 6H and 6I). Moreover, VEGFR1 deletion and Vegfb gene therapy alleviated ectopic lipid accumulation and dyslipidemia (Figures 6J and 6K), further highlighting the therapeutic potential of VEGFB/VEGFR1 pathway in obesity.
DISCUSSION
Here, we demonstrate that in adipose tissue, VEGFB/VEGFR1 provides a decoy pathway for VEGF/VEGFR2, which can be engaged to restore metabolic balance in obesity and holds promise for treating obesity. Vegfb gene transfer increased adipose tissue vascularity and perfusion, which was sufficient to enhance insulin delivery and function in adipose tissue, resulting in reduced inflammation and improvement of metabolic health in obesity. The surprising finding that Vegfb can enhance adipose tissue vascularity provides a safer therapeutic alternative to VEGF, because even high doses of VEGFB are well tolerated in preclinical models (Bry et al., 2014). Although these results are in line with evidence from VEGF transgenic mice, which show that angiogenesis is a promising therapeutic strategy to treat obesity, VEGF is difficult to dose due to its severe side effects that can adversely affect whole-body vasculature and metabolism (Elias et al., 2012; Sun et al., 2012; Sung et al., 2013). It is important to use species-specific VEGF in such experiments to properly evaluate its therapeutic potential (Mujagic et al., 2013).
It is well established that adipose tissue inflammation is a major contributor to systemic insulin resistance in obesity and type 2 diabetes (Glass and Olefsky, 2012). Suboptimal vascularization and perfusion of adipose tissue can limit adipose tissue expansion capacity and provoke inflammation, thus promoting the systemic metabolic complications (Virtue and Vidal-Puig, 2010). The finding that VEGFB increased vascular perfusion and improved insulin delivery and function in obese adipose tissue could prove important for the treatment of type 2 diabetes, because increased availability of insulin to target organs improves insulin sensitivity. Insulin is also a vasoactive hormone, which induces capillary recruitment and increases blood flow to facilitate glucose uptake in target tissues. Recent data show that deficient vascularization of skeletal muscle induces insulin resistance in lean mice (Bonner et al., 2013). In humans, a meta-analysis of genome-wide association studies showed a relationship between angiogenesis and insulin resistance, further supporting the notion that chronic tissue ischemia has an important role in insulin resistance (Shungin et al., 2015). Engaging VEGFB/VEGFR1 to improve perfusion in skeletal muscle and liver, in addition to adipose tissue, could be of further benefit for type 2 diabetic patients (Zheng and Liu, 2015).
VEGFB can be considered a VEGFR1 antagonist, because the primary physiological role of the soluble and membrane-bound VEGFR1 is to serve as a sink for VEGF (Shibuya, 2013). The finding that partial deletion of VEGFR1 enhanced the angiogenic potential of VEGFB is consistent with the concept that VEGFR1 downregulation further increased VEGF availability for VEGFR2 activation (Anisimov et al., 2013; Bry et al., 2014). In addition, VEGFR1 deletion in endothelial cells may increase VEGFR2 homodimers by decreasing VEGFR1/VEGFR2 heterodimers, thereby enhancing the angiogenic signaling activity of VEGF (Cudmore et al., 2012). However, additional mechanisms cannot be fully excluded, because angiogenesis is regulated by highly complex interactions among VEGFRs, VEGFs, extracellular matrix components, and co-receptors, as well as by alternative splicing, heterodimer formation, and proteolytic processing of the signaling components.
Downregulation of VEGFR1, together with increased VEGFB levels, unleashed the thermogenic capacity of subcutaneous adipose tissue, providing a promising therapeutic strategy for counteracting obesity (Bartelt and Heeren, 2014; Harms and Seale, 2013; Rosen and Spiegelman, 2014). An important further step is to identify the mechanism or mechanisms of how vascular remodeling and increased tissue perfusion induce the heat-dissipating beige adipocytes in the subcutaneous adipose tissue.
