
Figure 5.
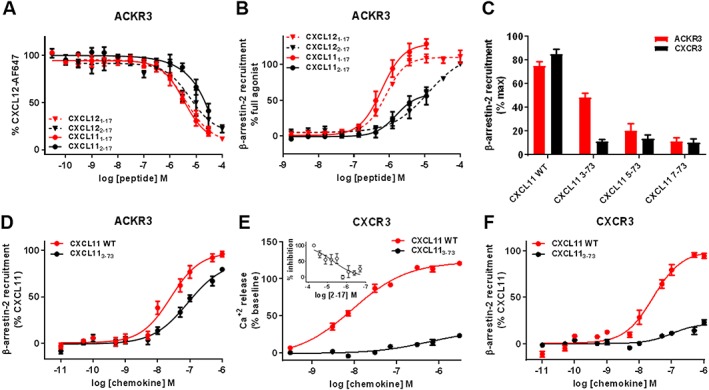
β‐arrestin‐2 recruitment and G protein signalling induced by N‐terminally truncated CXCL11 variants towards ACKR3 and CXCR3. (A and B) Comparison of the effects of N‐terminal residue truncation in CXCL12‐ and CXCL11‐derived peptides on (A) binding to ACKR3 assessed by competition studies with Alexa Fluor 647‐coupled CXCL12 and (B) β‐arrestin‐2 recruitment to ACKR3 monitored by NanoLuc complementation in U87 cells. (C) Impact of progressive N‐terminal truncation on the ability of CXCL11 (100 nM) to recruit β‐arrestin‐2 to ACKR3 and CXCR3. Values are expressed as percentage of the maximum β‐arrestin‐2 recruitment monitored with saturating concentrations of CXCL11WT. (D and F) Comparison of β‐arrestin‐2 recruitment to ACKR3 (D) and CXCR3 (F) induced by CXCL11WT and variants lacking the first two residues (CXCL113–73) monitored by BRET. (E) Comparison of maximum CXCR3 mediated intracellular calcium release induced by CXCL11WT and CXCL113–73 variant in HEK cells using FLIPR 4 Calcium Flux kit dye. (E‐inset) Antagonist properties of peptide CXCL112–17 monitored in calcium assay. Each experiment was performed five times, and the data shown are means ± SEM.