There are several possible reasons for the discrepancies between our results and those published by Hagberg et al. (2010, 2012). The authors do not mention whether littermates were used as a control group. Replication of these studies by Dijkstra et al. (2014) using wild-type littermates from the same Vegfb gene-targeted mouse strain also failed to reproduce the results. Furthermore, the 2H10 antibody used in mice and rats has been demonstrated to inhibit human VEGFB binding to VEGFR1 in vitro but not in vivo, and it is not known whether this antibody inhibits both isoforms of VEGFB in mice. If 2H10 disrupts the VEGFB/VEGFR1 interaction in vivo, this would facilitate VEGF binding to VEGFR1, inhibiting the vascular VEGFR2 signals that modulate glucose metabolism in the liver, as shown recently (Wei et al., 2013). Because of the rapid fatty acid turnover, analysis performed by Hagberg et al. (2010) 2 and 24 hr after administration should have employed fatty acid analogs resistant to the metabolic breakdown by β-oxidation (Shearer et al., 2008). Furthermore, administration of fatty acids by oral gavage results in their incorporation into lipoproteins, which may complicate the interpretation of the data, because the rate-limiting step for fatty acid release from triglyceride is lipoprotein lipase activity, while CD36 is the primary cellular fatty acid translocase (Goldberg et al., 2009).
Thiazolidinediones are the only current anti-diabetic agents that function primarily by increasing tissue insulin sensitivity (Soccio et al., 2014). Thiazolidinediones also increase VEGF levels, which could be one reason for the fluid retention and congestive heart failure associated with these drugs (Baba et al., 2001; Gealekman et al., 2008; Soccio et al., 2014; Sotiropoulos et al., 2006). In contrast, VEGFB would provide a cardiovascular protective effect while maintaining the stability of blood vessels (Aase et al., 2001; Bellomo et al., 2000; Bry et al., 2010; Huusko et al., 2012; Kivelä et al., 2014; Lähteenvuo et al., 2009; Li et al., 2008a; Pepe et al., 2010; Serpi et al., 2011; Zentilin et al., 2010). In preclinical studies, VEGFB was identified as a promising candidate for the treatment of myocardial ischemia (Kivelä et al., 2014; Lähteenvuo et al., 2009; Pepe et al., 2010). However, in humans, low levels of VEGFB predict left ventricular remodeling after acute myocardial infarction, and VEGFB expression is reduced in human cardiomyopathy (Devaux et al., 2012; Kivelä et al., 2014). VEGFB was also shown to induce survival of neurons and to reduce stroke volume in the middle cerebral artery ligation model (Li et al., 2008b; Sun et al., 2004). Because individuals with type 2 diabetes have a higher incidence of myocardial infarction and stroke, with worse outcomes than those for individuals without type 2 diabetes, it is exciting to find that VEGFB can also act as an insulin-sensitizing factor.
In conclusion, the VEGFB/VEGFR1 pathway can be employed to enhance the adipose tissue vascularity to improve metabolic health and combat obesity.
EXPERIMENTAL PROCEDURES
Animals
All animal experiments were approved by the Regional State Administrative Agency for Southern Finland. Mice were housed in individually ventilated cages with enrichment materials in a facility monitored by the Federation of European Laboratory Animal Science Associations guidelines and recommendations. Mice were fed a standard diet or a HFD containing 60% calories from fat (Research Diets, D12492). C57BL/6JOlaHsd male mice were used for AAV experiments. Flt1-fl/fl;Pdgfb-CreERT2 mice were maintained on a C57BL/6N background.
aP2-genB Mice
aP2-genB mice were generated by transgene microinjection into C57BL/6JOlaHsd zygotesand maintained on a C57BL/6JOlaHsd background by mating a transgenic with a wild-type mouse.
AVV Transduction
AAVs were administered by intraperitoneal (i.p.) injections at doses between 0.4 × 1011 and 2 × 1011 viral particles per mouse.
VEGF/VEGFR2 Blockade
Male mice were transduced with AAV-Ctrl or AAV-B186 and injected four times every 3–4 days with 0.8 mg per mouse of DC101 or vehicle. Analyzes were performed 2 weeks after transduction with AAVs.
Body Composition
Body composition measurements were performed using Lunar PIXImus dualenergy x-ray absorptiometry.
2DG Uptake
2DG uptake was determined in conscious and unrestrained mice by i.p. injection of 50 μCi/kg of2-[1,2-3H(N)]-deoxy-D-glucose, together with 0.75 U/kg insulin. Radioactivity from perfused tissues was measured by liquid scintillation.
In Vivo Adipose Tissue Lipolysis
Inhibition of adipose tissue lipolysis by insulin was assessed by quantification of serum glycerol levels before and 30 min after insulin injection (0.75 U/kg).
Whole-Body Glucose Test
[U-13C]-glucose (0.1 g/kg) was injected i.p., and blood glucose and tracer levels were monitored in blood to determine glucose fluxes, as described previously (van Dijk et al., 2013).
Analysis of Whole-Body Metabolism
Oxygen consumption, carbon dioxide production, and locomotor activity were recorded by the Oxymax Lab Animal Monitoring System at +22°C, +30°C, and +4°C.
IP-GTT and IP-ITT
Mice were fasted for 4–7 hr. For IP-GTT, glucose was injected i.p. to male mice fed a HFD (1 g/kg glucose) or a standard diet (2 g/kg glucose). For IP-ITT, 0.75 U/kg insulin was injected i.p. to HFD-fed mice. Glucose levels were measured from the tail tip puncture.
Hyperinsulinemic-Euglycemic IC
Catheters were implanted in a carotid artery and a jugular vein of mice for sampling and intravenous infusions 5 days before hyperinsulinemic-euglycemic IC (Ayalaet al., 2006). IC (2.5 mU·kg−1·min−1)was performed on mice that fasted for 5 hr, as previously described (Ayala et al., 2011).
ELISA
ELISA was used to quantify protein levels of mouse insulin (Crystal Chem), human insulin (10-1132-01, Mercodia), mouse VEGFR1 (DuoSet, DY471, R&D Systems), mouse PlGF (DY465, R&D Systems), and mouse VEGFB186 (inhouse ELISA based on R&D Systems antibodies; it does not recognize mouse VEGFB167).
Histology
Paraformaldehyde (PFA)-fixed tissues were analyzed from cryo-sections, from paraffin-sections, or as whole mounts. Images acquired with bright-field, slide scanner, epifluorescence, or confocal microscopes were analyzed using ImageJ software.
Vessel Perfusion and Leakage
Vessel perfusion and leakage were analyzed by intravenous injections of Labeled Tomato Lectin and FluoSpheres Carboxylate-Modified Microspheres, 0.1 μm. Optical sections from PFA-fixed tissues were taken with a confocal microscope.
Intravital Two-Photon Microscopy and Blood Flow Measurement
Animals were anesthetized with isoflurane, and respiration rate, heart rate, and body temperature were monitored and kept stable. eWAT was attached to a holder, and intravenously injected Texas Red 70 kDa dextran conjugate was imaged using a Zeiss Axio Examiner LSM7 MP microscope. Blood flow was measured with line scans at a 0.003–0.01 Hz repetition rate.
Lipid Analysis
Tissue lipid were extracted using a scaled-down Bligh & Dyer method and quantified with commercially available kits. For mass spectrometry, hepatic total lipids were extracted using the Folch method and analyzed using a triple quadrupole mass spectrometer.
Real-Time PCR
Real-time PCR was carried out following standard procedures using SYBR green or TaqMan primer-probe sets, and quantification was performed using the 2−ΔΔCT method.
Western Blotting and Immunoprecipitation
Partially defatted protein lysates were analyzed by a standard western blot procedure. VEGFB was immunoprecipitated from serum using purified hVEGFR1-Fc and protein G sepharose.
Statistics
The results are shown as mean ± SEM. All statistical analyses were performed by two-tailed unpaired t test for two-group comparisons (or a paired t test where appropriate) and one-way ANOVA for multiple groups (Prism, Graph-Pad). A p value of less than 0.05 was considered significant.
A detailed description of the methods is provided in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
VEGFB induces adipose tissue angiogenesis and improves glucose metabolism
VEGFB binding to VEGFR1 activates angiogenesis via the VEGF/VEGFR2 pathway
Increased adipose tissue perfusion improves insulin supply and function
VEGFB/VEGFR1 can promote weight loss and restore metabolic health in obese mice
Acknowledgments
We thank Andrey Anisimov and Harri Nurmi for valuable discussions and help with some experiments. Georgia Zarkada is acknowledged for her kind help with the acquisition and management of several mouse lines used in this study. We acknowledge Seppo Kaijalainen for cloning the aP2-genB construct and Dr. Napoleone Ferrara/Genentech for providing the Flt1-flox mice. Drs. Patrick Rensen and Ko Willems van Dijk (Leiden University Medical Centre, the Netherlands) are acknowledged for providing the apoE3*Leiden;hCETP-Tg mice. We acknowledge Mari Jokinen, Maija Atuegwu, Jarmo Koponen, Maria Arrano, Kirsi Lintula, Tanja Laakonen, Tapio Tainola, Sanna Sihvo, and Jaana Träskelin for expert technical assistance and Markus Räsänen and Shuo Chen for assistance in some experiments. We also thank the Biomedicum Imaging Unit, the AAV Gene Transfer and Cell Therapy Core Facility of Biocenter Finland, and Electron Microscopy Unit, Genome Biology Unit, and the Laboratory Animal Centre of the University of Helsinki for professional services. The Biocenter Oulu Transgenic Core Facility helped in generating the aP2-VEGFB mouse model. Funding was provided by the Wihuri Foundation, Academy of Finland (including TCB-CoE grant 271845), Cancer Society of Finland, Sigrid Juselius Foundation, European Research Council (ERC-2010-AdG-268804), Leducq Foundation (11CVD03), and European Union, FP7-HEALTH (305707). Support for the hyperinsulinemic-euglycemic IC in aP2-genB mice was provided by NIH grants U24 DK059637 and RO1 DK054902.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, sixteen figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.03.004.
AUTHOR CONTRIBUTIONS
M.R.R. designed and performed most of the experiments. R. Kivelä initially identified the VEGFB effect on adipose tissue vasculature, participated in study design, and helped in several experiments. I.M.W. and D.H.W. coordinated, designed, and performed the hyperinsulinemic-euglycemic IC in aP2-genB mice. J.F.d.B., T.H.v.D., and A.K.G. coordinated, designed, and performed the experiments using apoE3*Leiden;hCETP-Tg mice. H.E. and L.E. helped with generating the aP2-genB mice and initial characterization of the model. F.T.-S. and R. Käkelä generated lipidomic data from the liver. D.M. performed intravital microscopy and blood flow analysis. V.-M.L. produced the recombinant mouse albumin and VEGFB and contributed with important ideas. K.A. conceived and supervised the project.
References
- Aase K, von Euler G, Li X, Ponten A, Thorén P, Cao R, Cao Y, Olofsson B, Gebre-Medhin S, Pekny M, et al. Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation. 2001;104:358–364. doi: 10.1161/01.cir.104.3.358. [DOI] [PubMed] [Google Scholar]
- Anisimov A, Leppänen VM, Tvorogov D, Zarkada G, Jeltsch M, Holopainen T, Kaijalainen S, Alitalo K. The basis for the distinct biological activities of vascular endothelial growth factor receptor-1 ligands. Sci Signal. 2013;6:ra52. doi: 10.1126/scisignal.2003905. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, Malabanan C, James FD, Ansari T, Fueger PT, McGuinness OP, Wasserman DH. Hyperinsulinemic-euglyce-mic clamps in conscious, unrestrained mice. J Vis Exp. 2011;57:e3188. doi: 10.3791/3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Shimada K, Neugebauer S, Yamada D, Hashimoto S, Watanabe T. The oral insulin sensitizer, thiazolidinedione, increases plasma vascular endothelial growth factor in type 2 diabetic patients. Diabetes Care. 2001;24:953–954. doi: 10.2337/diacare.24.5.953. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- Bellomo D, Headrick JP, Silins GU, Paterson CA, Thomas PS, Gartside M, Mould A, Cahill MM, Tonks ID, Grimmond SM, et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res. 2000;86:E29–E35. doi: 10.1161/01.res.86.2.e29. [DOI] [PubMed] [Google Scholar]
- Bonner JS, Lantier L, Hasenour CM, James FD, Bracy DP, Wasserman DH. Muscle-specific vascular endothelial growth factor deletion induces muscle capillary rarefaction creating muscle insulin resistance. Diabetes. 2013;62:572–580. doi: 10.2337/db12-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bry M, Kivelä R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- Bry M, Kivelä R, Leppänen VM, Alitalo K. Vascular endothelial growth factor-B in physiology and disease. Physiol Rev. 2014;94:779–794. doi: 10.1152/physrev.00028.2013. [DOI] [PubMed] [Google Scholar]
- Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- Cudmore MJ, Hewett PW, Ahmad S, Wang KQ, Cai M, Al-Ani B, Fujisawa T, Ma B, Sissaoui S, Ramma W, et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat Commun. 2012;3:972. doi: 10.1038/ncomms1977. [DOI] [PubMed] [Google Scholar]
- Devaux Y, Vausort M, Azuaje F, Vaillant M, Lair ML, Gayat E, Lassus J, Ng LL, Kelly D, Wagner DR, Squire IB. Low levels of vascular endothelial growth factor B predict left ventricular remodeling after acute myocardial infarction. J Card Fail. 2012;18:330–337. doi: 10.1016/j.cardfail.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Dijkstra MH, Pirinen E, Huusko J, Kivelä R, Schenkwein D, Alitalo K, Ylä-Herttuala S. Lack of cardiac and high-fat diet induced metabolic phenotypes in two independent strains of Vegf-b knockout mice. Sci Rep. 2014;4:6238. doi: 10.1038/srep06238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias I, Franckhauser S, Ferré T, Vilà L, Tafuro S, Muñoz S, Roca C, Ramos D, Pujol A, Riu E, et al. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61:1801–1813. doi: 10.2337/db11-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gealekman O, Burkart A, Chouinard M, Nicoloro SM, Straubhaar J, Corvera S. Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am J Physiol Endocrinol Metab. 2008;295:E1056–E1064. doi: 10.1152/ajpendo.90345.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50(Suppl):S86–S90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsäter H, Scotney P, Nyqvist D, Samén E, Lu L, et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426–430. doi: 10.1038/nature11464. [DOI] [PubMed] [Google Scholar]
- Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6:209. doi: 10.1186/gb-2005-6-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko J, Lottonen L, Merentie M, Gurzeler E, Anisimov A, Miyanohara A, Alitalo K, Tavi P, Ylä-Herttuala S. AAV9-mediated VEGF-B gene transfer improves systolic function in progressive left ventricular hypertrophy. Mol Ther. 2012;20:2212–2221. doi: 10.1038/mt.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivelä R, Bry M, Robciuc MR, Räsänen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mäyränpää MI, et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307–321. doi: 10.1002/emmm.201303147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2:a006502. doi: 10.1101/cshperspect.a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- Lähteenvuo JE, Lähteenvuo MT, Kivelä A, Rosenlew C, Falkevall A, Klar J, Heikura T, Rissanen TT, Vähäkangas E, Korpisalo P, et al. Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis viavascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation. 2009;119:845–856. doi: 10.1161/CIRCULATIONAHA.108.816454. [DOI] [PubMed] [Google Scholar]
- Li X, Tjwa M, Van Hove I, Enholm B, Neven E, Paavonen K, Jeltsch M, Juan TD, Sievers RE, Chorianopoulos E, et al. Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler Thromb Vasc Biol. 2008a;28:1614–1620. doi: 10.1161/ATVBAHA.107.158725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, et al. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. 2008b;118:913–923. doi: 10.1172/JCI33673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen T, Olofsson B, Karpanen T, Hellman U, Soker S, Klagsbrun M, Eriksson U, Alitalo K. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J Biol Chem. 1999;274:21217–21222. doi: 10.1074/jbc.274.30.21217. [DOI] [PubMed] [Google Scholar]
- Mujagic E, Gianni-Barrera R, Trani M, Patel A, Gurke L, Heberer M, Wolff T, Banfi A. Induction of aberrant vascular growth, but not of normal angiogenesis, by cell-based expression of different doses of human and mouse VEGF is species-dependent. Hum Gene Ther Methods. 2013;24:28–37. doi: 10.1089/hgtb.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellet-Many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem J. 2008;411:211–226. doi: 10.1042/BJ20071639. [DOI] [PubMed] [Google Scholar]
- Pepe M, Mamdani M, Zentilin L, Csiszar A, Qanud K, Zacchigna S, Ungvari Z, Puligadda U, Moimas S, Xu X, et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ Res. 2010;106:1893–1903. doi: 10.1161/CIRCRESAHA.110.220855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpi R, Tolonen AM, Huusko J, Rysä J, Tenhunen O, Ylä-Herttuala S, Ruskoaho H. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc Res. 2011;89:204–213. doi: 10.1093/cvr/cvq267. [DOI] [PubMed] [Google Scholar]
- Shearer J, Coenen KR, Pencek RR, Swift LL, Wasserman DH, Rottman JN. Long chain fatty acid uptake in vivo: comparison of [125I]-BMIPP and [3H]-bromopalmitate. Lipids. 2008;43:703–711. doi: 10.1007/s11745-008-3183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13–19. doi: 10.1093/jb/mvs136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. ADIPOGen Consortium; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GEFOS Consortium; GENIE Consortium; GLGC; ICBP; International Endogene Consortium; LifeLines Cohort Study; MAGIC Investigators; MuTHER Consortium; PAGE Consortium; ReproGen Consortium New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos KB, Clermont A, Yasuda Y, Rask-Madsen C, Mastumoto M, Takahashi J, Della Vecchia K, Kondo T, Aiello LP, King GL. Adipose-specific effect of rosiglitazone on vascular permeability and protein kinase C activation: novel mechanism for PPARgamma agonist’s effects on edema and weight gain. FASEB J. 2006;20:1203–1205. doi: 10.1096/fj.05-4617fje. [DOI] [PubMed] [Google Scholar]
- Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, Scherer PE. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci USA. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Luby-Phelps K, Spurgin SB, An YA, Wang QA, Holland WL, Scherer PE. Brown adipose tissue derived VEGF-A modulates cold tolerance and energy expenditure. Mol Metab. 2014;3:474–483. doi: 10.1016/j.molmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA. Increased severity of cerebral ischemic injury in vascular endothelial growth factor-B-deficient mice. J Cereb Blood Flow Metab. 2004;24:1146–1152. doi: 10.1097/01.WCB.0000134477.38980.38. [DOI] [PubMed] [Google Scholar]
- Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, Nelson SM, Cowling R, Nagy K, Michael IP, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17:61–72. doi: 10.1016/j.cmet.2012.12.010. [DOI] [PubMed] [Google Scholar]
- van den Hoek AM, van der Hoorn JW, Maas AC, van den Hoogen RM, van Nieuwkoop A, Droog S, Offerman EH, Pieterman EJ, Havekes LM, Princen HM. APOE*3Leiden.CETP transgenic mice as model for pharmaceutical treatment of the metabolic syndrome. Diabetes Obes Metab. 2014;16:537–544. doi: 10.1111/dom.12252. [DOI] [PubMed] [Google Scholar]
- van Dijk TH, Laskewitz AJ, Grefhorst A, Boer TS, Bloks VW, Kuipers F, Groen AK, Reijngoud DJ. A novel approach to monitor glucose metabolism using stable isotopically labelled glucose in longitudinal studies in mice. Lab Anim. 2013;47:79–88. doi: 10.1177/0023677212473714. [DOI] [PubMed] [Google Scholar]
- Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome—an allostatic perspective. Biochim Biophys Acta. 2010;1801:338–349. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Wei K, Piecewicz SM, McGinnis LM, Taniguchi CM, Wiegand SJ, Anderson K, Chan CW, Mulligan KX, Kuo D, Yuan J, et al. A liver Hif-2alpha-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat Med. 2013;19:1331–1337. doi: 10.1038/nm.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, Coday M, Crow RS, Curtis JM, Egan CM, Espeland MA, et al. Look AHEAD Research Group Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145–154. doi: 10.1056/NEJMoa1212914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentilin L, Puligadda U, Lionetti V, Zacchigna S, Collesi C, Pattarini L, Ruozi G, Camporesi S, Sinagra G, Pepe M, et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 2010;24:1467–1478. doi: 10.1096/fj.09-143180. [DOI] [PubMed] [Google Scholar]
- Zheng C, Liu Z. Vascular function, insulin action, and exercise: an intricate interplay. Trends Endocrinol Metab. 2015;26:297–304. doi: 10.1016/j.tem.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.